

Abstract
Early-life respiratory viral infections are linked to subsequent development of allergic asthma in children. We assessed the underlying immunological mechanisms in a novel model of the induction phase of childhood asthma. BALB/c mice were infected neonatally with pneumonia virus of mice, then sensitized intranasally with ovalbumin following recovery. Animals were challenged with low levels of aerosolized ovalbumin for 4 weeks to induce changes of chronic asthma, then received a single moderate-level challenge to elicit mild acute allergic inflammation. To inhibit the initial induction of a T helper type 2 (Th2) response, we administered neutralizing antibodies against interleukin (IL)-4 or IL-25, then assessed development of airway inflammation and remodelling. Anti-IL-4 administered during chronic challenge prevented development of chronic and acute allergic inflammation, as well as goblet cell hyperplasia/metaplasia, but features of remodelling such as subepithelial fibrosis and epithelial hypertrophy were unaffected. In contrast, anti-IL-25 had limited effects on the airway inflammatory response but prevented key changes of remodelling, although it had no effect on goblet cells. Both antibodies suppressed development of a Th2 response, while anti-IL-25 also promoted a Th17 response. In further experiments, anti-IL-25 was administered in early life alone, and again had limited effects on airway inflammation, but prevented development of airway wall remodelling. We conclude that in this murine model of childhood asthma, administration of anti-IL-4 or anti-IL-25 prevents development of some key features of asthma, suggesting that suppression of development of a Th2 response during the neonatal period or later in childhood could be effective for primary prevention.
Keywords: airway inflammation, airway remodelling, allergy, Th17 response, Th2 response
Introduction
Asthma is one of the most common chronic diseases affecting children, particularly in industrialized societies. Epidemiological studies have identified a number of factors which increase the likelihood of developing asthma, notably early-life respiratory viral infections and repeated exposure to allergens, which appear to be synergistic [1]. Importantly, although this association is well documented, the pathogenesis of childhood asthma remains largely unexplained.
Viral infections in early life, notably with rhinovirus and respiratory syncytial virus (RSV), are associated strongly with the subsequent development of allergic asthma [2–4]. Despite significant investigation, the underlying immunological mechanisms remain unknown, due in large part to the lack of suitable animal models. To address this, we have recently described a model of the interaction between early-life respiratory viral infection and subsequent allergen exposure in the development of an asthmatic phenotype in mice [5]. This model uses pneumonia virus of mice (PVM), which belongs to the same family/genus as RSV but is a natural rodent pathogen, unlike human RSV, which exhibits limited replication in mice [6,7]. It employs intranasal sensitization and low-level, long-term aerosol challenge to elicit very mild changes of allergic inflammation of the airways, consistent with the induction phase of childhood asthma. We showed that while some features of asthma developed in response to either viral infection or allergen challenge, a complete asthmatic phenotype was evident only in animals that had recovered from neonatal infection with PVM and then received chronic allergen challenge. Furthermore, development of allergic inflammation with recruitment of eosinophils was dependent on the accumulation and activation of pulmonary T cells, with induction of a T helper type 2 (Th2)-biased immunological response, because the inflammatory response was abrogated in mice that did not express the interleukin (IL)-4 receptor α chain [5].
In this study, we have employed our novel murine model to further investigate the immunological mechanisms underlying the initial phase of development of allergic asthmatic inflammation of the airways in childhood, focusing specifically on mediators which play a crucial role in generating a Th2-biased immunological response, rather than on cytokines involved in the effector phase of allergic inflammation. Of particular interest in this context are IL-4 and IL-25. IL-4 has long been recognized as having a crucial role in the induction of Th2 responses [8], while IL-25, which is expressed by airway epithelial cells (AECs), can initiate Th2 differentiation in an IL-4-dependent manner [9]. To assess the role of these cytokines in the development of the asthmatic response, we administered neutralizing antibodies during the period of chronic allergen challenge. Additionally, because we have previously reported increased expression of IL-25 in the lungs immediately following PVM infection [5], we also administered neutralizing antibodies against IL-25 in early life. Collectively, this work has established that in this model of childhood asthma, treatment with anti-IL-4 or anti-IL-25 can prevent the development of some key features of asthma.
