

Abstract
There are currently no drugs to treat neurodegeneration in Parkinson’s disease (PD), and all existing medications only treat symptoms, lose efficacy over time, and produce untoward side effects. In the current work, we report the first highly selective, orally bioavailable c-jun-N-terminal kinase (JNK) inhibitor for protection of dopaminergic neurons in vitro and in vivo. At 300 nM, this compound showed statistically significant protection of primary dopaminergic neurons exposed to 1-methyl-4-phenylpyridinium (MPP+), had pharmacokinetic properties in rodents consistent with twice daily (b.i.d.) dosing, and was orally efficacious at 30 mg/kg in a mouse 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) model of Parkinson’s disease. Moreover, a dose-dependent target modulation of c-jun phosphorylation served as a biomarker for demonstrating on-target inhibition of JNK as the mechanism of action for this compound. Collectively, these results suggest that this JNK inhibitor could be a promising therapeutic neuroprotective agent in the treatment of Parkinson’s disease.
Keywords: JNK, MPTP, neuroprotection, Parkinsonʼs disease
Parkinson’s disease (PD) is a neurodegenerative disorder, whose cardinal features include bradykinesia, rigidity, and tremor.1−3 Pathologically, it is characterized by the loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) and loss of projecting nerve fibers in the striatum. Currently, all therapies for Parkinson’s disease are symptomatic treatment, with the mainstay being the use of levodopa (L-DOPA) which initially has response rates nearing 90%.2,3 This drug unfortunately loses effect over time and produces dyskinesias which are problematic. Importantly, however, there are no treatments, including L-DOPA, which have been shown to be neuroprotective. Indeed, a recent double blind, delayed-start phase III clinical trial utilizing a 2 mg dose of rasagiline in 1176 subjects with untreated Parkinson’s disease failed to show neuroprotective disease-modifying effects, whereas 1 mg showed possible disease-modifying effects, thereby leaving great caution in the positive interpretation of these results.4 Thus, any orally available compounds, which demonstrate neuroprotective disease-modifying effects, would be a significant innovation in the treatment of Parkinson’s disease.
The mitogen-activated protein (MAP) kinase family member c-jun-N-terminal kinase (JNK) is a promising target that has emerged over the past decade and may be a unique candidate for neuroprotection in PD. Indeed, Flavell and colleagues found increased levels of phospho-c-jun (p-c-jun) in post mortem analysis of PD patient brains compared to matched control patients.5 In 2001, Schulz and colleagues validated JNK as a target for PD in the chronic 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model by utilizing adenovirally mediated gene transfer of a 154 amino acid portion of JNK-interacting protein (JIP) by stereotactic injection into the left striatum. This JNK inhibitor was found to have neuroprotective benefit by increasing the number of tyrosine hydroxylase (TH) positive neurons in the SNpc to ∼86% of the saline control.6 In 2004, Flavell colleagues showed that both jnk2 and jnk3 knockout (KO) mice demonstrated a protective effect against acute MPTP-intoxication compared to both wild type and jnk1 knockout mice which showed no protective effect against MPTP-intoxication. The most compelling data in the report showed that jnk2/3−/− mice were dramatically protected against acute MPTP-induced nigrostriatal pathway injury compared to wild-type mice. This protective effect manifested in a 3-fold increase in the number of TH-positive neurons, again increasing the number of TH-positive neurons in the SNpc to ∼83% of the saline control.5 In the same year, Wang et al. showed that intraperitoneal injection of a nonselective JNK inhibitor, SP600125, also was protective in the chronic MPTP mouse model.7
We recently reported the synthesis, biological characterization, and pharmacokinetics of a series of aminopyrimidine JNK inhibitors which are highly brain penetrant.8 A new species from this class, SR-3306, is presented in this work, having improved drug metabolism properties compared to those previously reported. Crucially, this compound showed protection of primary dopaminergic neurons exposed to MPP+ in vitro and protection of dopaminergic neurons in the SNpc as measured by TH-immunoreactivity in vivo. Moreover, a dose-dependent target modulation of c-jun phosphorylation served as a biomarker for demonstrating on-target inhibition of JNK as the mechanism of action for this compound. Finally, it should be noted that numerous compounds from various mechanisms (e.g., nitric oxide synthase,9,10 caspase,11 and calpain12) of action have been shown to be efficacious in MPTP models, yet none of them were delivered orally. Thus, this is the first JNK inhibitor to show oral efficacy in a model of PD, and perhaps it represents one of the few orally available compounds showing efficacy in PD models.
Results
Biochemical and Cell-Based Potency and Selectivity of SR-3306
The synthesis of SR-3306 is described in Figure 1 in the Supporting Information. Table 1 presents the structure of SR-3306, biochemical IC50 values against human JNK3, JNK2, JNK1, and p38, along with the cell-based IC50 for inhibition of c-jun phosphorylation in INS-1 cells treated with streptozotocin (STZ). SR-3306 was a modestly potent ATP-competitive inhibitor (as determined by X-ray crystallography, data not shown) of JNK3 and JNK2 with an IC50 value near 200 nM and had >100-fold selectivity over p38, the most closely related MAP kinase family member (Table 1). Moreover, the cell-based IC50 was also near 200 nM, suggesting good cell penetration and potency. To assess the kinase selectivity of our compound, the interaction of 3 μM SR-3306 was profiled in an Ambit panel screen13,14 against 347 kinases and the compound was shown to be highly selective (Supporting Information Table 1). Indeed, only 35 out of the 347 (∼10%) had any hit at all, while only four kinases (KIT, KIT V559D, PDGFR-β, TYK2) (∼1.1%) had potency suggesting Kd values of <1 μM. Moreover, leucine rich repeat kinase 2 (LRRK2) and G2019S LRRK2 were not inhibited by up to 10 μM SR-3306. SR-3306 was also evaluated for its ability to inhibit the potassium channel, hERG, as well as nine cytochrome (CYP) P450 enzymes (1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, and 3A4). The results showed that SR-3306 had IC50 values of >30 μM versus hERG and >50 μM versus each of the nine CYP450s. This was also true for monoamine oxidase A (MAO-A) and monoamine oxidase B (MAO-B), where the IC50 values for each were >50 μM. Finally, a panel of 67 receptors, ion channels, and transporters (MDS Pharma lead profiling screen) were also assessed to determine the broad selectivity and drug development desirability of the amino pyrimidine class. An analogue of SR-3306 (compound 9f from Kamenecka et al.8) was assessed. A total of 65 of the 67 receptors, ion channels, and transporters (including the dopamine, serotonin, and norepinephrine transporters) showed less than 30% inhibition when challenged with 3 μM compound 9f (also referred to as SR-2502) (Supporting Information Table 2). Only the adenosine A3 receptor and the adenosine A2A receptor showed >50% inhibition at 3 μM (93% and 55%, respectively).
Table 1. Structure and Biochemical and Cell-Based Inhibition for SR-3306a.
