

Abstract
Immune-mediated cancer regression requires tumor infiltration by Ag-specific effector T cells, but lymphocytes are commonly sparse in melanoma metastases. Activated T cells express CXCR3, whose cognate chemokines are CXCL9/MIG, CXCL10/IP-10 and CXCL11/I-TAC. Little is known about expression of these chemokines in lymph node (LN) metastases of melanoma. We evaluated whether metastatic melanoma induces these CXCR3-cognate chemokines in human LN-derived tissues. Also, because these chemokines can be induced by interferon (IFN), we evaluated whether type I or II IFNs (IFN-α or IFN-γ, respectively) can modulate chemokine expression in an in vitro model of the human tumor microenvironment. Production of CXCL9-11 by melanoma-infiltrated nodes (MIN) was no different than tumor-free nodes (TFN); both produced less chemokine than activated LN (sentinel immunized nodes, SIN). These data suggest melanoma infiltration into LN neither induces nor reduces CXCL9-11. Stimulation with IFN-α or IFN-γ increased production of CXCL10-11 from MIN, but not TFN or SIN. IFN-γ also increased production of CXCL9 in MIN. In IFN-treated SIN, CD14+ cells were the primary source of CXCL9-11, whereas melanoma cells were the source of chemokine in MIN. Melanoma cells in MIN express IFN receptors. Consistent with these observations, multiple human melanoma lines expressed IFN receptors and produced CXCL9-11 in response to IFN treatment. Thus, melanoma infiltration of LN is insufficient to induce the production of CXCL9-11, but melanoma may be a significant source of IFN-induced chemokines. Collectively, these data suggest that IFN-α or IFN-γ may act in the tumor microenvironment to increase the chemotactic gradient for CXCR3+ T cells.
Keywords: melanoma/skin cancer, interferon-alpha, interferon-gamma, chemokines, chemokine receptors, CXCR3
Introduction
Effective immune therapy of cancer, whether using cancer vaccines or adoptive cellular therapy, arguably requires infiltration of antigen-specific effector T cells into the tumor microenvironment. However, metastatic melanomas often lack significant numbers of tumor-infiltrating lymphocytes (TIL)1, regardless of the presence of circulating tumor Ag-specific T cells. Lymphocyte extravasation and homing into tumor compartments is mediated, in part, by tissue-derived chemotactic molecules (chemokines) acting on surface-expressed G-protein-coupled receptors (chemokine receptors, CCRs). The absence of TIL may reflect inappropriate CCR expression by tumor-specific lymphocytes, or the lack of a T cell-chemotactic gradient at the site of tumor.
We demonstrated an association between duration of survival in patients with regionally metastatic melanoma and the presence of circulating tumor-reactive CD8+ T cells expressing the tissue-homing receptor (CXC chemokine receptor 3, CXCR3)2, suggesting that CXCR3 may facilitate or enhance T cell entry and control of melanoma metastases. CXCR3 is expressed on activated effector lymphocytes3;4, and vaccination with synthetic peptides in adjuvant5 induces populations of CXCR3+ melanoma Ag-specific CD8+ T cells*. T cells expressing CXCR3 and tissue-specific integrins achieve firm adhesion to vascular endothelium and extravasate into tissues rich in the CXCR3-cognate chemokines (CXCL9/MIG, CXCL10/IP-10 and CXCL11/I-TAC)6-9, and CXCL10 expression has been specifically associated with T cell recruitment to metastatic tumors10-12. Kunz and colleagues10 demonstrated lymphocytic clustering in areas of metastatic melanoma with high staining for CXCL10, suggesting that a subset of melanoma lesions attract CXCR3+ T cells through production of CXCL10, and possibly other chemokines, in the tumor microenvironment. CXCR3 has also been demonstrated to mediate lymph node (LN) infiltration by CD62L-CCR7- effector T cells6, suggesting that immune control of lymphatic metastases may require the local expression of CXCR3-cognate chemokines. However, the production of these chemokines has not been evaluated in melanoma-infiltrated human lymphoid compartments.
In a variety of cells and tissues, type I and II interferons (IFN-α/β and IFN-γ, respectively) induce the expression of CXCL98, CXCL1013, and CXCL1114, and IFNs are being investigated therapeutically in a variety of clinical settings, including melanoma15. IFN-α (Intron A®) is an approved agent for adjuvant systemic therapy in advanced melanoma16, and neoadjuvant therapy with high dose IFN-α was recently reported to induce objective tumor regression and to increase CD3+ cell infiltration in regional node metastases of melanomas17 through an undetermined biologic mechanism. IFN-γ (IFN-γ Ib, Actimmune), is FDA-approved for the treatment of chronic granulomatous disease and severe malignant osteopetrosis18-20. Systemic IFN-γ was previously evaluated in adjuvant therapy studies for melanoma without demonstrable clinical benefit 21;22. Arguably, a major limitation of systemic administration of IFN is its potential to induce chemokines systemically, rather than to induce local chemokine gradients. We are not aware of any prior studies of the local effects of IFN on chemokine production by melanoma or melanoma-infiltrated lymph nodes. Also, in anticipation of clinical studies of local or regional IFN therapy, we are interested in defining the relative effects of type I and type II interferons on induction of the CXCR3-cognate chemokines, a question that has not previously been addressed systematically.
In the present study, we demonstrate that infiltration by melanoma is not sufficient to induce the production of CXCL9-11 in human LN, but that chemokine production by melanoma cells – but not melanoma-associated lymphoid cells – can be induced by treatment with recombinant IFN. These data demonstrate a fundamental defect in CXCR3 chemotactic activity in melanoma-infiltrated LN and suggest a novel approach for combination vaccine/cytokine therapy to restore the chemotactic gradient and to enhance T cell infiltration of melanoma metastases.
Materials and Methods
Reagents
Recombinant human IFN-α (Intron A®) was purchased from Schering Corporation. Recombinant human IFN-γ, Abs for flow cytometric detection, and reagents for ELISA-based quantification of CXCL9, CXCL10, CXCL11, and CCL21 were purchased from R&D Systems.
Human tissue samples
Peripheral whole blood samples were collected from nine healthy donors under informed consent, as approved by the University of Virginia Human Investigation Committee (HIC, protocol 10598). Whole blood was heparinized during collection. Melanoma involved lymph nodes (MIN) and tumor-free nodes (TFN) were collected with informed consent from patients as part of standard clinical care or under a previous clinical trial protocol (HIC protocol 8380). Sentinel immunized LN (SIN) were collected from patients vaccinated with 4, 6 or 12 peptides under clinical trial protocols (HIC protocols 8878 & 10464)23;24. Independent pathology review of specimens (University of Virginia Department of Clinical Pathology) confirmed the presence of melanoma in MIN and the absence of melanoma in TFN and SIN. Tissue from MIN and TFN were first rendered into single cell suspensions by mechanical dissociation, and remaining solid fragments were enzymatically digested with collagenase, deoxyribonuclease I and hyaluronidase for 2-24 hours, then viably cryopreserved in 10% DMSO and fetal calf serum or human AB serum and stored in liquid nitrogen until evaluation. SIN samples were rendered into single cell suspensions by mechanical dissociation, without enzymatic digestion, then viably cryopreserved in 10% DMSO and fetal calf serum or human AB serum and stored in liquid nitrogen until evaluation.