Materials and methods
Infection, sensitization and challenge
PVM infection and ovalbumin sensitization were performed as described previously [5] and summarized in Fig. 1. Briefly, on days 1 and 2 of life, BALB/c mice were inoculated intranasally with 2 plaque-forming units (pfu) PVM (J3666 strain ∼1 × 105 pfu/ml). Subsequently, intranasal sensitization with 100 µg chicken egg ovalbumin (OVA, grade V, ≥ 98% pure; Sigma Australia, Castle Hill, NSW, Australia) was performed on days 28 and 29 of life.
Fig. 1.
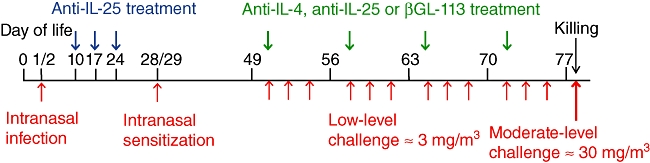
Diagrammatic representation of protocol for infection, sensitization and inhalational challenge. Antibody administration was either during the 3 weeks following pneumonia virus of mice (PVM) infection or the 4 weeks of low-level inhalational challenge.
Inhalational challenge with aerosolized OVA was performed as described previously [10,11]. Commencing at 7 weeks of age, mice were exposed to aerosolized OVA, ≈ 3 mg/m3, 30 min/day, 3 days/week for 4 weeks in flow-through wire cage racks in a 0·5 m3 inhalation chamber (Unifab Corporation, Kalamazoo, MI, USA). Particle concentration within the chamber was monitored continuously using a DustTrak 8520 instrument (TSI, St Paul, MN, USA). At the end of the period of chronic challenge (i.e. day 78 of life) mice received a single moderate challenge of ≈ 30 mg/m3 for 30 min, to induce changes of acute allergic inflammation simulating an acute exacerbation of asthma [11]. Animals were killed 4 h later.
All experimental procedures complied with the requirements of the Animal Care and Ethics Committee of the University of New South Wales (ref no. 06/119B) and the University of Newcastle (ref no. 1026).
Antibody neutralization
Mice were treated with anti-IL-4 (11B11) [12], anti-IL-25 (35B) [9] or an isotype control monoclonal antibody to β-galactosidase (βGl-113) for the positive control group. Intraperitoneal injections were administered once weekly (0·5 mg per injection) for 4 weeks during the period of inhalational challenge. Antibody preparations contained no detectable endotoxin by Limulus lysate assay. Additional groups of mice were administered 25 mg/kg of anti-IL-25 antibody on days 10, 17 and 24 of life (0·15, 0·22 and 0·3 mg, respectively) by intraperitoneal injection. Because we have demonstrated previously that the responses of sham-infected and sham-sensitized animals are indistinguishable from those of naive animals [5], the latter were used as the negative control group for these experiments.
Airway inflammation and remodelling
Bronchoalveolar lavage (BAL) cells were counted in Leishman-stained smears. Relative numbers of eosinophils recruited into the lungs were quantified using a colorimetric assay for eosinophil peroxidase, modified from Schneider and Issekutz [13]. Numbers of chronic inflammatory cells in the lamina propria, comprising predominantly CD3+ T cells and plasma cells [14,15], as well as the thickness of airway epithelium and subepithelial collagen, were quantified as described previously [5,10]. In brief, images were captured from longitudinally orientated sections of trachea, using a Spot Cooled Color Digital camera (Diagnostic Instruments, Sterling Heights, MI, USA) calibrated with a reference measurement slide. Nuclear profiles were counted in every second consecutive microscopic field in the lamina propria of haematoxylin and eosin-stained sections and the length of the epithelial basement membrane was measured by tracing using the Spot software. The data were used to calculate the mean number of cells per 100 µm of epithelial basement membrane in the lamina propria for individual animals. Reticulin-stained sections were used for assessment of subepithelial collagenization. The thickness of the reticulin-stained zone underneath the tracheal epithelium was measured and the mean thickness for each animal was calculated. The thickness of the epithelial layer was also measured in these sections and the mean thickness for each animal was calculated.