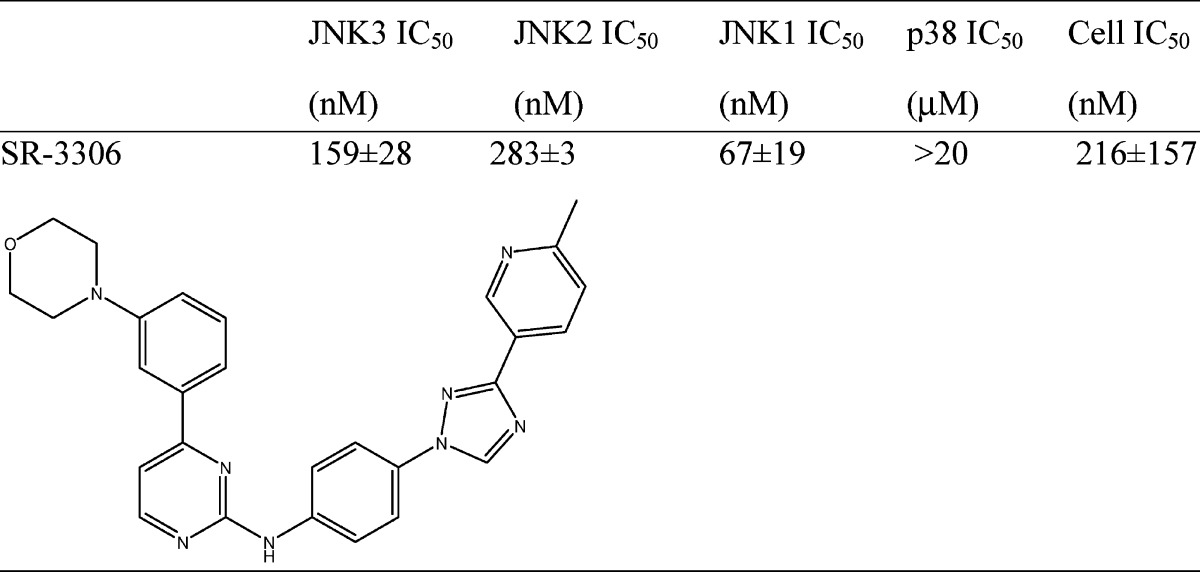 |
The biochemical IC50 values ± standard deviation are the average of 16 experiments for JNK3, 2 experiments for JNK2, 6 experiments for JNK1, 2 experiments for p38, and 6 experiments for the cell-based IC50 (p-c-jun inhibition). All enzymes used were human recombinant enzymes.
Drug Metabolism and Pharmacokinetics of SR-3306
In addition to the biochemical and cell-based inhibition parameters for SR-3306, we also evaluated the drug metabolism and pharmacokinetic (DMPK) properties of the compound. The microsomal stability in mouse, rat, and human liver microsomes was about the same for each species (26, 30, and 28 min, respectively) and was suggestive of a compound with a reasonable metabolism profile. We also determined the plasma protein binding in mouse, rat, monkey, and human and these values were found to be (86%, 97%, 96%, and 88%, respectively). Finally, the pharmacokinetic parameters for SR-3306 dosed at 1 mg/kg i.v. and 2 mg/kg p.o. in rats is presented in Supporting Information Table 3. SR-3306 had a relatively low clearance rate of 14 mL/min/kg and reasonable oral bioavailability (%F = 31). Collectively, these data suggest SR-3306 is a highly selective drug development candidate, with reasonable DMPK properties in rodents.
In Vitro Neuroprotection of SR-3306 in Primary Cultures of Mesencephalic Dopaminergic Neurons
To assess the ability of SR-3306 to be an effective agent in neuroprotective strategies in PD, we first tested to see if the compound would be able to protect primary dopaminergic neurons in culture against MPP+-induced cell death. Primary cultures of mesencephalic dopaminergic neurons treated with MPP+ have been successfully used to evaluate the protective effect of caspase inhibitors,15,16 and this method was employed in our work. Figure 1 presents the number of TH-positive neurons (a measure of viable dopaminergic neurons) in the absence (control) and presence of 10 μM MPP+, along with cells treated with 10 μM MPP+ in the presence of 10−1000 nM SR-3306. Addition of 10 μM MPP+ reduced the number of TH-positive neurons by approximately 50%, and 1000 nM SR-3306 alone had no effect compared to control (Figure 1). A statistically significant (p < 0.01), dose-dependent protection was seen in the presence of 10 μM MPP+ when SR-3306 was present at 300−1000 nM (Figure 1). Doses of 300 and 1000 nM showed greater than 90% neuroprotective effect (Figure 1). 4',6-Diamidino-2-phenylindole (DAPI) staining of total nuclei showed total cell counts were unaffected by 10 μM MPP+ (data not shown).
Figure 1.

SR-3306 protects mesencephalic dopaminergic neurons in vitro from MPP+-induced neurotoxicity. Primary dopaminergic cultures from E14 rat embryos were plated at 200 000 cells/well in 8-well chamber slides and exposed to 10 μM MPP+ for 48 h either in the presence or in the absence of varying concentrations of SR-3306 (10−1000 nM). SR-3306 was dosed 15 min prior to addition of MPP+. SR-3306 at 1000 nM was also dosed in the absence of MPP+. Cultures were fixed and immunostained for TH. Data points shown are from the average of three independent biological experiments, each consisting of four independent wells for each dosing regimen. Data are expressed as the number of TH+ neurons surviving as detected by TH immunocytochemistry (**p < 0.01; established by one-way ANOVA).
Brain Penetration for SR-3306
To assess the brain and plasma concentrations of SR-3306 at various time points, and relate these data to cell-based IC50 and cell-based neuroprotective concentrations, mice were given a single oral dose of SR-3306 at 30 mg/kg. Figure 2 presents the brain concentration of SR-3306 at 1, 2, 4, 8, and 24 h after dosing and relates this to the p-c-jun IC50 (INS-cell assay, from Table 1) and the lowest >90% neuroprotective concentration for primary dopaminergic neurons (Figure 1). The results show that the concentration of SR-3306 remained well above the cell-based IC50 out to 8 h and was 7-fold above the >90% neuroprotective concentration at 8 h. At 4 h, the brain concentration was 3 μM, and the 24 h brain concentration was 230 nM, the latter of which was approximately the cell-based IC50 as well as near the lowest concentration that showed >90% protection in primary dopaminergic neurons exposed to MPP+. It should be noted that we also found that the concentration of SR-3306 in the whole brain, as presented in Figure 2, was the same as it was in the isolated mesencephalon (data not shown). The overall brain/plasma ratio of this compound was approximately 30−40% at all time points and related well to that of other compounds in this class (9f (SR-2502) and 9l (SR-3562)).8 It should be noted that SR-3306 was not a P-glycoprotein (PGP) substrate.
Figure 2.

Pharmacokinetic brain concentration time course for SR-3306. Three mice/time points were used, and plasma and brain concentrations of SR-3306 were determined by LC-MS/MS analysis as described.8 Comparison of the brain concentration of SR-3306 at various time points (1, 2, 4, 8, and 24 h) to the cell-based IC50 and in vitro neuroprotective effect are shown. The cell-based IC50 for inhibition of c-jun phosphorylation (solid black line) was determined as described8,27−29 and is taken from Table 1. The >90% neuroprotective concentration (dashed line) is taken from the MPP+-induced neurotoxicity data presented in Figure 1.