Melanoma cell lines
Melanoma cells lines were obtained from Duke University (DM93, DM331) or established from patients at the University of Virginia (VMM12, VMM15, VMM18, VMM19, VMM39), with IRB approval or waiver (HIC protocols 5202 and 6346). MV3 cells were provided by Dr. Soldano Ferrone (University of Pittsburgh). Melanoma cells were maintained in RPMI-1640 media supplemented with 5% FBS, and cells were >98% viable by Trypan blue staining at time of assay.
Chemokine production assays in LN samples
Cryopreserved viable single-cell suspensions (SCS) of human LN (5×105 mononuclear immune cells per mL) were plated in 48 well tissue culture cluster plates (Corning), then rested at 37°C in a humidified 5% CO2 atmosphere for 12 h. Cell were then cultured for 24h either 1) left untreated or 2) treated with 1×105 International Units (IU) of IFN-α or IFN-γ, unless noted otherwise. Samples were then centrifuged, and cell-free supernatants were analyzed in triplicate for chemokine concentration by specific ELISA (R&D Systems), per the manufacturer's protocol. Some cells were also analyzed for intracellular chemokine expression by permeabilization with BD Biosciences Cytofix/Cytoperm and staining with specific chemokine mAb or isotype control mAb (R&D Systems), including anti-CD45, anti-CD14, anti-CD11c, and anti-S100. Stained cells were analyzed using a BD FacsCalibur or Dako Cyan cytometer. Dead cells were excluded by staining with 7-AAD, and doublets were excluded by FSC/SSC gating.
Assays for chemokine production by human melanoma cell lines
Established melanoma cells lines were cultured at 1×106 cells/well in 24-well tissue culture plates and rested for 24 h, then cultured for 24 h either 1) untreated or 2) with 1×105 IU of IFN-α or IFN-γ. Cell-free supernatants were evaluated for CXCL9-11, as described.
Evaluating effects of melanoma supernatants on whole blood
Melanoma cell supernatant was collected from 24h cultures of DM331 and DM93 cells plated at 5×106 cells/ml. The cell-free supernatant from the melanoma cell lines was added to one mL of whole blood samples from six normal donors at escalating doses from 0 to 1000 μL of melanoma supernatants. Additional medium was added to each sample as needed to bring the total volume to 2 mL. The samples were incubated with 1) no treatment, 2) 1×105 IU of IFN–α, or 2) 1×105 IU IFN–γ. After 24 hours of incubation, the samples were centrifuged and the supernatants were analyzed for production of CXCL9-11 via by ELISA.
Evaluation of human melanoma cells for expression of IFN receptors
Melanoma cell lines were stained with antibodies MAB 245 (clone 85228, R&D Systems) for IFN-αR1, MAB 6731 (clone 92101, R&D systems) for IFN-γR1, and an isotype-matched control, and evaluated by flow cytometry. MIN were stained with the same IFN receptor-specific Abs, as well as anti-CD45, anti-CD11c, anti-CD14, and anti-S100.
Statistical analyses
ELISA data are presented as mean ± standard deviation of triplicate determinations in a single assay, and data are representative of at least 3 independent experiments. The significance of difference in the production of CXCL9-11 was determined using a Student's t test, or a Wilcoxon-Mann-Whitney test when the data did not meet normality criteria, using SigmaStat (Systat Software). Pairwise analysis of variance (ANOVA with 95% Tukey's simultaneous confidence interval) was used assess differences in chemokine production in response to cytokine treatments and calculated using Minitab 15 (Minitab Inc.). P values of <0.05 were considered significant.
Results
Vaccination, but not infiltration by malignant melanoma, conditions lymph nodes for production of CXCL9-11 and CCL21
Inflammation induces production of CXCR3-cognate chemokines on the luminal side of LN HEVs6 through a TNF-α-dependent mechanism25. We anticipated that inflammation (manifest by CCL21 production26;27) would be associated with production of the CXCR3-cognate chemokines in human LN. Further, because of the growing link between inflammation and cancer, we hypothesized that infiltration by melanoma may induce, directly or indirectly, the production of those chemokines in human LN. To test these possibilities, we measured CXCL9, CXCL10, CXCL11, and CCL21 production by single-cell suspensions (SCS) of MIN and TFN from melanoma patients and SIN from patients receiving experimental multipeptide vaccines with Montanide ISA-51 adjuvant 23;24. During short-term (24h) culture, CXCL9-11 production by TFN and MIN samples was not different (Fig. 1A-C, p>0.05, corrected for multiple comparisons). However, SIN samples produced significantly (p<0.05) greater quantities of CXCL9, CXCL10, and CXCL11 protein than TFN or MIN samples (Fig. 1A-C). SIN drain inflamed skin at the site of the melanoma vaccine and thus are expected to be reactive nodes26, and SIN produced significantly more CCL21 than TFN and MIN (Fig. 1D). In additional studies, chemokine production by MIN was independent of the method of tissue disaggregation (enzymatic or mechanical, Supplemental Fig. 1 Supplemental digital content 1 http://links.lww.com/JIT/A81). Thus, in primary human LN suspensions, we do not observe evidence of CXCR3-cognate chemokine production or induction of conditioning as a consequence of melanoma infiltration, whereas all three CXCR3-cognate chemokines are induced in the vaccine-reactive lymph node, in parallel with CCL21 production.
Figure 1. SIN, but not TFN or MIN, produce CXCL9, CXCL10, and CXCL11.

Viable cell suspensions from three tumor-free nodes (TFN), nine melanoma-infiltrated nodes (MIN) and three sentinel immunized lymph nodes (SIN) were cultured in 48 well plates at 5×105 mononuclear inflammatory cells per mL and incubated for 24 hours. The supernatants were then assessed for CXCL9 (A), CXCL10 (B), CXCL11 (C), and CCL21 (D) production by ELISA. Data are individual ELISA results from one of three independent assays with similar results. The horizontal line represents the mean of the population.