Metaplasia and/or hyperplasia of mucus-secreting goblet cells was assessed in sections of lung tissue stained with Alcian blue–periodic acid-Schiff (PAS). The percentage of cells staining positive for mucins was enumerated in the largest visible airway and each animal was categorized according to a semilogarithmic scale as grade 0 = < 1% positive cells, grade 1 = 1–3%, grade 2 = 4–10%, grade 3 = 11–30% and grade 4 = ≥ 31%, as described previously [5,10]. CD3+ T cells were counted in immunostained frozen sections of lung tissue [5]. Active TGF-β1 was demonstrated by immunostaining with an affinity-purified rabbit polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA), as described previously [16]. Intensity of immunoreactivity was scored semi-quantitatively as grade 0 = no staining, grade 1 = weak staining, grade 2 = moderate staining and grade 3 = strong staining. The validity and reliability of the morphometric techniques we employed have been established in our previous reports.
Cell isolation
For isolation of CD4+ T cells, two pairs of lungs were pooled for each sample and diced into fine fragments (< 0·3 mm3). Tissue was dispersed in ≈ 3 ml of a mixture of Type IV collagenase with minimal non-specific protease activity (Worthington Biochemical, Lakewood, NJ, USA) and DNase (Roche Diagnostics, Sydney, Australia). After incubation at 37°C on a roller mixer for 40 min, samples were vortexed and passed through a 70-µm cell strainer to release cells. Cells were resuspended in erythrocyte lysis buffer, washed with phosphate-buffered saline (PBS), and CD4+ T cells were purified by positive selection using a FlowComp magnetic bead isolation kit (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocols.
Cell purity was assessed by flow cytometry, staining with conjugated antibodies to CD3 [fluorescein isothiocyanate (FITC)], CD4 [peridinin chlorophyll (PerCP)] and CD8 [phycoerythrin (PE)] or with appropriate isotype controls (BD Pharmingen, San Jose, CA, USA). Lymphocytes were selected by gating the population on the basis of forward- and side-scatter, and were confirmed by CD3+ staining. Analysis was on a fluorescence activated cell sorter (FACSort) (Becton Dickinson, Mountain View, CA, USA) using CellQuest Pro software (BD Pharmingen). As reported previously, the isolated lymphocyte population comprised ≈ 98·0% CD3+/CD4+ cells [17].
Polymerase chain reaction (PCR)
RNA was isolated from CD4+ T cells using TriReagent (Sigma) according to the manufacturer's instructions. Following DNase treatment (Turbo DNase; Ambion, Scoresby, Australia), samples were reverse-transcribed (RT) into cDNA using Superscript III (Invitrogen). Expression of cytokines and transcription factors was assessed via quantitative RT–PCR, with SYBR green used to detect amplified products. Primers designed to allow the use of identical thermocyler conditions were kindly provided by Dr Ken Hsu (University of New South Wales), or taken from an external reference [18], or designed using Primer Express software (Applied Biosystems, Australia). Reactions were performed using an ABI Prism 7700 Sequence Detector (Applied Biosystems, Melbourne, Australia) and gene expression was normalized to hypoxanthine phosphoribosyltransferase (HPRT).
Statistical analysis
Results are presented as arithmetic mean ± standard error of the mean (s.e.m.) or as medians where appropriate. Analysis was by one-way analysis of variance (anova) or a Kruskal–Wallis test, followed by a Dunnett's or Dunn's test as appropriate. GraphPad Prism (GraphPad Software, San Diego, CA, USA) was used for all data analysis and preparation of graphs.
