

Abstract
Long-range comparative sequence analysis provides a powerful strategy for identifying conserved regulatory elements. The stem cell leukemia (SCL) gene encodes a bHLH transcription factor with a pivotal role in hemopoiesis and vasculogenesis, and it displays a highly conserved expression pattern. We present here a detailed sequence comparison of 193 kb of the human SCL locus to 234 kb of the mouse SCL locus. Four new genes have been identified together with an ancient mitochondrial insertion in the human locus. The SCL gene is flanked upstream by the SIL gene and downstream by the MAP17 gene in both species, but the gene order is not collinear downstream from MAP17. To facilitate rapid identification of candidate regulatory elements, we have developed a new sequence analysis tool (SynPlot) that automates the graphical display of large-scale sequence alignments. Unlike existing programs, SynPlot can display the locus features of more than one sequence, thereby indicating the position of homology peaks relative to the structure of all sequences in the alignment. In addition, high-resolution analysis of the chromatin structure of the mouse SCL gene permitted the accurate positioning of localized zones accessible to restriction endonucleases. Zones known to be associated with functional regulatory regions were found to correspond precisely with peaks of human/mouse homology, thus demonstrating that long-range human/mouse sequence comparisons allow accurate prediction of the extent of accessible DNA associated with active regulatory regions.
One of the major challenges currently facing biological science concerns the characterization of transcriptional networks in higher organisms. The regulatory information must be present in the primary DNA sequence, but deciphering the code remains a formidable challenge. Long-range comparative sequence analysis provides an attractive strategy for the identification of functionally important gene regulatory regions, on the basis that their sequences are highly conserved during evolution (Hardison et al. 1997). Early studies used functional assays to identify homologous regulatory elements and subsequently used local sequence comparisons to identify conserved transcription factor binding sites (Aparicio et al. 1995; Popperl et al. 1995; Nonchev et al. 1996). More recently, the increasing availability of large tracts of genomic sequence allows a shift to long-range comparisons, and it has been suggested that the identification of regulatory elements through human/mouse sequence comparisons is sufficient justification for sequencing the entire mouse genome (Hardison et al. 1997). However, only a limited number of long-range comparisons have been reported so far. Human/mouse comparisons were shown to be an efficient approach for the reliable prediction of coding exons (Ansari-Lari et al. 1998; Endrizzi et al. 1999; Jang et al. 1999). Besides, analysis of the human and murine Bruton's tyrosine kinase loci revealed a new enhancer with activity in transfection assays (Oeltjen et al. 1997). Comparison of human and murine β-globin loci showed that peaks of high homology corresponded with functional regulatory regions, including the well-characterized DNaseI hypersensitive sites within the locus control region (Jackson et al. 1996; Hardison et al. 1997). Comparative sequence analysis of the human and mouse adenosine deaminase genes showed that several known regulatory regions displayed higher sequence conservation than some of the coding exons (Brickner et al. 1999). Most recently, long-range sequence comparisons of the human and mouse IL4/IL13/IL5 gene clusters led to the identification of a putative chromatin regulatory region, the activity of which was assayed in vivo using transgenic mice (Loots et al. 2000).
The SCL gene encodes a bHLH transcription factor with a critical role in hemopoiesis and vasculogenesis. It was identified by virtue of its disruption in T-cell acute leukemia, and rearrangements of the SCL locus are perhaps the most frequent molecular pathology associated with this tumor (Barton et al. 1999; Begley and Green 1999). Targeted mutation of the SCL gene has shown that it is essential for the development of all hemopoietic lineages (Porcher et al. 1996; Robb et al. 1996) and also for normal yolk sac angiogenesis (Visvader et al. 1998). Ectopic SCL expression in zebrafish embryos specifies hemangioblast development from early mesoderm, results in disproportionate production of blood and endothelial progenitors, and can partially rescue endothelial and hemopoietic phenotypes of the cloche mutant (Gering et al. 1998; Liao et al. 1998).
SCL is normally expressed in hemopoietic cells, endothelium, and within specific regions of the CNS. This pattern of expression is highly conserved throughout vertebrate evolution from mammals to teleost fish (Green et al. 1992; Kallianpur et al. 1994; Gering et al. 1998; Mead et al. 1998; Sinclair et al. 1999; Drake and Fleming 2000). SCL expression is tightly regulated and involves two alternative promoters with lineage-specific activity in distinct hemopoietic cell types (Lecointe et al. 1994; Bockamp et al. 1995, 1997, 1998). In addition, a detailed analysis of the chromatin structure of the mouse SCL locus identified a number of DNaseI hypersensitive sites associated with enhancer or silencer activity (Göttgens et al. 1997). More recently, studies using transgenic mice have identified five separate enhancers, which direct reporter gene expression in vivo to endothelium, midbrain, hindbrain/spinal cord, or hemopoietic progenitor cells, all subdomains of the normal SCL expression pattern (Sanchez et al. 1999; Sinclair et al. 1999; Göttgens et al. 2000).
We have recently cloned and sequenced the SCL locus from human, mouse, and chicken (Göttgens et al. 2000), but only limited sequence was available from the region downstream of the mouse SCL locus. To permit long-range human/mouse sequence comparisons, we have isolated and sequenced an additional 148 kb from the 3′ region of the mouse SCL locus. The structures of the human and mouse SCL loci are very similar in the immediate vicinity of the SCL gene, but substantial differences are present downstream of the SCL flanking gene, MAP17. We have developed a new sequence analysis tool (SynPlot) to allow rapid identification of candidate regulatory regions. Finally, we show that for known regulatory regions, the extent of homology within an individual peak corresponds precisely with sites sensitive to restriction endonuclease digestion.
RESULTS
Structure of Human and Mouse SCL Loci
Our previous analysis of the human SCL locus showed that human SCL was flanked upstream by the SIL gene and downstream by the MAP17 gene (Göttgens et al. 2000). However, the relative position of the original human and mouse genomic clones restricted the overlap between the human and mouse SCL loci to only 55.8 kb with only 11.0 kb of 3′ flanking sequence, which did not extend to the 3′ flanking gene. A new mouse SCL genomic clone was therefore isolated and completely sequenced (see Methods). This allowed, for the first time, a complete comparative analysis of 193 kb of the human and 234 kb of the mouse SCL loci, which extended to the 5′ and 3′ flanking genes in both species (Fig. 1).
Figure 1.