Type 1 and Type 2 interferons induce CXCR3 chemokine production in melanoma-infiltrated lymph node samples
IFN priming has been shown to induce the production of CXCL9-11 in LN25. We hypothesized that addition of type I or type 2 IFNs would induce or increase the production of these chemokines, both in MIN and TFN. Optimization assays, to determine the optimal IFN dose and timing of IFN treatment, were performed using whole blood from normal donors (Supplemental Fig. 2 supplemental digital content 2 http://links.lww.com/JIT/A82). Neither IFN-α nor IFN-γ induced CXCR3 ligand production by TFN samples (Fig. 2 A-C). However, in MIN samples, IFN-γ induced significant CXCL9-11 production, compared with untreated, and IFN-α treatment significantly increased CXCL10 and CXCL11 production (Fig. 2 B-C). Interestingly, the SIN cells, already producing CXCL9-11, were not responsive to IFNs for any further increase in chemokine production. Whereas melanoma infiltration is insufficient to induce CXCR3-cognate chemokine production, IFN treatment induces MIN samples, but not TFN, to produce CXCL9-11. Further, IFN-γ is more potent than IFN-α, on a unit-equivalency basis, for the induction of total CXCR3-cognate chemokine production.
Figure 2. IFN treatment induced or increased the production of CXCL9, CXCL10, and CXCL11 in MIN, but not TFN or SIN.

Viable cell suspensions from tumor-free nodes (TFN), melanoma-infiltrated (MIN), and sentinel immunized nodes (SIN) were plated in 48 well plates at 5×105 mononuclear inflammatory cells per mL and incubated with media alone, 1×105 IU of IFN-α, or 1×105 IU of IFN-γ for 24 hours. The supernatants were then assessed for CXCL9-11 production by ELISA. Data are means ± standard deviation of ELISA results from one of three independent assays. *, p < 0.05 vs. untreated; +, p < 0.01 vs. untreated.
Melanoma cells are the major contributors of CXCR3 ligands in the tumor involved lymph node
After finding that presence of tumor, rather than activation status, was associated with IFN-responsive chemokine production in human LN samples, we hypothesized that melanoma cells may be the source of IFN-induced CXCL9, CXCL10, and CXCL11 in MIN samples. We first evaluated IFN-γ-treated LN samples by intracellular flow cytometric staining for chemokines (Fig. 3). Melanoma cells were identified as S100+ CD45neg CD11cneg cells, in order to exclude S100+ DC. CXCR3-cognate chemokines were low or absent in S100neg cells, but the majority of melanoma cells (CD11cneg S100+) were strongly positive for each of the three chemokines tested (Fig. 3A). In the CD45+ population, which includes CD14+ cells, there was no significant staining for CXCL9-11 (Fig. 3B). There was no detectable CXCL9-11 in TFN samples (Fig. 3C), and CXCL9-11 staining in SIN was restricted to CD14+ populations (Fig. 3D).
Figure 3. Melanoma cells, but not CD14+ cells, produce CXCL9, CXCL10, and CXCL11 in response to IFN-γ treatment.
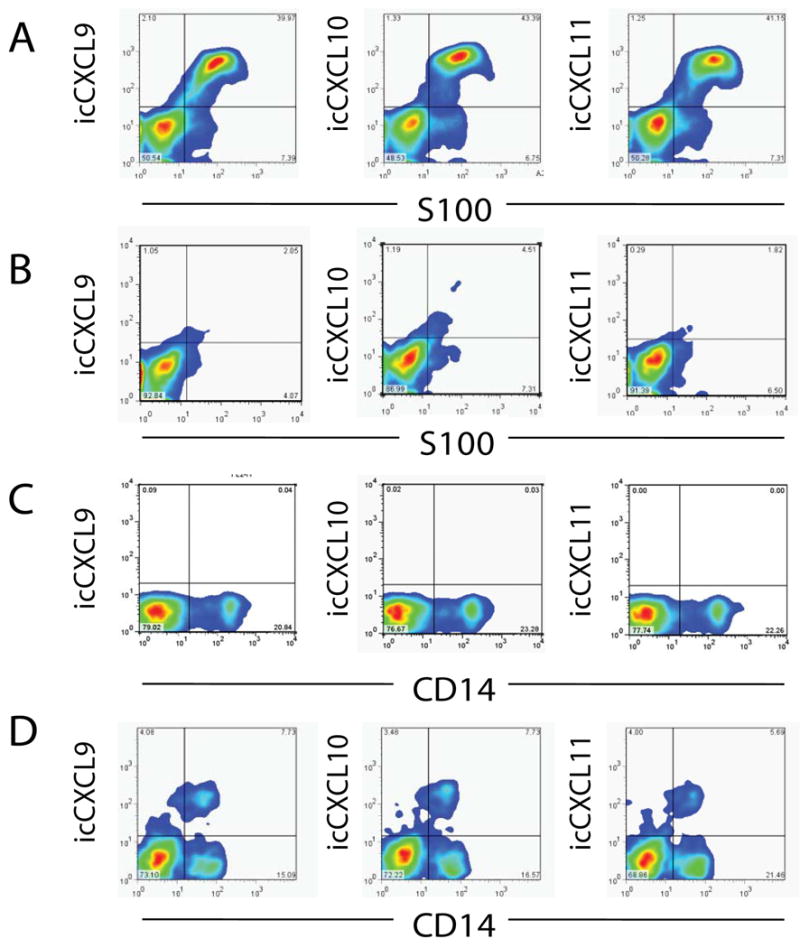
A) Staining for S100 and CXCL9, CXCL10, or CXCL11 in melanoma-involved node (MIN) cell suspensions, gated on CD45-CD11c- populations. B) Staining for S100 and CXCL9, CXCL10, or CXCL11 in MIN suspensions gated on CD45+ cells. C) Staining for CD14 and CXCL9, CXCL10, or CXCL11 in tumor-free node (TFN) cell suspensions, gated to exclude dead cells and doublets. D) Staining for CD14 and CXCL9, CXCL10, or CXCL11 in sentinel immunized node (SIN) cell suspensions, gated to exclude dead cells and doublets. Data are representative from one of three similar experiments.
To validate further that melanoma cells are capable of IFN-induced chemokine production, we evaluated established melanoma cell lines for IFN-α– and IFN-γ–induced production of CXCL9-11, and for expression of IFN receptors. In seven of seven cell lines tested, IFN treatment induced the production of at least one of the CXCR3-cognate chemokines (Fig. 4). Similar data were obtained for MV3 (Supplemental Fig. 3 supplemental digital content 3 http://links.lww.com/JIT/A83), a cell line previously reported to lack constitutive CXCL10 gene expression10. All human melanoma cell lines tested (n=16) expressed both IFN-α and IFN-γ receptors in a homogenous manner on the majority of cells (Fig. 5A-B). Further, in six of six MIN samples evaluated, melanoma (CD45neg CD11cneg CD14neg S100+ cells) expressed IFN receptors (Fig. 5C) in a pattern consistent with that observed in cultured melanoma cell lines.
Figure 4. Melanoma cell lines produce CXCL9, CXCL10, and CXCL11 in response to IFN-α or IFN-γ treatment.