Results
Effects of treatment with anti-IL-4 or anti-IL-25 during chronic allergen challenge
Inflammatory response
PVM-infected, ovalbumin-sensitized and challenged animals that were treated during challenge with a control antibody to β-galactosidase (βGl-113) (Fig. 1) are referred to hereafter as the control group. In these control mice, following the final single moderate challenge there was an increased yield of total numbers of BAL cells, especially lymphocytes and eosinophils (Table 1). Consistent with our previous report [5], and with the pattern of inflammation in clinical acute exacerbations of asthma [19], the inflammatory cells also included moderate numbers of neutrophils, although the increase was not statistically significant. Anti-IL-4-treated mice also exhibited increased numbers of cells in lavage fluid, although proportions of lymphocytes and eosinophils were lower (P < 0·01 for both), as were proportions of neutrophils (not significant). Compared to the control group, BAL cells were decreased markedly following treatment with anti-IL-25 and this antibody also reduced the proportion of lymphocytes and eosinophils (P < 0·01 for both) significantly in the population of recovered cells. In striking contrast, anti-IL-25 caused an increase in the percentage of neutrophils recovered by BAL (P < 0·001 compared to naive animals and P < 0·05 compared to control animals). However, in animals treated with this antibody there was no histopathological evidence of neutrophilic pulmonary inflammation, nor of any other changes consistent with pulmonary infection.
Table 1.
Bronchoalveolar lavage (BAL) cellular response following antibody treatment during chronic antigen challenge
| Groups | Naive | Control | Anti-IL-4 | Anti-IL-25 |
|---|---|---|---|---|
| Total | 121 (14·8) | 220 (43·9) | 199 (28·5) | 141 (19·7) |
| Macrophages | 94·5% (0·8) | 86·6% (1·7)* | 91·8% (2·2) | 84·0% (2·5)## |
| Lymphocytes | 4·4% (0·9) | 12·7% (1·6)*** | 6·0% (1·5)## | 5·0% (1·0)## |
| Neutrophil | 0·6% (0·2) | 3·0% (0·3) | 0·4% (0·2) | 8·5% (2·1)*** ## |
| Eosinophils | 0% (0·0) | 0·3% (0·1)** | 0% (0·0)## | 0% (0·0)## |
Data are presented as mean numbers of cells (thousands) ± standard error of the mean and as percentages for individual cell types (n = 8). Significant differences compared to naive animals shown as * P < 0·05, **P < 0·01 and ***P < 0·001, compared to control animals shown as ##P < 0·01. IL: interleukin.
Following the final single moderate challenge, there was also significant accumulation of intrapulmonary eosinophils in control antibody-treated mice, as assessed by eosinophil peroxidase activity (Table 2). This eosinophil accumulation decreased following treatment with anti-IL-4, but not in anti-IL-25 treated animals.
Table 2.
Inflammatory and epithelial changes in the airways and lungs following antibody treatment during chronic antigen challenge
| Experimental groups | ||||
|---|---|---|---|---|
| Naive | Control | Anti-IL-4 | Anti-IL-25 | |
| Intrapulmonary eosinophils (OD450) | 0·07 (0·02) | 0·21 (0·05)* | 0·05 (0·01)## | 0·38 (0·11)** |
| Intrapulmonary CD3+ T cells/mm2 | 6·7 (0·6) | 22·8 (2·5)*** | 7·1 (1·0)## | 45·3 (4·1)*** ### |
| Lamina propria cells/100 µm basement membrane | 20·3 (1·5) | 34·0 (1·2)*** | 20·9 (1·3)### | 34·0 (1·1)*** |
| Thickness of subepithelial collagen zone (µm) | 1·9 (0·1) | 3·7 (0·2)*** | 3·7 (0·3)*** | 1·8 (0·1)### |
| Thickness of epithelial cell layer (µm) | 12·9 (0·6) | 18·0 (0·7)*** | 15·9 (0·4)**# | 11·7 (0·2)### |
| Grade of goblet cell hyperplasia/metaplasia† | 0·0 (1·0) | 2·0 (2·0)* | 0·0 (1·0)# | 3·0 (0·0)** |
Data are presented as mean ± standard error of the mean, except †which are median (interquartile range). Significant differences compared to naive animals shown as *P < 0·05, **P < 0·01 and ***P < 0·001, compared to control animals shown as #P < 0·05, ##P < 0·01 and ###P < 0·001. IL: interleukin.