In Vivo Target Modulation of c-jun Phosphorylation by SR-3306
It has been established by Western blot and immunohistochemical analysis that MPTP intoxication in mice induces phosphorylation of c-jun5,6 and that this phosphorylation is colocalized with dopaminergic neurons in the SNpc.6 To show that SR-3306 could inhibit c-jun phosphorylation in the SNpc and to establish a biomarker that could quantitatively measure this inhibition, we utilized Western blot analysis, immunohistochemical analysis, and developed an ELISA assay for p-c-jun. Figure 3A presents the Western blot analysis for phospho-c-jun immunoreactivity of two mice treated with saline, three mice treated with MPTP, and two mice treated with MPTP + 20 mg/kg SR-3306. All three mice treated with MPTP showed an increase in phospho-c-jun signal compared to saline-treated mice, and MPTP-treated mice dosed with 20 mg/kg SR-3306 showed a decrease in the phospho-c-jun signal (Figure 3A). TATA binding protein (TBP) is shown as a loading control. Figure 3B−J presents the immunohistochemical staining of dopaminergic neurons by TH (green) and p-c-jun (red) under various treatment conditions. Phospho-c-jun (red, Figure 3B) was not detected in the dopaminergic neurons (green, Figure 3C) present in the vehicle-treated animal group (overlay, Figure 3D). MPTP-treated mice showed a large increase in the amount of p-c-jun (arrows Figure 3E), whereas the TH-positive neuron (green) number remained about the same in this 7 h treatment paradigm (Figure 3F). The overlay presentation shows that the p-c-jun (arrows) was localized to the nucleus of the TH neurons (Figure 3G). When the MPTP-treated mice were also treated with a single oral dose of 30 mg/kg SR-3306, the number of p-c-jun containing cells (red, Figure 3H) decreased compared to the MPTP-treated group, the number of TH-positive neurons (green, Figure 3I) was unchanged, and the number of TH-positive neurons containing p-c-jun (red) decreased compared to the MPTP-treated group with very few found in the nucleus of TH neurons (Figure 3J). Insets to Figures 3G and J show the 40× magnification of the nuclear p-c-jun in the dopaminergic neuron. The number of TH+ neurons containing p-c-jun in the nucleus was counted (Figure 3K). MPTP-treated mice had a greater than 10-fold increase in the number of TH and p-c-jun positive cells compared to saline-treated mice (Figure 3K). MPTP-treated mice that also received 30 mg/kg SR-3306 showed a dramatic decrease (4-fold) in the number of p-c-jun positive cells compared to MPTP-treated mice (Figure 3K). To quantitatively assess the effects of SR-3306 on in vivo target modulation, we established an ELISA assay to measure phospho-c-jun. Figure 3L presents the nanograms (ng ± SEM) of p-c-jun in 30 μg substantia nigra tissue lysate for vehicle-treated mice, MPTP-treated mice, and MPTP-treated mice in the presence of 10, 20, and 30 mg/kg SR-3306. The results showed a dose-dependent reduction in p-c-jun levels for mice treated with SR-3306. SR-3306 at 10 mg/kg showed no effect, whereas both 20 mg/kg (p < 0.01) and 30 mg/kg (p < 0.001) showed a statistically significant reduction in p-c-jun levels in the ventral midbrain (Figure 3L) when treated with SR-3306.
Figure 3.

SR-3306 inhibits MPTP-induced phosphorylation of c-jun in the substantia nigra pars compacta. (A) Western blot analysis for phospho-c-jun in the mouse mesencephalon after MPTP injection for two vehicle mice, three MPTP-treated mice, and two MPTP/20 mg/kg SR-3306-treated mice. (B−K) Double immunofluorescent staining for p-c-jun (red), tyrosine hydroxylase (green) on ventral midbrain sections. 16 μm sections from the substantia nigra were taken from bregma −3.0 and incubated with sheep anti-TH and rabbit anti-p-c-jun Ser73 under various treatments: vehicle (B−D), vehicle/MPTP (E−G), and 30 mg/kg SR-3306/MPTP (F−J). No expression of p-c-jun (red) was seen in the vehicle-treated group (B), with high levels of TH immunoreactivity (green) seen for this group (C). Increased expression of p-c-jun (red) was seen in the MPTP-treated group indicated by the arrows (E), with similar TH immunoreactivity (green) seen for this group as the vehicle group (F). Decreased expression of p-c-jun (red) was seen in the SR-3306/MPTP-treated group (H) compared to the MPTP-treated group (E), with TH immunoreactivity (green) remaining constant (I) between all groups. The overlays of p-c-jun and TH staining for the three treatment groups (D, vehicle; G, MPTP; J, MPTP/30 mg/kg SR-3306) showed localization of p-c-jun to the nucleus (arrow). Insets in (G) and (J) are shown at 40× magnification. DAPI staining for total cells was equivalent in all treatment groups (data not shown). (K) Number of TH and p-c-jun positive cells for three treatment groups (saline (D), MPTP (G), MPTP/30 mg/kg SR-3306 (J))) in the displayed focal plane. (L) Dose−response for inhibition of p-c-jun generation (ng p-c-jun ± SEM) in 30 μg SNpc tissue lysate after various treatments (vehicle (n = 6/group), vehicle/MPTP (n = 8/group), 10 mg/kg SR-3306/MPTP (n = 8/group), 20 mg/kg SR-3306/MPTP (n = 6/group), 30 mg/kg SR-3306/MPTP (n = 8/group)) as measured by ELISA using p-c-jun Ser63 antibody. *p < 0.01 for 20 mg/kg SR-3306/MPTP and **p < 0.001 for 30 mg/kg SR-3306/MPTP compared to vehicle, as determined by two-tailed, unpaired t test. 20 mg/kg MPTP-HCl was dosed i.p. three times at 2 h intervals. For SR-3306-treated groups, SR-3306 was dosed orally 30 min prior to the first MPTP dose. Mice were sacrificed at 7 h after the first MPTP dose, and brains were harvested for SNpc isolation, nuclear fractionation, immunohistochemistry, and ELISA analysis. Nuclear fractions were isolated from the SNpc as described in the Methods section.