Melanoma cell lines were cultured in 24 well plates at 1×106 cells per mL, either without or with 1×105 IU of IFN–α or IFN–γ for 24 hours. The supernatants were assayed in triplicate by ELISA for CXCL9 (A), CXCL10 (B), and CXCL11 (C). Data are means ± standard deviation of ELISA results from one of three independent assays.
Figure 5. Human melanoma cells express IFN-αR1 and IFN-γR1.
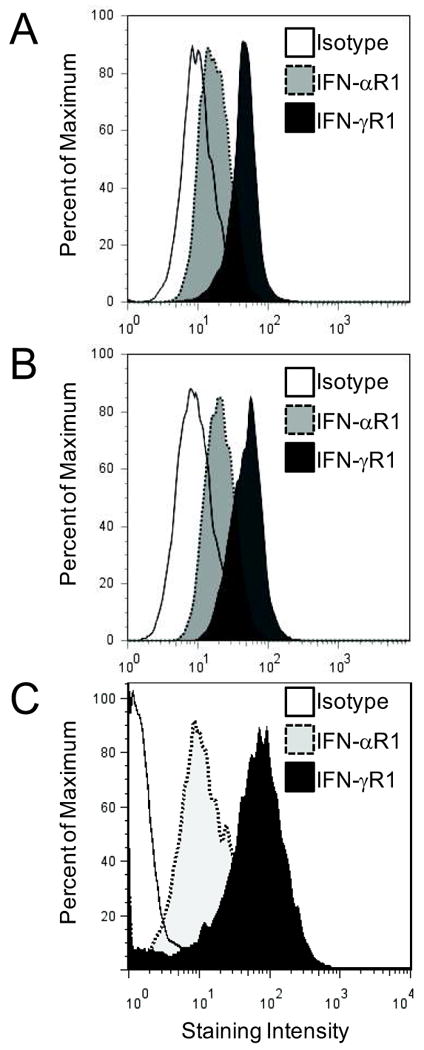
A and B) Expression of IFN receptors on human melanoma cell lines DM93 (A) and DM331 (B). Data are representative of staining for 16 human melanoma cell lines evaluated (VMM5; VMM12; VMM15; VMM18; VMM19; VMM39; VMM150; MV3; SKMel2; DM6; DM13; DM14; DM122; and DM281, not shown) from one of three similar experiments. C) Representative data for IFN receptor expression on melanoma (CD45neg CD11cneg CD14neg S100+) cells in MIN, from one of six MIN samples tested.
Melanoma-derived factors do not suppress IFN-mediated chemokine production in whole blood
Having found that the CD14+ population in IFN-treated SIN produced CXCL9-11, but that IFN-treated MIN cells were negative for CXCL9-11 staining, we questioned whether melanoma-derived factors may suppress IFN-induced chemokine production by IFN-responsive lymphocytes. To test this possibility, we evaluated the effect of supernatants from two melanoma cell lines (DM93 and DM331) on cells in whole blood from six healthy volunteers. We have observed that DM93 and DM331 constitutively produce soluble immunosuppressive factors, including transforming growth factor β1 (TGF-β1) and IL-10†. Cells from all six donors produced CXCL10 and CXCL11 in response to IFN-α treatment, and cells produced CXCL9, CXCL10, and CXCL11 in response to IFN-γ treatment (Fig. 6). CD14+ cells were the primary source of IFN-induced chemokine (Fig. 7A). Melanoma supernatant failed to quantitatively suppress CXCR3 ligand production (Fig. 6; pairwise ANOVA p > 0.05 vs. untreated), regardless of IFN treatment. Addition of supernatant to cultures did not reduce the percentage of CD14+ cells producing CXCL9 (Fig. 7B-C) in response to IFN signaling (Fig. 7). Similar data were obtained for CXCL10 and CXCL11 production (not shown). These data support the hypothesis that IFN non-responsiveness of CD14+ cells in MIN is not a consequence of soluble mediators produced by melanoma cells.
Figure 6. Melanoma-derived factors fail to block IFN-induced production of CXCL9, CXCL10, and CXCL11 by whole blood cells.

A-C) Chemokine production by whole blood samples following co-culture with melanoma supernatant and treatment with IFNs. Whole blood from a normal donor was incubated with escalating quantities of supernatant from a cultured melanoma cell line, either in media alone or with 1×105 IU of IFN–α or IFN–γ for 24h. The supernatants were assayed in triplicate by ELISA for CXCL9 (A), CXCL10 (B), and CXCL11 (C). Data are means ± standard deviation of pooled ELISA results from one of three independent assays using 6 normal donor patient samples.
Figure 7. IFN-γ-induced CXCL9 in peripheral blood-derived CD14+ cells is refractory to melanoma-derived factors.
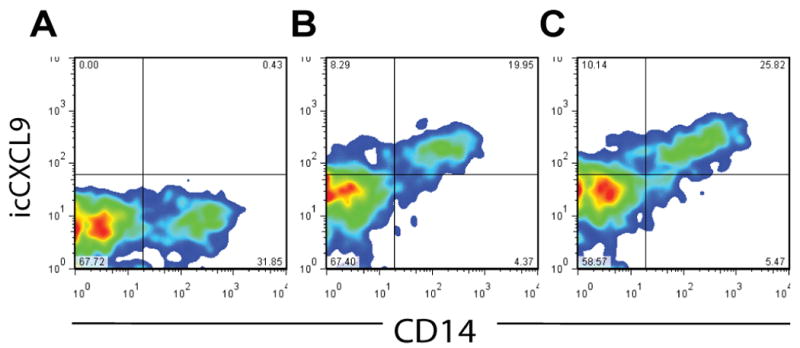
Normal donor PBL were incubated for 24h in media or with 1×105 IU of IFN–γ, and with or without supernatant (50% of culture) from the DM93 melanoma cell line. Cells were stained for CD14 and intracellular CXCL9. A) Staining for CD14 and CXCL9 in untreated PBL. B) Staining for CD14 and CXCL9 in IFN-γ-treated PBL. C) Staining for CD14 and CXCL9 in IFN-γ-treated PBL cultured with 50% melanoma supernatant. Data are representative from one of three similar experiments.
Discussion
The clinical value of vaccination-based active immunotherapy for cancer has been challenged28 due to the relatively rare occurrence of objective clinical responses29. However, immunogenicity has been demonstrated in a wide range of trial settings23;30-32. The incongruity of progressive disease in patients harboring large populations of circulating tumor Ag-specific lymphocytes likely reflects a qualitative rather than quantitative failure of vaccine-induced T cells to mediate anti-tumor immunity. With regard to malignant melanoma, the majority of tumors are poorly infiltrated by lymphocytes33-35, suggestive of trafficking defects or tumor-mediated impediments to T cell access that limit the clinical efficacy of immunogenic vaccination.