Structure of the human and murine SCL loci. The gene structure of the human and mouse SCL loci is shown above a profile displaying the respective G/C content. Arrows indicate the direction of genes. M1 and M2 refer to the sequences homologous to mitochondrial DNA, and (pCYP1) and (pCYP2) refer to partial segments of CYP4 genes present in the mouse locus.
Complete annotation of the two sequences led to the description of three new murine genes: mouse MAP17 and two members of the cytochrome p450 Cyp4 family of genes. In addition, examination of the human SCL sequence downstream of CYP4A11 revealed the 3′ half of another member of the CYP4 gene family, the as-yet-unpublished CYP4Z1 gene (D. Bell, pers. comm.; Fig. 1). Comparison of the human and mouse loci showed that the order of the CYP4 genes was not conserved, even though the SCL locus on human chromosome 1 forms part of a long region of synteny with mouse chromosome 4. The CYP4 family of cytochrome p450 genes is divided into subfamilies based on sequence homology (i.e., CYP4A, CYP4B, etc.). The two new mouse genes have been allocated to subfamilies and named Cyp4x1 and Cyp4a21 (see Cytochrome p450 Nomenclature Committee at http://drnelson.utmem.edu/CytochromeP450.html). Surprisingly, the 5′ human gene, CYP4A11, is more similar to the 3′ mouse gene Cyp4a21 than to the 5′ mouse gene Cyp4x1. Moreover, a human ortholog to mouse Cyp4x1 has recently been mapped to the same region of human chromosome 1 and presumably lies further into the CYP4 gene cluster (D. Bell, pers. comm.). These results show that the order of individual genes within the CYP4 locus is not conserved in man and mouse. Consistent with this observation, the number of genes within each CYP4 subfamily differs between mammals (see http://drnelson.utmem.edu/CytochromeP450.html), indicating that the CYP4 locus is evolving by gene duplication. This view is further supported by the presence in 3′ of mouse Cyp4a21 of two regions homologous with the last exon and exons 4–5 of CYP4 family members. These two regions (pCyp1 and pCyp2, respectively in Fig. 1) were both in the opposite transcriptional orientation to Cyp4a21, and are likely to represent remnants of Cyp4 genes resulting from partial gene duplications/inversions. Therefore, our analysis showed that the gene structure and order of the SIL, SCL, and MAP17 genes was highly conserved, whereas 3′ of MAP17 of the human and mouse loci were not colinear.
The G/C content of the human and mouse sequences was 44.5% and 43%, respectively, which is typical of the relatively gene-poor isochore H1 (Bernardi 2000), and consistent with the location of the human SCL gene within a Giemsa light band. Neither the human nor the murine G/C profile followed the regular sinusoidal pattern described for the human α-globin locus (Flint et al. 1997), but the SCL and MAP17 genes were in regions of high G/C content in both species. CpG islands were found to be associated with the promoters of SIL and SCL, but not the MAP17 or CYP4 genes (data not shown). Indeed, the CpG content of a 50-kb segment containing the SCL and MAP17 genes is so high, that it would be part of the gene-rich isochore H2 (Bernardi 2000). This finding contrasts with the suggestion that isochores are units greater than 200 kb in size and characterized by a high compositional homogeneity (above a 3 kb size level) (Bernardi 2000).
A Mitochondrial Genome Insertion in the Human but Not the Mouse Locus
Sequence database searches with the human SCL locus sequence revealed the presence of two regions on either side of the CYP4A11 gene that had high homology with segments of the mammalian mitochondrial genome (Fig. 2). The first region (112028–112388 and 113106–113425) matches the human mitochondrial genome from 2932 to 3651. The match is flanked by Alu and LTR repeats at the 5′ and 3′ ends, respectively, and the gap interrupting the match contains two partial Alu repeats. The second region (140989–141045 and 141372–141429) matches the human mitochondrial genome from 12213 to 12327, and was flanked by LINE repeats (L2 and L1PA16 repeats, respectively). No similarity to these or any other regions of vertebrate mitochondrial genomes were detected in the sequence of the mouse SCL locus.
Figure 2.


A transposition of mitochondrial DNA 3′ of the human MAP17 gene. (A) Diagram showing the position of the two mitochondrial homology regions relative to the MAP17 and CYP4A11 genes. Boxes labeled M correspond to the mitochondrial homology regions shown in (B), and boxes labeled R show the position of repeat elements. (B) Alignment of mitochondrial homology regions 1 and 2 to mitochondrial sequence of cow and human.
The serial endosymbiosis theory postulates that symbiotic organelles such as mitochondria gradually transferred a large proportion of their genes to the eukaryotic/nuclear genome during early evolution. Remnants of more recent transposition events of mitochondrial DNA into the nuclear genome are also detectable by Southern hybridization (du Buy and Riley 1967; Tsuzuki et al. 1983). However, this is the first report describing the sequence of such an insertion. The two segments of nuclear-mitochondrial DNA described here were codirectional, consistent with the suggestion that they represent the remnants of a single integration event. Two observations indicate the relatively ancient nature of this mitochondrial-to-nuclear transposition. First, the transposed mitochondrial DNA has been disrupted through the integration of repeat elements and even an entire gene (CYP4A11). Second, the nuclear sequence is no more similar to human than to other mammalian mitochondrial sequences, and it is the most divergent sequence when aligned to mammalian mitochondrial DNA (Fig. 2B; data not shown). BLAST scores were higher for alignments with horse, cow, and sheep mitochondrial DNA (BLASTX scores ∼180) than for alignments with human or mouse mitochondrial DNA (BLASTX scores ∼160). Taken together, these data indicate that the transposition event occurred before the divergence of human and horse, cow or sheep.
Sequence Variation of Mouse Strains
The sequence of the murine SCL locus used in this study was assembled from P1 clone ICRFP703C1281Q and PAC clone PAC129.0.726 from the genetic backgrounds C57/Bl6 and 129SV/SvEvTac, respectively. We analyzed the overlap of 23.8 kb of the two insert sequences to assess the frequency of sequence length and single nucleotide polymorphisms (SNPs) between these two important laboratory strains. We found 18 sequence length polymorphisms, 10 of which were just one nucleotide, with the remaining eight covering 33 bp in total. The number of C↔T transitions (11) exceeded the number of transversions (6) consistent with findings from human SNP analysis. However, all six transversions were of the G↔T type, with no occurrence of the G↔C or A↔T alternatives, whereas the three possible transversions occur at similar frequencies in human (Brookes 1999). Therefore, it will be interesting to see if other regions of the mouse genome show a preference for the G↔T transversion. The total number of 17 SNPs in 23.8 kb is in good agreement with the observed SNP frequency of 1:884 bp derived from a recent analysis of 3884 sequence-tagged sites (STSs) from the mouse strains, C57Bl/6J and 129/Sv (Lindblad-Toh et al. 2000), and for the expected frequency of one human SNP in every 1000 bp (Taillon-Miller et al. 1998).