Dopaminergic Neurons in Mice Treated with SR-3306 Are Protected Against MPTP Intoxication
To test the hypothesis that small molecule JNK inhibitors could be effective in mouse models of PD-associated neurodegeneration, we compared the toxicity of MPTP in mice not treated or treated with 30 mg/kg SR-3306. Immunohistochemical staining with TH of representative sections in the SNpc showed that SR-3306 was indeed protective against MPTP-induced neurodegeneration (Figure 4A−C). Sections of 40 μm taken at −2.92 from bregma in vehicle-treated mice showed substantial TH immunoreactivity indicative of plentiful, healthy dopaminergic neurons (Figure 4A). Mice treated with MPTP showed a significant decrease in the number of TH-positive neurons (Figure 4B), whereas mice treated with MPTP and 30 mg/kg SR-3306 showed neuroprotection (Figure 4C), with TH immunoreactivity similar to that found in the vehicle-treated group. To quantitate the neuroprotective effect of SR-3306, stereological counts of the TH-positive cells in the SNpc at 7 days after MPTP intoxication was carried out for all animals in all groups. Figure 4D presents the number of TH-positive cells for the three treatment groups (vehicle, MPTP, MPTP + 30 mg/kg SR-3306). The results showed that MPTP intoxication decreased the number of TH-positive cells by 46% compared to vehicle (p < 0.01). Addition of 30 mg/kg SR-3306 to MPTP-treated animals showed a neuroprotective effect increasing the number of TH-positive cells to 72% of the vehicle group (p < 0.05). DAPI staining revealed an equal number of total nuclei in all treatment groups, indicating selective toxicity of MPTP to the dopaminergic neurons (data not shown). While SR-3306 was efficacious in sparing the nigral dopaminergic cell bodies, we were not able to demonstrate protection of striatal dopamine and homovanillic acid (HVA) levels, although there did appear to be some protection of 3,4-dihydroxyphenylacetic acid (DOPAC) levels (Suppporting Information Figure 2).
Figure 4.

JNK inhibition by SR-3306 prevents dopaminergic cell loss in the SNpc after MPTP treatment. (A−C) Tyrosine hydroxylase immunohistochemistry for TH on midbrain sections from (A) vehicle, (B) MPTP, and (C) MPTP + 30 mg/kg SR-3306-treated mice. A 4 s exposure was used. (D) Unbiased stereological counts of TH-positive cells in the SNpc at 7 days after MPTP intoxication. Beginning at bregma −2.70, brains were sectioned at 40 μm for 48 sections, discarding every other section until bregma −4.04. Of the 28 sections, 15 sections between bregma −2.80 and −3.80 were counted. Inhibition of JNK by 30 mg/kg SR-3306 dosed orally, b.i.d on day one and q.d. on days 2−6 showed sparing of TH-positive neurons. Three groups were analyzed: vehicle/vehicle (n = 4/group), vehicle/MPTP (n = 8/group), and 30 mg/kg SR-3306/MPTP (n = 10/group). 18 mg/kg MPTP-HCl was dosed i.p. four times at 2 h intervals in one day. For SR-3306-treated groups, SR-3306 was dosed orally 30 min prior to the first MPTP dose. Data are expressed as the number of TH+ neurons (±SEM) surviving 7 days after the MPTP treatment as detected by TH immunohistochemistry (*p < 0.05; established by Mann−Whitney U test).
Discussion
Given the absence of any approved neuroprotective drugs for the treatment of PD, the recent failure of rasagiline as a neuroprotective agent,4 and the paucity of new drugs in development as neuroprotective agents, there is a great void to fill for this unmet medical need. Indeed, a recent search using the terms neuroprotection and Parkinson’s disease on www.clinicaltrials.gov revealed only five ongoing trials, none of which were investigating a new chemical entity. We have chosen JNK as a potential target for a neuroprotective strategy, given the strong validation from KO5 and peptide inhibitor studies,6 and have embarked on developing compounds which are orally bioavailable and efficacious in animal models of PD. To accomplish this, numerous challenges must be overcome and blended into one molecule. An ideal candidate must have potency on the enzyme target, have good selectivity over other related family members as well as other receptors and ion channels, have good cellular potency, and produce an in vitro functional effect. In addition to these attributes, the compound must also have good DMPK properties, have good brain penetration, and be neuroprotective in vivo. SR-3306 met these criteria.
The structure−activity relationships (SAR) affecting selectivity, biochemical and cell-based potency, DMPK properties, and brain penetration for the aminopyrimidines has already been described.8 While none of those compounds had all of the attributes for a potential development candidate, SR-3306 did, showing improved P450 inhibition profiles compared to compound 9l (SR-3562) and improved brain penetration compared to compound 9j.8 The lack of P450 inhibition for SR-3306 can be attributed to the p-methyl-3-pyridyl-substituted triazole, compared to the morpholino-substituted triazole in compound 9l. The enhanced brain penetration for SR-3306 over compound 9j can be attributed to the lower polar surface area (PSA = 94 for SR-3306 versus PSA = 118 for compound 9j).8 Brain penetration has been shown to be directly correlated with the polar surface area (PSA) of a compound (i.e., the lower the PSA, the greater the brain penetration).17 Indeed, it has been shown that polar surface area is positively correlated with both CNS exposure and oral bioavailability,18 and hence, it is likely that SR-3306 strikes the balance needed for good permeability to cross the gut and blood-brain barrier, be stable against liver metabolism, as well as have potent cell inhibition of JNK.
A cell-based assay from primary dopaminergic neurons utilizing the neurotoxin MPP+ showing functional neuroprotection is essential for selecting compounds to advance to in vivo studies, and a few studies have evaluated the effects of caspase inhibitors in such cultures.15,16 Moreover, it has been shown that MPP+ induces phosphorylation of c-jun in dopaminergic neurons from mesencephalic cultures,19 further suggesting this as a good model for developing JNK inhibitors. Comparison of the neuroprotective effect seen with SR-3306 in these assays (Figure 1) with that seen for caspase inhibitors such as z-VAD suggests that JNK inhibition may be a superior way of protecting dopaminergic neurons from cell death, as greater than 90% of the primary dopaminergic neurons were protected at 300 nM SR-3306 (Figure 1) whereas it required concentrations of >100 μM of z-VAD to evoke the same effect.15 Since there are no published reports of small molecule inhibitors of caspase to compare in this assay format, it is difficult to say if the superior effect seen with JNK inhibition was due to the better druglike properties of SR-3306 compared to z-VAD, or if this improvement was due to inhibition of JNK as opposed to caspase. Given the peptidic nature of z-VAD, this difference may be attributed to cell-permeability issues. However, numerous studies have shown z-VAD to be cell permeable and effective in cell-based assays at nanomolar to micromolar concentrations, making the former possibility less likely.20,21 Unfortunately, the aforementioned calpain12 and iNOS9,10 inhibitors were not evaluated in these models, so no comparisons could be made between these mechanisms and JNK. Thus, SR-3306 stands as one of the few small molecule inhibitors to show potent neuroprotective effects in functional cell-based assays.
To try to establish if a single dose of SR-3306 would have significant brain exposure, we related the cell-based inhibition of c-jun phosphorylation and the >90% neuroprotective effect to the brain concentration of SR-3306 over a 24 h time period (Figure 2). We hypothesized that the ideal compound would have a once daily dosing regimen where the brain concentration of the compound was at least two times the cell-based IC50 for 24 h. However, it is unclear whether >99% inhibition is needed for 24 h or if as little as 50% inhibition for 12 h would be sufficient for full efficacy. Based on the JNK KO data, it would appear that full inhibition of JNK2 and JNK3 may give up to 83% neuroprotection. How this translates to percent enzyme inhibition is a little difficult to estimate and will be discussed further in the efficacy section.