A potential barrier to T cell access of tumor or tumor-infiltrated LN may be a diminished or deficient chemotactic gradient. Several lines of evidence have suggested that the CXCR3 axis is a critical component of T cell access of tumors. Rejection of tumors is significantly enhanced36; infiltrating T cells co-localize with CXCL10 expression in malignant melanoma10; and enhanced survival in patients with advanced metastatic melanoma correlates with the presence of CXCR3-expressing tumor Ag-reactive CD8 T cells in circulation2. In the present study, we evaluated human lymphoid compartments to determine the impact of infiltration by malignant melanoma on production of CXCR3-cognate chemokines. Our observation that production of CXCL9-11 by MIN is not different than TFN strongly suggests that the presence of melanoma in LN is insufficient to generate the signals that create or maintain an inflammatory environment. In murine and primate models, CXCL9 and CXCL10 expression in draining lymphatics and luminal surfaces of HEV is limited to inflammatory settings6;37 and may require priming by PAMPS or expression of TNF-α38;39. Likely, TNF-α or other pro-inflammatory cytokines released by activated DC are necessary to induce the expression of IFN receptors by LN stroma or resident immune cells in order to achieve IFN-mediated production of CXCL9, CXCL10, and CXCL11. Infiltration of melanoma into LN may be inadequate to activate resident DC. Although we observed that IFN-induced chemokine production by PBL is refractory to melanoma-derived supernatant, the presence of melanoma in the LN may yet impede DC activation, thus ablating LN inflammation and CXCL9-11 production. These possibilities remain under investigation.
Induction of a CXCR3 ligand chemotactic gradient at the tumor may facilitate successful active immune therapy of LN-infiltrating metastatic melanoma. A variety of cells produce CXCL9-11 in response to IFN signaling, and we hypothesized that IFN treatment of LN cells may induce chemokine expression. Interestingly, among the LN compartments evaluated, only MIN-derived cells produced CXCL9-11 in response to IFN treatment, and melanoma cells were the source of these chemokines. Even lymphocytes in the SIN, which produced CXCL9-11 constitutively, did not increase production of those chemokines in the presence of IFN. These observations suggest that the chemokine gradient in melanoma-infiltrated LN may be uniquely amenable to pharmacologic modulation to induce or to improve infiltration of effector T cells into the tumor microenvironment.
The paucity of chemotactic molecule production by MIN is consistent with the need for an additional inflammatory agonist38;39. Untreated SIN samples produced CXCL9-11 but were recalcitrant to IFN treatment, suggesting either that vaccination with adjuvant had induced optimal chemokine production, or that requisite soluble inflammatory co-factors were lost in the preparation of single cell suspensions, or that the chemokine response is differentially controlled in the CD14+ cells of the SIN compared to the melanoma cells of the MIN. Beyond the tumor environment, this observation has possible implications for immune therapy. Activated SIN may harbor high levels of CXCR3 chemokines, chemoattracting the exact antigen-specific, vaccine-derived T cells one hopes would go to tumor. It is conceivable that extended vaccination schedules may induce this paradoxical effect to a greater degree than vaccines administered once or twice, and this concept is worthy of further study. From another perspective, it also may be feasible to target vaccine-induced T cells to lymph nodes with microscopic tumor involvement by repeated vaccination at the primary tumor site, with resultant expression of these chemokines in the nodes at most risk for tumor. Ohtani and colleagues40 recently demonstrated that CXCR3+ T cells were enriched in LN draining tumors that expressed CXCL9 and efficiently recruited CXCR3+ CTL. Further investigation into the source and duration of the CXCL9-11 production in activated lymph nodes is needed.
The present study is unique in comparing the effect of these two FDA-approved IFNs on production of CXCL9-11, and the data suggest the superiority of IFN-γ for this purpose. Previous studies comparing the effects of IFN-α versus IFN-γ have been limited to murine models41 or single cell suspensions of human neutrophils42 and myeloid-derived DC43. We extend the literature by demonstrating that the CXCR3 chemokine axis in MIN is inducible by IFN treatment, and that IFN-γ more potently induces the expression of CXCL9-11. These data support the re-examination of IFN-γ as a therapeutic means to establish locally-active chemotactic gradients at sites of melanoma metastases.
Our data lend a possible explanation for one of the mechanisms of the clinical effect of IFN-α in the treatment of metastatic melanoma. CXCL9-11 were not upregulated with IFN-α in normal LN or inflamed LN, suggesting that systemic therapy with IFN-α may be able to induce chemokines selectively in the melanoma-infiltrated lymph node environment. We have not evaluated whether other tissues (such as visceral resident macrophages or endothelium) may also upregulate CXCL9-11 during IFN-α treatment. Further investigations into the effects of systemic interferon treatment are needed to better understand the impact of IFN therapy on the chemokine gradient. Should IFN-α induce a tumor-directed chemokine gradient in situ, it would support combining vaccines or adoptive T cell transfer with IFN therapy as a means of directing CTL migration to tumors.
The paucity of IFN-induced CXCR3-cognate chemokine production in MIN-resident monocytes remains unexplained. Possible explanations include tumor-suppression of macrophage responsiveness to IFN, or the sequestration of CXCR3 ligands by CXCR3-expressing melanoma cells acting as cytokine sinks. The former possibility is partially discounted by the experimental observation that addition of melanoma-derived supernatants to whole blood failed to block IFN-γ-induced production of CXCR3-cognate chemokines. The later possibility is unlikely to render chemokine-producing macrophages negative for intracellular chemokine staining. The MIN has been shown to be an immunosuppressive compartment44 that limits the activation and infiltration of tumor-specific effector T cells45. The paucity of T cells in MIN may be the consequence of suppressed chemokine production. While the precise mechanisms the limit chemokine production in MIN remain undefined, it may reflect a dysregulation of macrophage IFN receptor expression or function, consequent to the absence of TNF in these lymph nodes. The mechanism warrants further study.
With regard to IFN-induced chemokine production by melanoma, we are aware of only a single previous study that evaluated production of CXCL9-11 in human melanoma cells: Kunz and colleagues reported that CXCL10 gene message was absent in two melanoma cell lines studied (540 and MV3)10. However, in our hands, MV3 expressed both IFN-αR1 and IFN-γR1 and also produced significant CXCL9, CXCL10, and CXCL11 protein in response to IFN treatment. Our experimental measure is cumulative protein production over a 24 hour period, as opposed to the reported single time-point determination of mRNA levels that may have occurred after CXCL10 gene expression had ceased. We conclude that melanomas are capable of CXCR3-cognate chemokine production, likely as a consequence of IFN receptor expression, and future studies will evaluate chemokine production directly ex vivo. We noted that IFN-γ more potently stimulated melanoma production of chemokines than equivalent activities of IFN-α, but our studies stop short of determining whether this is a result of differential cytokine activity, signal intensity, or receptor expression.