Large-Scale Comparison of Human and Mouse SCL Loci
Although a potentially powerful approach for characterizing regulatory elements (Hardison et al. 1997), comparative analysis of large genomic sequences entails distinct and challenging problems. The most widely used sequence alignment algorithms, such as BLAST (Altschul et al. 1990) or CLUSTAL (Thompson et al. 1997), were originally developed to align coding regions. Therefore, the scoring schemes for calculating optimum alignments use gap and gap extension penalties. Because some repeat elements in human and mouse DNA are species-specific, such algorithms can only compute high-scoring local alignments, but can never produce a long-range global alignment. Hence, current programs for the display of comparative sequence analysis use one of the two sequences as the reference sequence and indicate the position of high-scoring local alignments with the test sequence (Jareborg et al. 1999; Schwartz et al. 2000). As a result, it is not possible to display the position of homology peaks within the test sequence relative to other sequence features (exons, repeats, other homology peaks, etc).
Therefore, we developed a new software tool (SynPlot), which uses the Dialign algorithm (Morgenstern et al. 1998) for computation of a long-range global alignment. Dialign scans sequences of unlimited length for areas of high local similarity and uses these as anchor points for a global alignment. The SynPlot graphical output includes a similarity profile of the long-range alignment together with a diagrammatic representation of both loci, including the position and size of all gaps inserted to permit optimum global alignment. Consequently, the positions of all features in the individual sequences have to be transformed into their new positions within the aligned file. This transformation is performed automatically by SynPlot, which calculates the new positions using the locations and lengths of all gaps. Moreover, feature files generated during annotation in ACeDB (Durbin and Mieg 1991), and that contain the positions of exons and repeat elements, can be directly imported into the graphical output. Therefore, the SynPlot output conveys comparative gene structure, repeat patterns (plus any other user-defined patterns), and relative sequence homology in a single linear plot with the upper limit being the length of the region of synteny.
The SynPlot output file (Fig. 3) displays a comparative analysis of the human and mouse SCL loci from within the SIL gene to beyond the CYP4A11 and Cyp4a21 genes, respectively. Although the latter two genes may not be orthologs (because there are three more murine members of the Cyp4a subfamily, Cyp4a10/12/14), their subfamily relationship is recognized by Dialign. As a result, the two genes are aligned and long gaps introduced in the human sequence to accommodate the region occupied by mouse Cyp4x1. Display of repeat elements in the profile shows that most other gaps inserted for optimum sequence alignment coincide with species-specific repeat elements in either locus.
Figure 3.
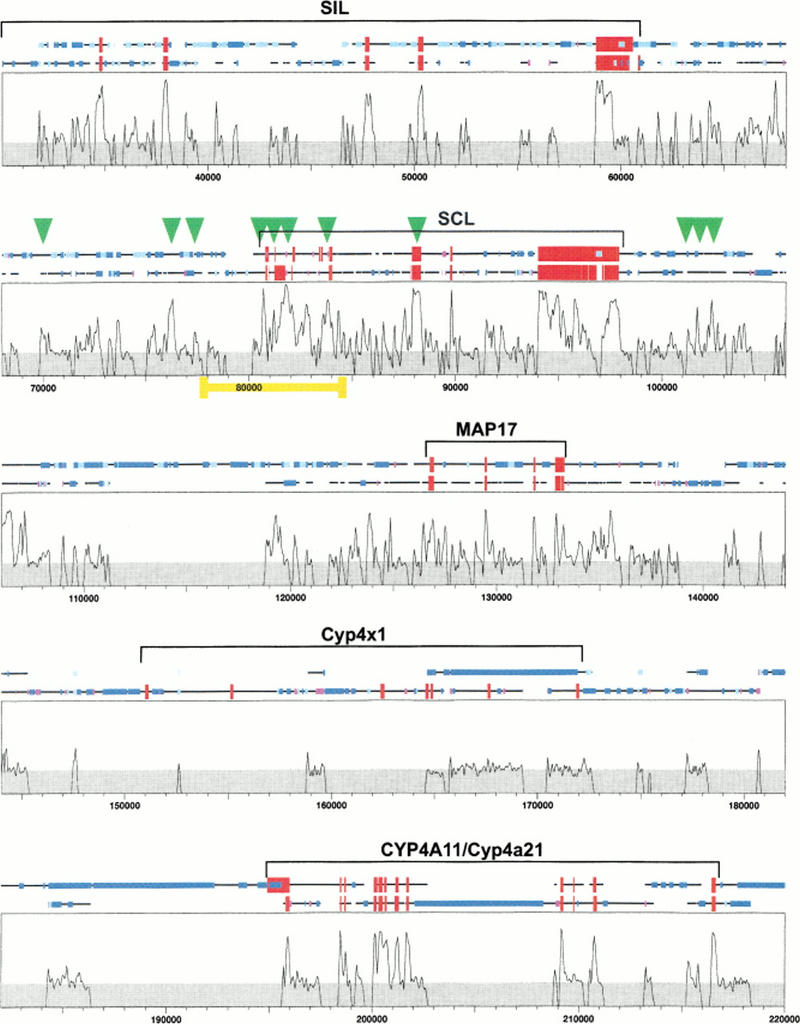
SynPlot analysis of the human and mouse SCL loci. Human and mouse clones starting with the last five exons of SIL, and ranging to beyond the CYP4A11/Cyp4a21 genes, were aligned using Dialign. The alignment, together with locus features, was displayed using SynPlot. Numbers on the horizontal axis represent distance (nucleotides) from the beginning of the aligned file. Numbers on the vertical axis represent the proportion of identical nucleotides within a 49 nt window, moved by 25 nt increments across the entire alignment. Hence, regions with gaps of >50 bp show 0% identity. The horizontal lines above the profile represent the human and mouse sequences and illustrate the position of gaps introduced to permit optimum alignment. Red boxes show exon positions, and the smaller boxes represent repeats as follows: (dark blue) LINEs,(light blue) SINEs, (magenta) tandem repeats. Green arrowheads indicate the positions of previously mapped DNaseI hypersensitive sites, and the yellow bar delimits the portion of the profile shown in Figure 4. Gray shading indicates background similarity of ≤25%.