The findings from the target modulation assays presented in Figure 3 were consistent with those previously published for both acute5 and chronic6 MPTP studies that monitored p-c-jun levels after intoxication. That is, p-c-jun increased after MPTP treatment. In the acute study using JNK KO mice, p-c-jun levels were assessed by Western blot analysis and were studied after MPTP intoxication only in wild-type mice. There was no analysis of the jnk2−/−, jnk3−/−, and jnk2/3−/− mice, thereby not allowing for comparisons of p-c-jun levels between the wild type group and the knockout mice after MPTP treatment.5 Thus, we are not able to compare our results utilizing SR-3306 directly with those found by Hunot et al.5 A more direct comparison can be made between our findings and those of Xia et al.6 Xia et al. showed by immunohistochemistry that p-c-jun levels were increased in the SNpc after MPTP treatment and that the p-c-jun was colocalized with dopaminergic neurons.6 They did not, however, show by this method that the adenovirally expressed JIP protein could decrease the p-c-jun levels. In a similar manner, we showed by immunohistochemistry that p-c-jun levels were increased in the SNpc after MPTP treatment and that the p-c-jun was colocalized with dopaminergic neurons (Figure 3G), but in addition we also showed that SR-3306 could reduce the levels of p-jun in dopaminergic neurons (Figure 3J−K). This finding, along with the Western blot and ELISA results (Figure 3), was consistent with the findings from Xia et al. where they showed by Western blot analysis that adenovirally expressed JIP protein could decrease p-c-jun levels.6 For ELISA analysis, we only analyzed cohorts that showed a greater than 2-fold difference between the vehicle-treated groups and the MPTP-treated groups. If those criteria were not met, we did not include that cohort for analysis. Thus, the novel aspect of our studies was that they showed both qualitatively and quantitatively that SR-3306 was able to inhibit c-jun phosphorylation in a dose dependent manner and that the likely mechanism of action which contributed to the efficacy in preserving dopaminergic neurons was inhibition of JNK. This was further supported by the finding that SR-3306 was not an inhibitor of MAO-B and thus did not interfere with lesion development in the MPTP model, as striatal MPP+ levels from the MPTP + SR-3306 group were equivalent to those in the MPTP group (data not shown).
The results presented in this work are the first to report an orally bioavailable, brain penetrant, small molecule JNK inhibitor to show efficacy in protecting dopaminergic neurons against neurodegeneration in an acute mouse model of Parkinson’s disease. Our efficacy measure, TH immunoreactivity in the SNpc for dopaminergic neurons, showed protection back to 72% of the vehicle levels (Figure 4D). This is less than what was seen for the jnk2/3−/− mice and the adenovirally expressed JIP protein which both showed about 85% return to vehicle levels.5,6 One potential reason for the lower degree of efficacy with the small molecule JNK inhibitor could be that that the brain exposure of the compound was not high enough to get complete inhibition of JNK after once daily dosing. For example, if >99% JNK inhibition is required for the 24 h period between dosing to achieve full protection, it may mean that b.i.d. dosing is needed for full efficacy. Greater than 99% inhibition for the entire dosing time course might be more akin to the jnk2/3−/− mice where presumably there is no JNK enzyme activity from these two isoforms. Even in that case, TH+ levels were only restored to 83% of the control, suggesting either a small contribution from JNK1 or some non-JNK-related effects for neuronal survival. Similarly, it is presumed that the exposure of the adenovirally expressed JIP protein is near 100%, and this may explain the higher efficacy levels in the 86% range. Nevertheless, the level of protection provided by SR-3306 at this dose and this dosing regimen still was within 15% of the knockout mice and the JIP protein. A second explanation for the lower degree of efficacy might be that the free brain concentration of SR-3306 may be lower than the whole brain concentration reported, thereby dropping the free brain concentration of SR-3306 lower than the cell-based IC50 values. Two potential solutions for achieving 85% or greater protection of dopaminergic neurons in mice with small molecule JNK inhibitors would be (a) to have a compound that when dosed q.d. would have >99% JNK inhibition for 24 h and/or have greater free brain concentrations, or (b) dose compounds such as SR-3306 at more frequent intervals such as b.i.d. or t.i.d. until sacrifice after 7 days. It should be noted that all of these interpretations for SR-3306 are based on mouse and rat PK data. It may be that the PK parameters for this compound in higher species such as monkey or man may be significantly better or worse than seen in the rodents, and therefore, it would be hard to extrapolate if q.d. or b.i.d. dosing would be appropriate in these species. Another potential reason for not seeing full protection of TH neurons is that SR-3306 was not potent enough. Perhaps a compound with greater cell-based potency would show greater efficacy in preserving dopaminergic neurons in the SNpc. Finally, it may merely be that a higher dose of SR-3306 would have given a higher level of neuroprotection. This hypothesis could be tested given the dose-linear pharmacokinetic profile of SR-3306, but it is not central to establishing this class of compounds as the first orally available efficacious JNK inhibitors in PD models.
While we did see protection of dopaminergic neurons in the SNpc with treatment of SR-3306, we did not see increases in striatal catecholamine levels. This finding is in contrast to that seen by both Hunot et al.5 and Xia et al.6 where the JNK KO mice and the adenovirally expressed JIP protein showed increases in dopamine, DOPAC, and HVA. One possible explanation for the protection of the dopaminergic neurons in the SNpc but not for sparing striatal dopamine levels is that dopamine levels are dependent on the TH-catalytic function, and as soon as TH activity is lost, striatal dopamine levels decrease. Hence, the cell body may likely be saved by JNK inhibition, but the projection to the striatum may be compromised causing the dopamine levels to be depressed. Another potential reason may be the slightly lower TH protection of SR-3306 compared to KO mice or JIP. While unexpected, our findings were not unprecedented and are consistent with those described in iNOS−/− mice.22 For example, the iNOS−/− mice were shown to demonstrate substantially increased numbers of TH-positive neurons in the SNpc, but this was not accompanied by striatal sparing of fibers.22 More recently, Li et al. observed the opposite effect in LRRK2R1441G BAC transgenic mice where they observed that the number of TH+ neurons in the SNpc was normal, but there was a significant reduction in extracellular striatal dopamine as measured by microdialysis indicating that these two measures may not always be in sync.23 Again, it will be interesting to see if either b.i.d. dosing or a compound with better brain exposure than SR-3306 would aid in increasing striatal dopamine levels. Finally, since behavioral end points which measure motor dysfunction in the Parkinsonian syndrome are not routinely done in the MPTP mouse model and are not well founded,24 models such as 6-hydroxy dopamine lesions in rats,25,26 where more reliable locomotor behavior can be assessed, should be tested with SR-3306. Those experiments are ongoing in our lab.
Through the translational research nature of our work, we have achieved the delicate balance of instilling potency, selectivity, good DMPK properties, and brain penetration into a small molecule neuroprotective agent for the treatment of Parkinson’s disease. Indeed, SR-3306 is the first orally available JNK inhibitor to demonstrate neuroprotection. While it is still unclear if SR-3306 would be a useful agent in higher species such as monkey or man to be neuroprotective, it presents, based on the rodent data, as a good candidate to consider for nonhuman primate MPTP studies.
Methods
Synthesis of SR-3306
The compound was synthesized by similar methods as described.8 Detailed synthesis information is found in the Supporting Information.