Our data suggest that IFN treatment may be a plausible maneuver to induce local CXCR3-cognate chemokine production, thus enhancing CXCR3+ T cell infiltration of MIN. Melanoma also expresses CXCR3 and utilizes this receptor for metastasis to LN46. Further, expression of CXCR3 in primary melanoma coincides with other clinical and pathologic markers of poor prognosis47. Therefore, amplification of the CXCR3 ligands may be called into question. Preclinical and clinical studies will be required to exclude the possibility that increased CXCL9-11 production in LN may chemoattract CXCR3-expressing melanoma cells and worsen patient outcomes. Further, the potential risk of newly-migrating tumor cells from the periphery into LN may be substantially offset by the increased migration of tumor-specific T cells into the same nodes. Collectively, these data support investigations of IFN, delivered in temporally-and spatially-constrained protocols, as biological mediators of chemokine production as a means to enhance T cell infiltration of metastatic melanomas.
Supplementary Material
Supplemental Figure 1. IFN-induced production of CXCL9, CXCL10, and CXCL11 by MIN is independent of the method of node disaggregation. Viable cell suspensions from three melanoma-infiltrated lymph nodes (MIN) were rendered into single cell suspensions by mechanical dissociation (non-digest) or enzymatically digested with collagenase, deoxyribonuclease I and hyaluronidase for 2-24 hours (digest). Samples were cultured in 48 well plates at 5×105 mononuclear inflammatory cells per mL and incubated with media alone, 1×105 IU of IFN-α, or 1×105 IU of IFN-γ for 24 hours. Supernatants were assessed for CXCL9-11 production by ELISA and summed. Data are mean the sum of these three CXCR3 chemokines, and data are means ± standard deviation of pooled ELISA results from two independent assays, each with two sets of tissues.
Supplemental Figure 2. Optimization of IFN-induced production of CXCL9, CXCL10, and CXCL11 by human whole blood cells. To determine the dose and timing of IFN treatment that induced maximal production of CXCR3-cognate chemokines, whole blood samples from three healthy donors were left untreated or treated with a range of IFN-α or IFN-γ doses, then incubated for 24 (A-F). Based on these data, a dose of 1 × 105 IU was chosen for the remaining studies. Cell-free supernatants were assayed in triplicate by ELISA for CXCL9-11. (G) Whole blood samples from three healthy donors were treated with 1×105 International Units (IU) of IFN-α or IFN-γ and incubated for 24, 48, or 72 h. Cell-free supernatants were assayed in triplicate by ELISA for CXCL9-11. Based on these data, cells were incubated in the presence of IFN for 24 h in the remaining studies.
Supplemental Fig. 3. MV3 human melanoma cell line expresses IFN receptors and produces CXCL9, CXCL10, and CXCL11 in response to IFN treatment. MV3 melanoma cell line was cultured in 24 well plates with at 1×106 cells per mL, either without or with 1×105 IU of IFN-α or IFN-γ for 24 hours. The supernatants were assayed in triplicate by ELISA for CXCL9, CXCL10, and CXCL11. Data are means ± standard deviation of ELISA results from one of three independent assays.
Acknowledgments
We thank Irene M. Mullins, Victor H. Engelhard, and Michael J. Weber for insightful comments.
Supported by awards from the Cardiovascular Surgery Research Training Grant (USPHS T32 HL007849, to LTD); Commonwealth Foundation for Cancer Research and matching gifts (James P. Craig, III and Rebecca T. Craig; Richard and Sherry Sharp; Alice T. and William H. Goodwin, Jr.; and George S. and Linda L. Suddock: to CLS); USPHS R01 CA57653 (to CLS); USPHS P30 CA044579 Cancer Center Support Grant Pilot Projects (to CLS and DWM); USPHS R01 CA134799 (to DWM); and the Melanoma Research Alliance (to DWM).
Footnotes
Financial Disclosure: All authors have declared there are no financial conflicts of interest in regards to this work.
Unpublished observation, V.N. Moore and D.W. Mullins.
Unpublished observation, J.L. Oliver and D.W. Mullins.
Disclosure of Potential Conflicts of Interest: The authors have no potential conflicts of interest.
References
- 1.Mihm MC, Jr, Murphy GF. Malignant melanoma in situ: an oxymoron whose time has come. Hum Pathol. 1998;29:6–7. doi: 10.1016/s0046-8177(98)90383-5. [DOI] [PubMed] [Google Scholar]
- 2.Mullins IM, Slingluff CL, Lee JK, et al. CXC chemokine receptor 3 expression by activated CD8+ T cells is associated with survival in melanoma patients with stage III disease. Cancer Res. 2004;64:7697–7701. doi: 10.1158/0008-5472.CAN-04-2059. [DOI] [PubMed] [Google Scholar]
- 3.Yamamoto J, Adachi Y, Onoue Y, et al. Differential expression of the chemokine receptors by the Th1- and Th2-type effector populations within circulating CD4+ T cells. J Leukoc Biol. 2000;68:568–574. [PubMed] [Google Scholar]
- 4.Rabin RL, Alston MA, Sircus JC, et al. CXCR3 is induced early on the pathway of CD4+ T cell differentiation and bridges central and peripheral functions. J Immunol. 2003;171:2812–2824. doi: 10.4049/jimmunol.171.6.2812. [DOI] [PubMed] [Google Scholar]
- 5.Slingluff CL, Jr, Yamshchikov G, Neese P, et al. Phase I trial of a melanoma vaccine with gp100(280-288) peptide and tetanus helper peptide in adjuvant: immunologic and clinical outcomes. Clin Cancer Res. 2001;7:3012–3024. [PubMed] [Google Scholar]
- 6.Guarda G, Hons M, Soriano SF, et al. L-selectin-negative CCR7- effector and memory CD8+ T cells enter reactive lymph nodes and kill dendritic cells. Nat Immunol. 2007;8:743–752. doi: 10.1038/ni1469. [DOI] [PubMed] [Google Scholar]
- 7.Cox MA, Jenh CH, Gonsiorek W, et al. Human interferon-inducible 10-kDa protein and human interferon-inducible T cell alpha chemoattractant are allotopic ligands for human CXCR3: differential binding to receptor states. Mol Pharmacol. 2001;59:707–715. doi: 10.1124/mol.59.4.707. [DOI] [PubMed] [Google Scholar]
- 8.Liao F, Rabin RL, Yannelli JR, Koniaris LG, Vanguri P, Farber JM. Human Mig chemokine: biochemical and functional characterization. J Exp Med. 1995;182:1301–1314. doi: 10.1084/jem.182.5.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Farber JM. Mig and IP-10: CXC chemokines that target lymphocytes. J Leukoc Biol. 1997;61:246–257. [PubMed] [Google Scholar]