All coding exons clearly stand out as homology peaks. Noncoding homology peaks are seen only in the region of the SIL, SCL, and MAP17 genes. These peaks represent candidate regulatory regions, or may serve other conserved aspects of genome function such as replication or domain structure. The distinct absence of noncoding homology peaks in the CYP4 gene region is consistent with our suggestion that the genes present in human and mouse are not orthologous.
Local Regions of Sensitivity to Restriction Endonucleases Correspond to Peaks of Human/Mouse Homology
Many active enhancers are associated with regions of hypersensitivity to DNaseI digestion. Therefore, analysis of chromatin structure is frequently used to identify the location of candidate regulatory regions. However, this strategy is constrained by the fact that the activity of individual enhancers may be restricted to rare cell types, thus precluding biochemical analysis. Furthermore the resolution of Southern blotting is limited. Therefore, we investigated whether the peaks of human/mouse homology can be used to predict the position and extent of localized zones of altered chromatin structure associated with regulatory regions of the murine SCL gene.
A systematic analysis of the chromatin structure of the mouse SCL locus previously identified a number of DNaseI hypersensitive sites associated with enhancer or silencer activity (Göttgens et al. 1997). However, DNaseI hypersensitive site mapping has a limited resolution and the precise location or extent of each DNaseI hypersensitive site was not known. To compare the extent of accessible DNA in each case with the human/mouse alignment, a restriction endonuclease accessibility assay (Boyes and Felsenfeld 1996) was modified to pinpoint the location of sites sensitive to restriction endonuclease digestion within the DNA sequence.
Restriction endonuclease accessibility studies were performed in two primitive myeloid cell lines (M1 and 416B) known to express the SCL gene (Fig. 4). Our results show that seven previously described DNaseI hypersensitive sites (promoter 1a, promoter 1b, +1, +3, Fig. 4; +7, −3, −4, data not shown; numbering represents the distance [kb] from the start of murine exon 1a) were associated with localized zones of increased endonuclease accessibility. All of these DNaseI hypersensitive sites have been shown to be associated with enhancer or silencer activity using transfection or transgenic reporter assays (Göttgens et al. 1997; Fordham et al. 1999; Sanchez et al. 1999; Sinclair et al. 1999). The use of multiple restriction endonucleases allowed the extent of the regions of endonuclease accessibility to be mapped in detail. In each case, these regions correlated precisely with a peak of homology in the human/mouse alignment (Fig. 4B), thus identifying these peaks as candidate regulatory regions.
Figure 4.

Localized regions of sensitivity to restriction endonucleases correspond to peaks of human/mouse homology. (A) Restriction endonuclease accessibility assay showing the mapping of the hypersensitive sites at promoter 1b, +1 (+1HS), and +3 (+3HS) (numbering corresponds to distances in kb from the start of exon 1a) in the 416B and M1 primitive myeloid cell lines. Nuclei were incubated with HaeIII. DNA was subsequently extracted, digested with SacI, and hybridized with probes indicated in B. The absence of the +3 hypersensitive site in M1 is consistent with previous DNaseI hypersensitive site analysis (Göttgens et al. 1997). (B) Summary of restriction endonuclease data for the 5′ region of the mouse SCL gene. The top part of the diagram shows the approximate locations of previously mapped DNaseI hypersensitive sites (gray arrowheads labeled P1a, P1b, +1HS, and +3HS) followed by the positions of mouse SCL exons 1a to 3 and the SacI sites used for the Southern blots shown in part A. This is followed by restriction maps for the three enzymes (AvaII, HhaI, and HaeIII) used to determine endonuclease sensitivity, and a summary of the endonuclease sensitivity experiments in which black and gray boxes represent the minimum and maximum regions of endonuclease accessibility in 416B and/or M1 cells. The profile of the mouse/human alignment underneath is a 6250 nucleotide section of the alignment from Fig. 3 (see yellow bar in Fig. 3) and shows the concordance of endonuclease accessibility and sequence conservation.
We have previously identified 11 DNaseI hypersensitive sites in a ∼40 kb portion of the mouse SCL locus (see arrowheads in Fig. 3) (Göttgens et al. 1997). All of these corresponded to regions of endonuclease accessibility and peaks in the homology profile. These 11 sites represent approximately one-third of all geographically distinct regions of high sequence similarity. A further four peak regions correspond to coding exons and the sequence surrounding the polyadenylation site. The significance of the remaining peaks of homology remains unclear. However, it is likely that at least some of these peaks will prove to mark the sites of enhancers associated with DNaseI hypersensitive sites in tissues in which the chromatin structure of the SCL locus has not yet been characterized. Alternatively, they may mark the sites of regulatory elements not linked with DNaseI hypersensitive sites, but which control other conserved processes such as domain structure, chromatin architecture, or DNA replication.
DISCUSSION
In this paper, we present a detailed comparison of 193 kb of the human to 234 kb of the mouse SCL locus. Four new genes and an ancient mitochondrial transposition have been identified, and a new program to support long-range comparative analysis has been developed. In addition, our results show for the first time that not only do long-range human/mouse sequence comparisons help locate chromatin sites associated with regulatory regions, but also that peaks of human/mouse homology accurately predict the extent of such regions of accessible DNA.
Correlation of Sequence Homology and Sites of Endonuclease Accessibility
A major objective of genome sequence analysis is to identify transcriptional regulatory elements in DNA sequences, and from this information to understand transcriptional networks in higher organisms. The SCL gene encodes a critical transcriptional regulator of hemopoiesis and vasculogenesis, and the molecular basis for the tissue-specific regulation of the murine SCL gene has been characterized in considerable detail (Göttgens et al. 1997; Sanchez et al. 1999; Sinclair et al. 1999; Göttgens et al. 2000). Here we describe detailed sequence comparisons of the human and mouse SCL loci, which together with a comprehensive chromatin structure analysis, have allowed us to draw several important conclusions.