Homogeneous Time-Resolved Fluorescence Biochemical Assays to Determine IC50's for JNK3, JNK2, JNK1, and p38
Cell-Based Assay Measuring JNK Activity
Inhibition of c-jun phosphorylation in INS-1 β-pancreatic cells was run as previously described.27−29
P450 Inhibition and Microsomal Stability Assays
P450 inhibition for the nine major human isoforms was evaluated as described.8,30 Microsome (mouse, rat, human; Xenotech, Lenexa, KS) stability was evaluated as previously described.8,30
Rat Pharmacokinetics and Mouse Brain Penetration
Pharmacokinetics of SR-3306 was assessed in Sprague−Dawley rats (n = 3). Compounds were dosed intravenously at 1 mg/kg and orally by gavage at 2 mg/kg. Blood was taken at eight time points (5, 15, 30 min; 1, 2, 4, 6, 8 h) and collected into EDTA containing tubes, and plasma was generated using standard centrifugation techniques. Plasma proteins were precipitated with acetonitrile, and compound concentrations were determined by LC-MS/MS. Data was fit by WinNonLin using a noncompartmental model and basic pharmacokinetic parameters including peak plasma concentration (Cmax), oral bioavailability, exposure (AUC), half-life (t1/2), clearance (CL), and volume of distribution (Vd) were calculated.
CNS exposure was evaluated in C57Bl6 mice (n = 3/time point). SR-3306 was dosed at 30 mg/kg orally and after blood and brain were collected at 1, 2, 4, 8, and 24 h. Plasma was generated, and the samples were frozen at −80 °C. The plasma and brain were mixed with acetonitrile (1:5 v/v or 1:5 w/v, respectively). The brain sample was sonicated with a probe tip sonicator to break up the tissue, and samples were analyzed for compound levels by LC-MS/MS. Plasma compound levels were determined against standards made in plasma, and brain levels against standards made in blank brain matrix. All procedures were approved by the Scripps Florida IACUC.
In Vitro Neuroprotection of SR-3306 in Primary Cultures of Mesencephalic Dopaminergic Neurons
The procedures used were similar to those of Bilsland et al.15 The ventral mesencephalon was dissected from 14 d gestation Sprague−Dawley rat embryos and placed in ice cold Hank’s buffered salt solution (HBSS). Tissues were incubated with 0.25% trypsin in HBSS for 5 min in 37 °C/5%CO2. Cells were dissociated by trituration through a glass Pasteur pipet and then passed through a 70 μm cell strainer (Falcon 352350). Cells were plated at a density of 200 000 cells/well onto poly-d-lysine/laminin-coated 8-well chamber slides (Invitrogen) in DMEM (Invitrogen) + 10% HIFBS (Invitrogen) + 1% glutamine +1% Pen-Strep (Invitrogen) and incubated for 2 h. The medium was aspirated after 2 h and then replaced with selection medium (DMEM +2.75% SATO serum +1% glutamine +1% Pen-Strep) as described.15 Cultures were incubated in the selection medium for 5 days, and the medium was changed daily to remove dead cells. Cells were treated with varying concentrations (10−1000 nM) of SR-3306 15 min prior to 10 μM MPP+ exposure. SR-3306 was added to four independent wells at each concentration, and three independent biological replicates were measured. Cultures were incubated at 37 °C/5%CO2 for a further 48 h without media change and then were fixed using 4% parformaldehyde in PBS and immunostained for TH. Determination of TH-immunoreactive neuronal survival was determined by a blinded investigator in a manner similar to that of Bilsland et al.15 utilizing rabbit anti-tyrosine hydroxylase polyclonal antibody (Abcam) at 1:100 dilution. The secondary antibody Alexa Fluor 488 goat anti-rabbit (Molecular Probes) was used at 1:1000 dilution. In order for a cell to be defined as surviving, the cell body had to be intact with at least one axonal projection. Cells were counted by a blinded investigator. Statistical analysis of the TH counting data was performed utilizing one-way ANOVA analysis followed by Dunnett’s post test.
Western Blot Analysis
Ventral midbrain was dissected and lysed as described by Hunot et al.5 Protein lysates (30 μg) from nuclear fractions were processed for Western blot analysis as described.5
Immunohistochemistry for TH and p-c-jun
Brains from three mice/group were subjected to immunohistochemistry for TH and phospho-c-jun. Sections of 16 μm were used for double-labeling experiments, and DAPI staining was utilized for normalization of total cells. Sections of 16 μm from the substantia nigra were taken from bregma −3.0 and incubated with sheep anti-TH (Abcam) diluted 1:50 and rabbit anti-p-c-jun Ser73 (Cell Signaling) at 1:100 dilution overnight at 4 °C. Slides were washed 3× with PBS for 10 min each. Slides were incubated in the dark for 2 h with goat anti-rabbit Alexa Fluor 594 (Invitrogen) diluted 1:500 and with donkey anti-sheep Alexa Fluor 488 diluted to 1:500.
ELISA Analysis for p-c-jun Target Modulation
Nuclear extracts were prepared from brain slices containing the substantia nigra by using the NE-PER reagents (Pierce) supplemented with protease (Sigma) and phosphatase inhibitors (Calbiochem). Total protein content in the nuclear extracts was determined by BCA (Pierce). The amount of phospho-c-jun Ser63 in 30 μg of total protein was determined by ELISA (Cell Signaling Technology) following the manufacturer’s protocol. The nuclear extract samples were assayed in duplicate, and quantification of p-c-jun in each sample was achieved by utilizing a standard curve with recombinant p-c-jun. Statistical significance between sample groups was determined with a two-tailed, unpaired t test.
MPTP Mouse Studies for Immunohistochemistry for TH and p-c-jun, and ELISA Analysis for p-c-jun Target Modulation
Eleven week old male C57BL/6J mice (Jackson Laboratories) weighing in the range of 25−30 g were used for all studies. Mice were acclimated for 1 week prior to initiation of all studies. Mice were injected intraperitoneally (i.p.) four times at 2 h intervals over 1 day with either 18 mg/kg MPTP-HCl (Sigma) dissolved in 0.9% saline or a corresponding volume of saline alone. For SR-3306 dosed groups, SR-3306 was orally dosed 30 min prior (t = −30 min) to the first MPTP injection. Mice were sacrificed 7 h after the MPTP administration and evaluated for counting of p-c-jun, TH staining in the SNpc, and ELISA analysis (n = 6 for vehicle group, n = 6 for vehicle/MPTP group, n = 8 for MPTP/10 mg/kg SR-3306 group, n = 6 for MPTP/20 mg/kg SR-3306 group, n = 8 for MPTP/30 mg/kg SR-3306 group).