- 10.Kunz M, Toksoy A, Goebeler M, Engelhardt E, Brocker E, Gillitzer R. Strong expression of the lymphoattractant C-X-C chemokine Mig is associated with heavy infiltration of T cells in human malignant melanoma. J Pathol. 1999;189:552–558. doi: 10.1002/(SICI)1096-9896(199912)189:4<552::AID-PATH469>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 11.Wenzel J, Bekisch B, Uerlich M, Haller O, Bieber T, Tuting T. Type I interferon-associated recruitment of cytotoxic lymphocytes: a common mechanism in regressive melanocytic lesions. Am J Clin Pathol. 2005;124:37–48. doi: 10.1309/4EJ9KL7CGDENVVLE. [DOI] [PubMed] [Google Scholar]
- 12.Gajewski TF, Harlan H. Chemokines expressed in melanoma metastases associated with T cell infiltration. J Clin Oncol. 2007;25:8501. [abstract] Gajewski TF, Harlan H. [Google Scholar]
- 13.Kaplan G, Luster AD, Hancock G, Cohn ZA. The expression of a gamma interferon-induced protein (IP-10) in delayed immune responses in human skin. J Exp Med. 1987;166:1098–1108. doi: 10.1084/jem.166.4.1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cole KE, Strick CA, Paradis TJ, et al. Interferon-inducible T cell alpha chemoattractant (I-TAC): a novel non-ELR CXC chemokine with potent activity on activated T cells through selective high affinity binding to CXCR3. J Exp Med. 1998;187:2009–2021. doi: 10.1084/jem.187.12.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller CH, Maher SG, Young HA. Clinical Use of Interferon-γ. Ann N Y Acad Sci. 2009;1182:69–79. doi: 10.1111/j.1749-6632.2009.05069.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kirkwood JM, Tarhini AA, Panelli MC, et al. Next generation of immunotherapy for melanoma. J Clin Oncol. 2008;26:3445–3455. doi: 10.1200/JCO.2007.14.6423. [DOI] [PubMed] [Google Scholar]
- 17.Moschos SJ, Edington HD, Land SR, et al. Neoadjuvant treatment of regional stage IIIB melanoma with high-dose interferon alfa-2b induces objective tumor regression in association with modulation of tumor infiltrating host cellular immune responses. J Clin Oncol. 2006;24:3164–3171. doi: 10.1200/JCO.2005.05.2498. [DOI] [PubMed] [Google Scholar]
- 18.Condino-Neto A, Newburger PE. Interferon-gamma improves splicing efficiency of CYBB gene transcripts in an interferon-responsive variant of chronic granulomatous disease due to a splice site consensus region mutation. Blood. 2000;95:3548–3554. [PubMed] [Google Scholar]
- 19.Ishibashi F, Mizukami T, Kanegasaki S, et al. Improved superoxide-generating ability by interferon gamma due to splicing pattern change of transcripts in neutrophils from patients with a splice site mutation in CYBB gene. Blood. 2001;98:436–441. doi: 10.1182/blood.v98.2.436. [DOI] [PubMed] [Google Scholar]
- 20.Interferon gamma-1b: new indication. Severe malignant osteopetrosis: too many unknowns. Prescrire Int. 2006;15:179–180. [PubMed] [Google Scholar]
- 21.Meyskens FL, Jr, Kopecky KJ, Taylor CW, et al. Randomized trial of adjuvant human interferon gamma versus observation in high-risk cutaneous melanoma: a Southwest Oncology Group study. J Natl Cancer Inst. 1995;87:1710–1713. doi: 10.1093/jnci/87.22.1710. [DOI] [PubMed] [Google Scholar]
- 22.Kleeberg UR, Suciu S, Brocker EB, et al. Final results of the EORTC 18871/DKG 80-1 randomised phase III trial. rIFN-alpha2b versus rIFN-gamma versus ISCADOR M versus observation after surgery in melanoma patients with either high-risk primary (thickness >3 mm) or regional lymph node metastasis. Eur J Cancer. 2004;40:390–402. doi: 10.1016/j.ejca.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 23.Slingluff CL, Jr, Petroni GR, Chianese-Bullock KA, et al. Immunologic and clinical outcomes of a randomized phase II trial of two multipeptide vaccines for melanoma in the adjuvant setting. Clin Cancer Res. 2007;13:6386–6395. doi: 10.1158/1078-0432.CCR-07-0486. [DOI] [PubMed] [Google Scholar]
- 24.Slingluff CL, Jr, Petroni GR, Olson W, et al. Helper T-cell responses and clinical activity of a melanoma vaccine with multiple peptides from MAGE and melanocytic differentiation antigens. J Clin Oncol. 2008;26:4973–4980. doi: 10.1200/JCO.2008.17.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bromley SK, Mempel TR, Luster AD. Orchestrating the orchestrators: chemokines in control of T cell traffic. Nat Immunol. 2008;9:970–980. doi: 10.1038/ni.f.213. [DOI] [PubMed] [Google Scholar]
- 26.Martín-Fontecha A, Sebastiani S, Hopken UE, et al. Regulation of dendritic cell migration to the draining lymph node: impact on T lymphocyte traffic and priming. J Exp Med. 2003;198:615–621. doi: 10.1084/jem.20030448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Manzo A, Bugatti S, Caporali R, et al. CCL21 expression pattern of human secondary lymphoid organ stroma is conserved in inflammatory lesions with lymphoid neogenesis. Am J Pathol. 2007;171:1549–1562. doi: 10.2353/ajpath.2007.061275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gattinoni L, Powell DJ, Jr, Rosenberg SA, Restifo NP. Adoptive immunotherapy for cancer: building on success. Nat Rev Immunol. 2006;6:383–393. doi: 10.1038/nri1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Terando AM, Faries MB, Morton DL. Vaccine therapy for melanoma: current status and future directions. Vaccine. 2007;25 2:B4–16. doi: 10.1016/j.vaccine.2007.06.033. [DOI] [PubMed] [Google Scholar]
- 31.Chianese-Bullock KA, Pressley J, Garbee C, et al. MAGE-A1-, MAGE-A10-, and gp100-derived peptides are immunogenic when combined with granulocyte-macrophage colony-stimulating factor and montanide ISA-51 adjuvant and administered as part of a multipeptide vaccine for melanoma. J Immunol. 2005;174:3080–3086. doi: 10.4049/jimmunol.174.5.3080. [DOI] [PubMed] [Google Scholar]
- 32.Yamshchikov GV, Barnd DL, Eastham S, et al. Evaluation of peptide vaccine immunogenicity in draining lymph nodes and peripheral blood of melanoma patients. Int J Cancer. 2001;92:703–711. doi: 10.1002/1097-0215(20010601)92:5<703::aid-ijc1250>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 33.Yagi H, Hashizume H, Horibe T, et al. Induction of therapeutically relevant cytotoxic T lymphocytes in humans by percutaneous peptide immunization. Cancer Res. 2006;66:10136–10144. doi: 10.1158/0008-5472.CAN-06-1029. [DOI] [PubMed] [Google Scholar]