First, all DNaseI hypersensitive sites linked to known SCL regulatory regions were associated with localized zones of restriction endonuclease accessibility that coincided with regions of high human/mouse sequence homology. It is not clear whether both DNaseI and restriction endonucleases reveal the same structural features, and the question remains regarding the pattern of endonuclease sensitivity that might be observed in a cell that does not express SCL. However, our data are consistent with a model in which both assays detect aspects of a localized region of an altered chromatin structure, which is associated with active SCL regulatory regions. There are several possible explanations for the peaks of homology that do not correspond to zones of DNaseI hypersensitivity or endonuclease accessibility. The chromatin studies have only been performed in a limited number of hemopoietic lineages and additional localized regions of altered chromatin structure may exist in other SCL-expressing cell types. Alternatively, some categories of enhancers may not be associated with such regions, as may other classes of regulatory regions that may have important roles in conserved processes such as gene silencing, domain structure, and chromosome architecture.
Second, high-resolution analysis of the chromatin structure of SCL regulatory regions showed that the extent of each individual homology peak corresponded precisely with the region accessible to endonucleases. This observation indicates that human/mouse sequence comparisons will greatly reduce the experimental studies required to define minimal functional units associated with enhancers. Moreover, incorporation of more distant species into the comparisons permits the generation of phylogenetic footprints (Aparicio et al. 1995; Popperl et al. 1995; Nonchev et al. 1996), which can identify conserved transcription factor binding sites, and thus provide rapid insight into the detailed architecture of an enhancer.
Third, our results confirm the utility of comparative sequence analysis as a way of identifying regulatory regions within the reams of sequence information generated by genome sequencing projects. The combination of comparative sequence analysis with a rapid and high throughput transgenic Xenopus assay (Kroll and Amaya 1996) provides a powerful strategy, particularly if focused on critical developmental genes with functions and expression patterns conserved throughout vertebrate evolution (Göttgens et al. 2000).
Interpretation of Comparative Sequence Analysis
Widespread application of long-range comparative sequence analysis will require the use of intuitive computational tools. At present, the most widely used approach is that of percentage identity plots (PIPs) generated by the PIPMaker program (Schwartz et al. 2000). These do not require a global alignment, but instead are based on local alignments and display areas of high local similarity between a reference and the test sequence, with the advantage that PIPs can display matches if the two sequences are not colinear. However, PIPs can display only the features (exons, repeats, etc.) of one of the two loci (i.e., the reference sequence), and the relative position of high-scoring local alignments within the test sequence cannot be displayed. A comparison of human, sheep, and mouse PrP loci (Lee et al. 1998) has provided an example of how locus features can be displayed in combination with a sequence similarity profile. However, this approach involves multiple steps, including the removal and reinsertion of species-specific repeats. Moreover, neither the alignment program nor the display tools have been published or been made available on the Internet.
Therefore, we have developed a new software tool (SynPlot), which is based on a global alignment and has several novel features. First, the nature and positions of features, such as exons, repeat elements, or CpG islands, can be shown for both sequences, thereby indicating the position of homology peaks relative to the gene structure of both loci. Second, SynPlot allows large-scale sequence alignments of multiple (i.e., >2) sequences to be presented in a single graphical display. Third, the strategy used by SynPlot to transform the coordinates of sequence features to their new positions in the global alignment can be incorporated into future sequence analysis applications. These coordinates are read into SynPlot from a file in General Feature Format or GFF (see http://www.sanger.ac.uk/Software/formats/GFF/), which will facilitate the display of sequence features from other analysis programs and databases (e.g., the ENSEMBL project; see http://www.ensembl.org/).
The Dialign algorithm (Morgenstern et al. 1998) used in this manuscript does not employ gap penalties, but instead collects small gap-free alignments, which are weighted depending on their percent identity and length. The global alignment is constructed by assembling these local alignments in an optimized colinear configuration. Whereas this approach overcomes many problems of generating global alignments, its utility is restricted to colinear loci. Importantly, however, SynPlot and PIPMaker produce similar results on such loci when used for the identification of conserved noncoding sequences (unpubl.) even though they are based on very different principles.
When performing comparative analysis, it is our practice to start by assessing collinearity, e.g., by dotter analysis (Sonnhammer and Durbin 1995). “Working draft” sequence is noncontiguous, and therefore not suitable for long-range global alignments. Consequently, in the absence of colinearity and for working draft sequence, PIPs remain the preferred approach. However, if colinearity is shown, SynPlot analysis is performed to take advantage of its ability to present the position of homology peaks relative to other features in all of the loci being compared, together with its ability to display comparisons between multiple sequences.
Comparative Analysis of Evolutionarily Active Loci
To date, long-range human/mouse comparative sequence analyses have focused on loci conserved during mammalian evolution. The SCL locus on human chromosome 1 is part of a long region of synteny with mouse chromosome 4. However, our results show that, within this long region, the CYP4 loci have evolved substantially because the divergence of man and mouse and are not colinear. The cytochrome P450 monooxygenases (CYPs) are a large and ancient superfamily of proteins, often involved in the metabolism of hormones or foreign compounds such as toxins or drugs. Therefore, the acquisition of different sets of CYP genes may partially explain the different sensitivity of humans and mice to drugs and carcinogens.
Rapidly evolving loci, such as the human and mouse CYP4 clusters, highlight two challenges for comparative analysis. First, unless the individual sequences are annotated very carefully, incorrect homology relationships between human and mouse genes may be inferred. Second, the usefulness of comparative analysis for the prediction of regulatory regions may be severely impaired. Such predictions are based on common ancestry, and true orthologs may be difficult to establish. Moreover, rapid evolution may be accomplished by the acquisition of distinct expression patterns by individual members of a gene cluster.
Interpreting the Genome Code
Genome projects are producing a vast data resource which will have to be made accessible to the scientific community. It is clear that, in addition to the primary DNA sequence, genome-wide data on gene expression patterns, protein structures, and protein-protein interactions will also be available in the near future. Our own interest lies in how gene regulatory networks are encoded in the primary DNA sequence, and we use comparative sequence analysis to address this interest. The data presented here indicate that a very careful analysis of individual loci is necessary to establish true homology relationships before proceeding with the comparative analysis. We also show that human/mouse comparisons can give insights into recent genome evolution. Most importantly, we show that comparative sequence analysis, combined with an intuitive graphical display, will be very useful for the prediction of the location and size of gene regulatory regions.