MPTP Mouse Studies and Stereology
The acute MPTP-lesion regimen, immunostaining of TH, stereological evaluation of TH+ cells in the SNpc, and safety precautions as outlined by Jackson-Lewis and Przedborski31 and Hunot et al.5 were followed. Eleven week old male C57BL/6J mice (Jackson Laboratories) weighing in the range of 25−30 g were used for all studies. Mice were acclimated for 1 week prior to initiation of all studies. Mice were injected intraperitoneally (i.p.) four times at 2 h intervals over 1 day with either 18 mg/kg MPTP-HCl (Sigma) dissolved in 0.9% saline or a corresponding volume of saline alone. For SR-3306 dosed groups, SR-3306 was orally dosed 30 min prior (t = −30 min) to the first MPTP injection. The dosing regimen for SR-3306 was b.i.d. on day 1 (t = −30 min before first MPTP dose and t = 5.5 h after first MPTP dose) and q.d. on days 2−6. On days 2−6, the SR-3306 dose was administered 24 h after the previous SR-3306 dose. SR-3306 was administered as the HCl-salt, and saline was used as the vehicle. Mice were sacrificed 7 days after the MPTP administration and evaluated for counting of SNpc dopaminergic neurons (n = 4 for vehicle group, n = 8 for vehicle/MPTP group, n = 10 for MPTP/SR-3306 group) and for striatal catecholamine levels (n = 4/group). Beginning at bregma −2.70, brains were sectioned at 40 μm for 48 sections, discarding every other section until bregma −4.04. Of the 28 sections, 15 sections between bregma −2.80 and −3.80 were counted. Unbiased stereological counting by a blinded investigator of TH+ cells in the SNpc was quantitated (±SEM) using the Stereo Investigator (v 8.0) software program (Microbrightfield) as described.5,6,9,10,22,24,32 Statistical analysis of the TH counting data was performed utilizing the Mann−Whitney U test.
Striatal Dopamine, DOPAC, and HVA Levels
Brain sections were mixed 1:50 w/v with 0.3 M perchloric acid and sonicated on ice. The samples were centrifuged at 16 000g for 5 min, and 10 μL of the supernatant was analyzed directly with an Agilent 1100 HPLC instrument using an ESA HR-80 80 × 4.6 mm column and ESA MD-TM mobile phase. HVA, DOPAC, and dopamine were detected with a Coulochem II electrochemical detector (ESA Inc.,) using a 5021A conditioning cell set to −100 mV and a 5011A analytical cell with E1 = −40 nM and E2 = +350 nM.
Acknowledgments
We are grateful to Layton H. Smith and Madeline McGill-Lopez for technical help with the primary dopaminergic neuronal assays and to Susan Khan for help with DMPK studies. We are also grateful to Yamille Del Rosario and Melissa Tellinghuisen for administrative assistance in preparing the manuscript. INS-1 cells were a kind gift from Dr. Christopher Newgard at Duke University.
Glossary
Abbreviations
- BAC
bacterial artificial chromosome
- b.i.d.
twice daily
- CYP450
cytochrome p450
- DAPI
4′,6-diamidino-2-phenylindole
- DMPK
drug metabolism and pharmacokinetics
- DOPAC
3,4-dihydroxyphenylacetic acid
- DMEM
Dulbecco’s modified Eagle's medium
- ELISA
enzyme-linked immunosorbent assay
- HBSS
Hank’s buffered salt solution
- HIFBS
heat-inactivated fetal bovine serum
- hERG
human-ether-a-go-go potassium channel
- HPMC
hydroxypropyl methyl cellulose
- HVA
homovanillic acid
- INS-1
insulinoma-1
- JIP
JNK-interacting protein
- JNK
c-jun-N-terminal kinase
- KO
knockout
- LRRK2
leucine rich repeat kinase 2
- MPP+
1-methyl-4-phenylpyridinium
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- MAO-A
monoamine oxidase A
- MAO-B
monoamine oxidase B
- PD
Parkinson’s disease
- p-c-jun
phospho c-jun
- q.d.
only daily
- ROS
reactive oxygen species
- SAR
structure−activity relationship
- SEM
standard error of the mean
- SNpc
substantia nigra pars compacta
- STZ
streptozotocin
- TBP
TATA binding protein
- TH
tyrosine hydroxylase
- t.i.d.
three times daily.
Supporting Information Available
Additional experimental details, figures, and tables. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
† These authors contributed equally to this work.
† A.P., M.D.C., and P.L. conceived and designed the experiments; S.H., M.G., D.H. Jr., T.K., X.S., D.D., W.C., Y.Y.L., J.W.C., L.C., L.L., and C.H.R. performed the experiments; A.P., M.D.C., D.D., J.W.C., L.C., and P.L. analyzed the data; and P.L. wrote the manuscript.
The authors declare that they have no competing financial interests.
This work was supported by NIH Grant U01-NS057153 awarded to P.L.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Dauer W.; Przedborski S. (2003) Parkinson’s disease: mechanisms and models. Neuron 39, 889–909. [DOI] [PubMed] [Google Scholar]
- Lang A. E.; Lozano A. M. (1998) Parkinson’s disease. First of two parts. N. Engl. J. Med. 339, 1044–1053. [DOI] [PubMed] [Google Scholar]
- Lang A. E.; Lozano A. M. (1998) Parkinson’s disease. Second of two parts. N. Engl. J. Med. 339, 1130–1143. [DOI] [PubMed] [Google Scholar]
- Olanow C. W.; Rascol O.; Hauser R.; Feigin P. D.; Jankovic J.; Lang A.; Langston W.; Melamed E.; Poewe W.; Stocchi F.; Tolosa E. (2009) A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N. Engl. J. Med. 361, 1268–1278. [DOI] [PubMed] [Google Scholar]
- Hunot S.; Vila M.; Teismann P.; Davis R. J.; Hirsch E. C.; Przedborski S.; Rakic P.; Flavell R. A. (2004) JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. U.S.A. 101, 665–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X. G.; Harding T.; Weller M.; Bieneman A.; Uney J. B.; Schulz J. B. (2001) Gene transfer of the JNK interacting protein-1 protects dopaminergic neurons in the MPTP model of Parkinson’s disease. Proc. Natl. Acad. Sci. U.S.A. 98, 10433–10438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Shi L.; Xie Y.; Ma C.; Li W.; Su X.; Huang S.; Chen R.; Zhu Z.; Mao Z.; Han Y.; Li M. (2004) SP600125, a new JNK inhibitor, protects dopaminergic neurons in the MPTP model of Parkinson’s disease. Neurosci. Res. 48, 195–202. [DOI] [PubMed] [Google Scholar]