- 34.Yazdi AS, Morstedt K, Puchta U, et al. Heterogeneity of T-cell clones infiltrating primary malignant melanomas. J Invest Dermatol. 2006;126:393–398. doi: 10.1038/sj.jid.5700082. [DOI] [PubMed] [Google Scholar]
- 35.Al-Batran SE, Rafiyan MR, Atmaca A, et al. Intratumoral T-cell infiltrates and MHC class I expression in patients with stage IV melanoma. Cancer Res. 2005;65:3937–3941. doi: 10.1158/0008-5472.CAN-04-4621. [DOI] [PubMed] [Google Scholar]
- 36.Gorbachev AV, Kobayashi H, Kudo D, et al. CXC chemokine ligand 9/monokine induced by IFN-gamma production by tumor cells is critical for T cell-mediated suppression of cutaneous tumors. J Immunol. 2007;178:2278–2286. doi: 10.4049/jimmunol.178.4.2278. [DOI] [PubMed] [Google Scholar]
- 37.Yoneyama H, Matsuno K, Matsushimaa K. Migration of dendritic cells. Int J Hematol. 2005;81:204–207. doi: 10.1532/IJH97.04164. [DOI] [PubMed] [Google Scholar]
- 38.Sanghavi SK, Reinhart TA. Increased expression of TLR3 in lymph nodes during simian immunodeficiency virus infection: implications for inflammation and immunodeficiency. J Immunol. 2005;175:5314–5323. doi: 10.4049/jimmunol.175.8.5314. [DOI] [PubMed] [Google Scholar]
- 39.Janatpour MJ, Hudak S, Sathe M, Sedgwick JD, McEvoy LM. Tumor necrosis factor-dependent segmental control of MIG expression by high endothelial venules in inflamed lymph nodes regulates monocyte recruitment. J Exp Med. 2001;194:1375–1384. doi: 10.1084/jem.194.9.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ohtani H, Jin Z, Takegawa S, Nakayama T, Yoshie O. Abundant expression of CXCL9 (MIG) by stromal cells that include dendritic cells and accumulation of CXCR3+ T cells in lymphocyte-rich gastric carcinoma. J Pathol. 2009;217:21–31. doi: 10.1002/path.2448. [DOI] [PubMed] [Google Scholar]
- 41.Meyer M, Hensbergen PJ, van der Raaij-Helmer EM, et al. Cross reactivity of three T cell attracting murine chemokines stimulating the CXC chemokine receptor CXCR3 and their induction in cultured cells and during allograft rejection. Eur J Immunol. 2001;31:2521–2527. doi: 10.1002/1521-4141(200108)31:8<2521::aid-immu2521>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 42.Gasperini S, Marchi M, Calzetti F, et al. Gene expression and production of the monokine induced by IFN-gamma (MIG), IFN-inducible T cell alpha chemoattractant (I-TAC), and IFN-gamma-inducible protein-10 (IP-10) chemokines by human neutrophils. J Immunol. 1999;162:4928–4937. [PubMed] [Google Scholar]
- 43.Padovan E, Spagnoli GC, Ferrantini M, Heberer M. IFN-alpha2a induces IP-10/CXCL10 and MIG/CXCL9 production in monocyte-derived dendritic cells and enhances their capacity to attract and stimulate CD8+ effector T cells. J Leukoc Biol. 2002;71:669–676. [PubMed] [Google Scholar]
- 44.Shu S, Cochran AJ, Huang RR, Morton DL, Maecker HT. Immune responses in the draining lymph nodes against cancer: implications for immunotherapy. Cancer Metastasis Rev. 2006;25:233–242. doi: 10.1007/s10555-006-8503-7. [DOI] [PubMed] [Google Scholar]
- 45.Huang RR, Paul E, Wang HJ, et al. Vascular activation and T lymphocyte transmigration vary between sentinel and non-sentinel nodes in melanoma patients. 1st International Symposium on Cancer Metastasis and the Lymphovascular System; San Francisco, CA. 2005. [abstract] Huang RR, Paul E, Wang HJ et al. [Google Scholar]
- 46.Kawada K, Sonoshita M, Sakashita H, et al. Pivotal role of CXCR3 in melanoma cell metastasis to lymph nodes. Cancer Res. 2004;64:4010–4017. doi: 10.1158/0008-5472.CAN-03-1757. [DOI] [PubMed] [Google Scholar]
- 47.Monteagudo C, Martin JM, Jorda E, Llombart-Bosch A. CXCR3 chemokine receptor immunoreactivity in primary cutaneous malignant melanoma: correlation with clinicopathologic prognostic factors. J Clin Pathol. 2006 doi: 10.1136/jcp.2005.032144. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. IFN-induced production of CXCL9, CXCL10, and CXCL11 by MIN is independent of the method of node disaggregation. Viable cell suspensions from three melanoma-infiltrated lymph nodes (MIN) were rendered into single cell suspensions by mechanical dissociation (non-digest) or enzymatically digested with collagenase, deoxyribonuclease I and hyaluronidase for 2-24 hours (digest). Samples were cultured in 48 well plates at 5×105 mononuclear inflammatory cells per mL and incubated with media alone, 1×105 IU of IFN-α, or 1×105 IU of IFN-γ for 24 hours. Supernatants were assessed for CXCL9-11 production by ELISA and summed. Data are mean the sum of these three CXCR3 chemokines, and data are means ± standard deviation of pooled ELISA results from two independent assays, each with two sets of tissues.
Supplemental Figure 2. Optimization of IFN-induced production of CXCL9, CXCL10, and CXCL11 by human whole blood cells. To determine the dose and timing of IFN treatment that induced maximal production of CXCR3-cognate chemokines, whole blood samples from three healthy donors were left untreated or treated with a range of IFN-α or IFN-γ doses, then incubated for 24 (A-F). Based on these data, a dose of 1 × 105 IU was chosen for the remaining studies. Cell-free supernatants were assayed in triplicate by ELISA for CXCL9-11. (G) Whole blood samples from three healthy donors were treated with 1×105 International Units (IU) of IFN-α or IFN-γ and incubated for 24, 48, or 72 h. Cell-free supernatants were assayed in triplicate by ELISA for CXCL9-11. Based on these data, cells were incubated in the presence of IFN for 24 h in the remaining studies.
Supplemental Fig. 3. MV3 human melanoma cell line expresses IFN receptors and produces CXCL9, CXCL10, and CXCL11 in response to IFN treatment. MV3 melanoma cell line was cultured in 24 well plates with at 1×106 cells per mL, either without or with 1×105 IU of IFN-α or IFN-γ for 24 hours. The supernatants were assayed in triplicate by ELISA for CXCL9, CXCL10, and CXCL11. Data are means ± standard deviation of ELISA results from one of three independent assays.