METHODS
Isolation and Sequencing of 129/SvEvTac Murine SCL Locus
The RPCI-21 (129S6/SvEvTac) mouse PAC library (segment 2) (Osoegawa et al. 2000) was screened with a probe from exon 6 of the mouse SCL gene. Eight positive clones were identified and further analyzed by PFGE and Southern analysis. One clone (PAC129.0.726) did not hybridize to a probe from the 5′ end of the mouse SCL gene and contained a large insert (>150 kb), thereby maximizing the extension of the known mouse SCL sequence. The insert of clone PAC129.0.726 was fully sequenced as described (Göttgens et al. 1998). The insert was found to be 171,863 bp, and it extended the previously known mouse SCL sequence by 148,000 bp. The human and mouse sequences used in this study, together with their respective feature files, are available at http://www.sanger.ac.uk/∼jgrg/SynPlot.
Sequence Analysis
Interactive annotation of genomic sequences was performed within ACeDB (Eeckman and Durbin 1995) as described (Göttgens et al. 1998). Global alignments were calculated using Dialign version 1 (Morgenstern et al. 1998) with the threshold for diagonals (T) set to 20. We wrote an application, SynPlot (see http://www.sanger.ac.uk/∼jgrg/SynPlot), to visualize global alignments of syntenic genomic sequence. SynPlot is a set of Perl modules and a driver script and is freely available from the SynPlot Web site. It takes as its input aligned sequences in fasta format, with gaps in the sequences introduced by the alignment represented by '-' characters. We have also written the necessary modules to feed the output of the recently published GLASS global sequence alignment algorithm (Batzoglou et al. 2000) into SynPlot (see http://www.sanger.ac.uk/∼jgrg/SynPlot). The percentage identity is calculated from the alignment within a sliding window, the width of which can be specified by the user. This information is used to draw a picture of the alignment in postscript format. The sequences are rendered as lines interrupted by spaces corresponding to the gaps introduced by the alignment, with a plot of the percentage identity underneath. Features can also be drawn on the sequence lines. This uses a GFF format file (see http://www.sanger.ac.uk/Software/formats/GFF) output by ACeDB from the annotated genomic sequence, and a configuration file, which specifies the color, height, and order in which the rectangles representing the features are drawn. Small-scale DNA alignments were performed and displayed as described (Göttgens et al. 1998). G/C profiles were calculated and displayed using the GCG sequence analysis package (Wisconsin package version 8.0, Genetics Computer Group) using a window size of 2000 bp with an incremental move of 320 bp. BLASTX scores for mitochondrial sequences were calculated using the vertebrate mitochondrial genetic code.
Restriction Endonuclease Accessibility Assay
M1 and 416B cells were maintained as described previously (Bockamp et al. 1997). For the restriction endonuclease assay, 2 × 108 cells were washed twice in PBS before resuspending in 8 mL of lysis buffer (50 mM KCl, 10 mM MgSO4, 3 mM DTT, 5 mM HEPES at pH 7.2, 0.05% NP-40, and 1 mM PMSF), followed by a 60 min incubation at room temperature. Nuclei were collected by 5 min centrifugation at 5000 g, washed in RSB (10 mM NaCl, 10 mM Tris at pH 7.4, 3 mM MgCl2), and resuspended in 10 mL restriction enzyme buffer 2 (New England Biolabs, Hitchin, UK). Four hundred μL aliquots were digested with 0, 80, 160, or 320 units of PalI (HaeIII), AvaII, or HhaI for 60 min at 37°C, followed by conventional DNA isolation and Southern blot analysis using SacI digestion and the following probes: a = 900 bp SmaI/SacI fragment; b = 180 bp SmaI/SacI fragment (Fig. 4B).
Acknowledgments
This work was supported by the Wellcome Trust and the Kay Kendall Leukemia Fund. We also acknowledge the support of Sequencing Team 47 at the Sanger Centre. We are also grateful for the help of D. Bell (Nottingham) with CYP4 nomenclature.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL bg200@cam.ac.uk; FAX 44-1223-762670.
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.153001.
REFERENCES
- Altschul SF, Gish W, Miller W, Meyers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Ansari-Lari MA, Oeltjen JC, Schwartz S, Zhang Z, Muzny DM, Lu J, Gorrell JH, Chinault AC, Belmont JW, Miller W, et al. Comparative sequence analysis of a gene-rich cluster at human chromosome 12p13 and its syntenic region in mouse chromosome 6. Genome Res. 1998;8:29–40. [PubMed] [Google Scholar]
- Aparicio S, Morrison A, Gould A, Gilthorpe J, Chaudhuri C, Rigby P, Krumlauf R, Brenner S. Detecting conserved regulatory elements with the model genome of the Japanese puffer fish, Fugu rubripes. Proc Natl Acad Sci. 1995;92:1684–1688. doi: 10.1073/pnas.92.5.1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton LM, Göttgens B, Green AR. The stem cell leukemia (SCL) gene: A critical regulator of haemopoietic and vascular development. Int J Biochem Cell Biol. 1999;31:1193–1207. doi: 10.1016/s1357-2725(99)00082-5. [DOI] [PubMed] [Google Scholar]
- Batzoglou S, Pachter L, Mesirov JP, Berger B, Lander ES. Human and mouse gene structure: Comparative analysis and application to exon prediction. Genome Res. 2000;10:950–958. doi: 10.1101/gr.10.7.950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begley CG, Green AR. The SCL gene: From case report to critical hematopoietic regulator. Blood. 1999;93:2760–2770. [PubMed] [Google Scholar]