- Kamenecka T.; Jiang R.; Song X.; Duckett D.; Chen W.; Ling Y. Y.; Habel J.; Laughlin J. D.; Chambers J.; Figuera-Losada M.; Cameron M. D.; Lin L.; Ruiz C. H.; LoGrasso P. V. (2010) Synthesis, biological evaluation, X-ray structure, and pharmacokinetics of aminopyrimidine c-jun-N-terminal kinase (JNK) inhibitors. J. Med. Chem. 53, 419–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przedborski S.; Jackson-Lewis V.; Yokoyama R.; Shibata T.; Dawson V. L.; Dawson T. M. (1996) Role of neuronal nitric oxide in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced dopaminergic neurotoxicity. Proc. Natl. Acad. Sci. U.S.A. 93, 4565–4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D. C.; Jackson-Lewis V.; Vila M.; Tieu K.; Teismann P.; Vadseth C.; Choi D. K.; Ischiropoulos H.; Przedborski S. (2002) Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J. Neurosci. 22, 1763–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L.; Sugama S.; Mischak R. P.; Kiaei M.; Bizat N.; Brouillet E.; Joh T. H.; Beal M. F. (2004) A novel systemically active caspase inhibitor attenuates the toxicities of MPTP, malonate, and 3NP in vivo. Neurobiol. Dis. 17, 250–259. [DOI] [PubMed] [Google Scholar]
- Crocker S. J.; Smith P. D.; Jackson-Lewis V.; Lamba W. R.; Hayley S. P.; Grimm E.; Callaghan S. M.; Slack R. S.; Melloni E.; Przedborski S.; Robertson G. S.; Anisman H.; Merali Z.; Park D. S. (2003) Inhibition of calpains prevents neuronal and behavioral deficits in an MPTP mouse model of Parkinson’s disease. J. Neurosci. 23, 4081–4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian M. A.; Biggs W. H.; Treiber D. K.; Atteridge C. E.; Azimioara M. D.; Benedetti M. G.; Carter T. A.; Ciceri P.; Edeen P. T.; Floyd M.; Ford J. M.; Galvin M.; Gerlach J. L.; Grotzfeld R. M.; Herrgard S.; Insko D. E.; Insko M. A.; Lai A. G.; Lelias J. M.; Mehta S. A.; Milanov Z. V.; Velasco A. M.; Wodicka L. M.; Patel H. K.; Zarrinkar P. P.; Lockhart D. J. (2005) A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 23, 329–336. [DOI] [PubMed] [Google Scholar]
- Karaman M. W.; Herrgard S.; Treiber D. K.; Gallant P.; Atteridge C. E.; Campbell B. T.; Chan K. W.; Ciceri P.; Davis M. I.; Edeen P. T.; Faraoni R.; Floyd M.; Hunt J. P.; Lockhart D. J.; Milanov Z. V.; Morrison M. J.; Pallares G.; Patel H. K.; Pritchard S.; Wodicka L. M.; Zarrinkar P. P. (2008) A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 26, 127–132. [DOI] [PubMed] [Google Scholar]
- Bilsland J.; Roy S.; Xanthoudakis S.; Nicholson D. W.; Han Y.; Grimm E.; Hefti F.; Harper S. J. (2002) Caspase inhibitors attenuate 1-methyl-4-phenylpyridinium toxicity in primary cultures of mesencephalic dopaminergic neurons. J. Neurosci. 22, 2637–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C. T.; Zhu J. H.; Cao G.; Signore A.; Wang S.; Chen J. (2005) Apoptosis inducing factor mediates caspase-independent 1-methyl-4-phenylpyridinium toxicity in dopaminergic cells. J. Neurochem. 94, 1685–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelder J.; Grootenhuis P. D.; Bayada D. M.; Delbressine L. P.; Ploemen J. P. (1999) Polar molecular surface as a dominating determinant for oral absorption and brain penetration of drugs. Pharm. Res. 16, 1514–1519. [DOI] [PubMed] [Google Scholar]
- Martin Y. C. (2005) A bioavailability score. J. Med. Chem. 48, 3164–3170. [DOI] [PubMed] [Google Scholar]
- Gearan T.; Castillo O. A.; Schwarzschild M. A. (2001) The parkinsonian neurotoxin, MPP+ induces phosphorylated c-Jun in dopaminergric neurons of mesencephalic cultures. Parkinsonism Relat. Disord. 8, 19–22. [DOI] [PubMed] [Google Scholar]
- Han B. S.; Noh J. S.; Gwag B. J.; Oh Y. J. (2003) A distinct death mechanism is induced by 1-methyl-4-phenylpyridinium or by 6-hydroxydopamine in cultured rat cortical neurons: degradation and dephosphorylation of tau. Neurosci. Lett. 341, 99–102. [DOI] [PubMed] [Google Scholar]
- Kunstle G.; Leist M.; Uhlig S.; Revesz L.; Feifel R.; MacKenzie A.; Wendel A. (1997) ICE-protease inhibitors block murine liver injury and apoptosis caused by CD95 or by TNF-α. Immunol. Lett. 55, 5–10. [DOI] [PubMed] [Google Scholar]
- Liberatore G. T.; Jackson-Lewis V.; Vukosavic S.; Mandir A. S.; Vila M.; McAuliffe W. G.; Dawson V. L.; Dawson T. M.; Przedborski S. (1999) Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat. Med. 5, 1403–1409. [DOI] [PubMed] [Google Scholar]
- Li Y.; Liu W.; Oo T. F.; Wang L.; Tang Y.; Jackson-Lewis V.; Zhou C.; Geghman K.; Bogdanov M.; Przedborski S.; Beal M. F.; Burke R. E.; Li C. (2009) Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson’s disease. Nat. Neurosci. 12, 826–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emborg M. E. (2004) Evaluation of animal models of Parkinson’s disease for neuroprotective strategies. J. Neurosci. Methods 139, 121–143. [DOI] [PubMed] [Google Scholar]
- Deumens R.; Blokland A.; Prickaerts J. (2002) Modeling Parkinson’s disease in rats: an evaluation of 6-OHDA lesions of the nigrostriatal pathway. Exp. Neurol. 175, 303–317. [DOI] [PubMed] [Google Scholar]
- Kirik D.; Rosenblad C.; Bjorklund A. (1998) Characterization of behavioral and neurodegenerative changes following partial lesions of the nigrostriatal dopamine system induced by intrastriatal 6-hydroxydopamine in the rat. Exp. Neurol. 152, 259–277. [DOI] [PubMed] [Google Scholar]
- Jiang R.; Duckett D.; Chen W.; Habel J.; Ling Y. Y.; LoGrasso P.; Kamenecka T. M. (2007) 3,5-Disubstituted quinolines as novel c-Jun N-terminal kinase inhibitors. Bioorg. Med. Chem. Lett. 17, 6378–6382. [DOI] [PubMed] [Google Scholar]
- Kamenecka T.; Habel J.; Duckett D.; Chen W.; Ling Y. Y.; Frackowiak B.; Jiang R.; Shin Y.; Song X.; LoGrasso P. (2009) Structure−activity relationships and X-ray structures describing the selectivity of aminopyrazole inhibitors for c-Jun N-terminal kinase 3 (JNK3) over p38. J. Biol. Chem. 284, 12853–12861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin Y.; Chen W.; Habel J.; Duckett D.; Ling Y. Y.; Koenig M.; He Y.; Vojkovsky T.; LoGrasso P.; Kamenecka T. M. (2009) Synthesis and SAR of piperazine amides as novel c-jun N-terminal kinase (JNK) inhibitors. Bioorg. Med. Chem. Lett. 19, 3344–3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsky R. L.; Obach R. S. (2004) Validated assays for human cytochrome P450 activities. Drug Metab. Dispos. 32, 647–660. [DOI] [PubMed] [Google Scholar]
- Jackson-Lewis V.; Przedborski S. (2007) Protocol for the MPTP mouse model of Parkinson’s disease. Nat. Protoc. 2, 141–151. [DOI] [PubMed] [Google Scholar]
- Cleren C.; Calingasan N. Y.; Chen J.; Beal M. F. (2005) Celastrol protects against MPTP- and 3-nitropropionic acid-induced neurotoxicity. J. Neurochem. 94, 995–1004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.