- Bernardi G. Isochores and the evolutionary genomics of vertebrates. Gene. 2000;241:3–17. doi: 10.1016/s0378-1119(99)00485-0. [DOI] [PubMed] [Google Scholar]
- Bockamp EO, McLaughlin F, Murrell AM, Göttgens B, Robb L, Begley CG, Green AR. Lineage-restricted regulation of the murine SCL/TAL-1 promoter. Blood. 1995;86:1502–1514. [PubMed] [Google Scholar]
- Bockamp EO, McLaughlin F, Göttgens B, Murrell AM, Elefanty AG, Green AR. Distinct mechanisms direct SCL/tal-1 expression in erythroid cells and CD34 positive primitive myeloid cells. J Biol Chem. 1997;272:8781–8790. doi: 10.1074/jbc.272.13.8781. [DOI] [PubMed] [Google Scholar]
- Bockamp EO, Fordham JL, Göttgens B, Murrell AM, Sanchez MJ, Green AR. Transcriptional regulation of the stem cell leukemia gene by PU.1 and Elf-1. J Biol Chem. 1998;273:29032–29042. doi: 10.1074/jbc.273.44.29032. [DOI] [PubMed] [Google Scholar]
- Boyes J, Felsenfeld G. Tissue-specific factors additively increase the probability of the all-or-none-formation of a hypersensitive site. EMBO J. 1996;15:2496–2507. [PMC free article] [PubMed] [Google Scholar]
- Brickner AG, Koop BF, Aronow BJ, Wiginton DA. Genomic sequence comparison of the human and mouse adenosine deaminase gene regions. Mamm Genome. 1999;10:95–101. doi: 10.1007/s003359900951. [DOI] [PubMed] [Google Scholar]
- Brookes AJ. The essence of SNPs. Gene. 1999;234:177–186. doi: 10.1016/s0378-1119(99)00219-x. [DOI] [PubMed] [Google Scholar]
- Drake CJ, Fleming PA. Vasculogenesis in the day 6.5 to 9.5 mouse embryo. Blood. 2000;95:1671–1679. [PubMed] [Google Scholar]
- du Buy HG, Riley FL. Hybridization between the nuclear and kinetoplast DNAs of Leishmania enrietti and between nuclear and mitochondrial DNA's of mouse liver. Proc Natl Acad Sci. 1967;57:790–797. doi: 10.1073/pnas.57.3.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin, R. and Mieg, J.T. 1991. A C. elegans database. Documentation, code and data available from anonymous FTP servers at http://lirmm.lirmm.fr, http://cele.mrc-lmb.cam.ac.uk, and http://ncbi.nlm.nih.gov
- Eeckman FH, Durbin R. ACeDB and macace. Methods Cell Biol. 1995;48:583–605. [PubMed] [Google Scholar]
- Endrizzi M, Huang S, Scharf JM, Kelter AR, Wirth B, Kunkel LM, Miller W, Dietrich WF. Comparative sequence analysis of the mouse and human Lgn1/SMA interval. Genomics. 1999;60:137–151. doi: 10.1006/geno.1999.5910. [DOI] [PubMed] [Google Scholar]
- Flint J, Thomas K, Micklem G, Raynham H, Clark K, Doggett NA, King A, Higgs DR. The relationship between chromosome structure and function at a human telomeric region. Nat Genet. 1997;15:252–257. doi: 10.1038/ng0397-252. [DOI] [PubMed] [Google Scholar]
- Fordham JL, Göttgens B, McLaughlin F, Green AR. Chromatin structure and transcriptional regulation of the stem cell leukemia (SCL) gene in mast cells. Leukemia. 1999;13:750–759. doi: 10.1038/sj.leu.2401420. [DOI] [PubMed] [Google Scholar]
- Gering M, Rodaway ARF, Göttgens B, Patient RK, Green AR. The SCL gene specifies haemangioblast development from early mesoderm. EMBO J. 1998;17:4029–4045. doi: 10.1093/emboj/17.14.4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göttgens B, McLaughlin F, Bockamp EO, Fordham JL, Begley CG, Kosmopoulos K, Elefanty AG, Green AR. Transcription of the SCL gene in erythroid and CD34 positive primitive myeloid cells is controlled by a complex network of lineage-restricted chromatin-dependent and chromatin-independent regulatory elements. Oncogene. 1997;15:2419–2428. doi: 10.1038/sj.onc.1201426. [DOI] [PubMed] [Google Scholar]
- Göttgens B, Gilbert JGR, Barton LM, Aparicio S, Hawker K, Mistry S, Vaudin M, King A, Bentley D, Elgar G, et al. The pufferfish SLP-1 gene, a new member of the SCL/TAL-1 family of transcription factors. Genomics. 1998;48:52–62. doi: 10.1006/geno.1997.5162. [DOI] [PubMed] [Google Scholar]
- Göttgens B, Barton LM, Gilbert JG, Bench AJ, Sanchez MJ, Bahn S, Mistry S, Grafham D, McMurray A, Vaudin M, et al. Analysis of vertebrate SCL loci identifies conserved enhancers. Nat Biotechnol. 2000;18:181–186. doi: 10.1038/72635. [DOI] [PubMed] [Google Scholar]
- Green AR, Visvader J, Lints T, Harvey R, Begley CG. SCL is co-expressed with GATA-1 in haemopoietic cells but is also expressed in developing brain. Oncogene. 1992;7:653–660. [PubMed] [Google Scholar]
- Hardison RC, Oeltjen J, Miller W. Long human-mouse sequence alignments reveal novel regulatory elements: A reason to sequence the mouse genome. Genome Res. 1997;7:959–966. doi: 10.1101/gr.7.10.959. [DOI] [PubMed] [Google Scholar]
- Jackson JD, Petrykowska H, Philipsen S, Miller W, Hardison R. Role of DNA sequences outside the cores of DNase hypersensitive sites (HSs) in functions of the beta-globin locus control region. Domain opening and synergism between HS2 and HS3. J Biol Chem. 1996;271:11871–11878. doi: 10.1074/jbc.271.20.11871. [DOI] [PubMed] [Google Scholar]
- Jang W, Hua A, Spilson SV, Miller W, Roe BA, Meisler MH. Comparative sequence of human and mouse BAC clones from the mnd2 region of chromosome 2p13. Genome Res. 1999;9:53–61. [PMC free article] [PubMed] [Google Scholar]
- Jareborg N, Birney E, Durbin R. Comparative analysis of noncoding regions of 77 orthologous mouse and human gene pairs. Genome Res. 1999;9:815–824. doi: 10.1101/gr.9.9.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallianpur AR, Jordan JE, Brandt SJ. The SCL/TAL-1 gene is expressed in progenitors of both the hematopoietic and vascular systems during embryogenesis. Blood. 1994;83:1200–1208. [PubMed] [Google Scholar]
- Kroll KK, Amaya E. Transgenic Xenopus embryos from sperm nuclear transplantations reveal FGF signaling requirements during gastrulation. Development. 1996;122:3173–3183. doi: 10.1242/dev.122.10.3173. [DOI] [PubMed] [Google Scholar]
- Lecointe N, Bernard O, Naert K, Joulin V, Larsen CJ, Romeo PH, Mathieu-Mahul D. GATA- and SP1-binding sites are required for the full activity of the tissue-specific promoter of the tal-1 gene. Oncogene. 1994;9:2623–2632. [PubMed] [Google Scholar]
- Lee IY, Westaway D, Smit AF, Wang K, Seto J, Chen L, Acharya C, Ankener M, Baskin D, Cooper C, et al. Complete genomic sequence and analysis of the prion protein gene region from three mammalian species. Genome Res. 1998;8:1022–1037. doi: 10.1101/gr.8.10.1022. [DOI] [PubMed] [Google Scholar]
- Liao EC, Paw BH, Oates AC, Pratt SJ, Postlethwait JH, Zon LI. SCL/Tal-1 transcription factor acts downstrem of cloche to specify hematopoietic and vascular progenitors in zebrafish. Genes & Devel. 1998;12:621–626. doi: 10.1101/gad.12.5.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindblad-Toh K, Winchester E, Daly MJ, Wang DG, Hirschhorn JN, Laviolette JP, Ardlie K, Reich DE, Robinson E, Sklar P, et al. Large-scale discovery and genotyping of single-nucleotide polymorphisms in the mouse. Nat Genet. 2000;24:381–386. doi: 10.1038/74215. [DOI] [PubMed] [Google Scholar]
- Loots GG, Locksley RM, Blankespoor CM, Wang ZE, Miller W, Rubin EM, Frazer KA. Identification of a coordinate regulator of interleukins 4, 13, and 5 by cross-species sequence comparisons. Science. 2000;288:136–140. doi: 10.1126/science.288.5463.136. [DOI] [PubMed] [Google Scholar]
- Mead PE, Kelley CM, Hahn PS, Piedad O, Zon LI. SCL specifies hematopoietic mesoderm in Xenopus embryos. Development. 1998;125:2611–2620. doi: 10.1242/dev.125.14.2611. [DOI] [PubMed] [Google Scholar]
- Morgenstern B, Frech K, Dress A, Werner T. DIALIGN: Finding local similarities by multiple sequence alignment. Bioinformatics. 1998;14:290–294. doi: 10.1093/bioinformatics/14.3.290. [DOI] [PubMed] [Google Scholar]
- Nonchev S, Maconochie M, Vesque C, Aparicio S, Arizamcnaughton L, Manzanares M, Maruthainar K, Kuroiwa A, Brenner S, Charnay P, et al. The conserved role of Krox-20 in directing Hox gene-expression during vertebrate hindbrain segmentation. Proc Natl Acad Sci. 1996;93:9339–9345. doi: 10.1073/pnas.93.18.9339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oeltjen JC, Malley TM, Muzny DM, Miller W, Gibbs RA, Belmont JW. Large-scale comparative sequence analysis of the human and murine Bruton's tyrosine kinase loci reveals conserved regulatory domains. Genome Res. 1997;7:315–329. doi: 10.1101/gr.7.4.315. [DOI] [PubMed] [Google Scholar]
- Osoegawa K, Tateno M, Woon PY, Frengen E, Mammoser AG, Catanese JJ, Hayashizaki Y, de Jong PJ. Bacterial artificial chromosome libraries for mouse sequencing and functional analysis. Genome Res. 2000;10:116–128. [PMC free article] [PubMed] [Google Scholar]
- Popperl H, Bienz M, Studer M, Chan SK, Aparicio S, Brenner S, Mann RS, Krumlauf R. Segmental expression of Hoxb-1 is controlled by a highly conserved autoregulatory loop dependent upon exd/pbx. Cell. 1995;81:1031–1042. doi: 10.1016/s0092-8674(05)80008-x. [DOI] [PubMed] [Google Scholar]
- Porcher C, Swat W, Rockwell K, Fujiwara Y, Alt FW, Orkin SH. The T cell leukemia oncoprotein SCL/tal-1 is essential for development of all hematopoietic lineages. Cell. 1996;86:47–57. doi: 10.1016/s0092-8674(00)80076-8. [DOI] [PubMed] [Google Scholar]
- Robb L, Elwood NJ, Elefanty AG, Köntgen F, Li R, Barnett LD, Begley CG. The SCL gene product is required for the generation of all hematopoietic lineages in the adult mouse. EMBO J. 1996;15:4123–4129. [PMC free article] [PubMed] [Google Scholar]
- Sanchez MJ, Göttgens B, Sinclair AM, Stanley M, Begley CG, Hunter S, Green AR. An SCL 3′ enhancer targets developing endothelium together with embryonic and adult haematopoietic progenitors. Development. 1999;126:3891–3904. doi: 10.1242/dev.126.17.3891. [DOI] [PubMed] [Google Scholar]
- Schwartz S, Zhang Z, Frazer KA, Smit A, Riemer C, Bouck J, Gibbs R, Hardison R, Miller W. PipMaker-a web server for aligning two genomic DNA sequences. Genome Res. 2000;10:577–586. doi: 10.1101/gr.10.4.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair AM, Göttgens B, Barton LM, Aparicio S, Bahn S, Sanchez MJ, Fordham J, Stanley ML, Green AR. Distinct 5′ SCL enhancers direct transcription to developing brain, spinal chord and endothelium; conserved neural expression is GATA factor dependant. Dev Biol. 1999;209:128–142. doi: 10.1006/dbio.1999.9236. [DOI] [PubMed] [Google Scholar]
- Sonnhammer EL, Durbin R. A dot-matrix program with dynamic threshold control suited for genomic DNA and protein sequence analysis. Gene. 1995;167:GC1–GC10. doi: 10.1016/0378-1119(95)00714-8. [DOI] [PubMed] [Google Scholar]
- Taillon-Miller P, Gu Z, Li Q, Hillier L, Kwok PY. Overlapping genomic sequences: A treasure trove of single-nucleotide polymorphisms. Genome Res. 1998;8:748–754. doi: 10.1101/gr.8.7.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuzuki T, Nomiyama H, Setoyama C, Maeda S, Shimada K. Presence of mitochondrial-DNA-like sequences in the human nuclear DNA. Gene. 1983;25:223–229. doi: 10.1016/0378-1119(83)90226-3. [DOI] [PubMed] [Google Scholar]
- Visvader JE, Fujiwara Y, Orkin SH. Unsuspected role for the T-cell leukemia protein SCL/tal-1 in vascular development. Genes & Dev. 1998;12:473–479. doi: 10.1101/gad.12.4.473. [DOI] [PMC free article] [PubMed] [Google Scholar]