As shown in Table 2, intrapulmonary CD3+ T cells were increased significantly in control animals (P < 0·001 compared to naive mice). Following administration of anti-IL-4, the number of CD3+ T cells in lung tissue was decreased significantly compared to the control group (P < 0·01). In contrast, anti-IL-25 treatment did not decrease the numbers of these cells; indeed, there was a significant increase compared to both naive and control antibody-treated animals (P < 0·001 for both). Similarly, accumulation of chronic inflammatory cells in the lamina propria of the conducting airways, a consistent feature of airway-specific chronic inflammation, was increased significantly in control antibody-treated animals (P < 0·001 compared to naive mice). A significant reduction was observed following treatment with anti-IL-4 (P < 0·001), whereas anti-IL-25 did not decrease the numbers of these cells (P < 0·001 compared to naive mice).
Airway wall remodelling
Significant subepithelial collagenization developed in the control antibody-treated animals compared to the naive group (P < 0·001) (Table 2). This was also evident in anti-IL-4 treated mice, with animals exhibiting a significant increase compared to naive animals (P < 0·001). In striking contrast, no subepithelial collagenization was apparent in mice treated with anti-IL-25 (P < 0·001 compared to control antibody-treated mice) (Table 2, Fig. 2). Similarly, control and anti-IL-4 treated animals exhibited increased thickness of the airway epithelium compared to naive mice (at least P < 0·01), whereas there was no evidence of epithelial hypertrophy following treatment with anti-IL-25 (P < 0·001 compared to control antibody-treated animals) (Table 2, Fig. 2).
Fig. 2.
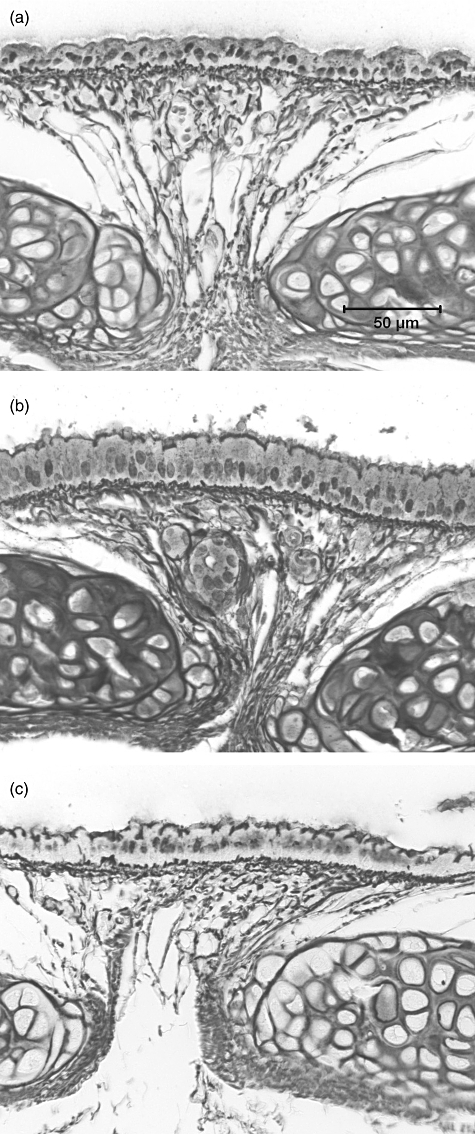
Reticulin-stained trachea from (a) a naive mouse, demonstrating normal thickness of epithelial and subepithelial regions, (b) an animal from the control antibody-treated group, demonstrating significant thickening of both the epithelium and the subepithelial collagenous zone and (c) an animal from the anti-interleukin (IL)-25-treated group.
Mucin-secreting goblet cells were increased significantly in control antibody-treated animals (P < 0·05), but were virtually absent following administration of anti-IL-4 (P < 0·05) (Table 2). Treatment with anti-IL-25 had no effect on the mucous cell response, with the numbers of goblet cells in the intrapulmonary airways equivalent to those of control mice (P < 0·01 compared to naive mice).
Pulmonary CD4+ T cell response and cytokine profile
Because we had shown previously that generation of a Th2-biased immunological response was a key feature of this model of the induction phase of childhood asthma [5], we specifically examined the effects of antibody treatment on the responses of pulmonary CD4+ T cells. Significantly increased numbers of CD4+ T cells were recovered from disaggregated lung tissue from control mice (P < 0·01 compared to naive animals) (Fig. 3a). Treatment with anti-IL-4 decreased the number of cells recovered (P < 0·001 compared to control animals) significantly, whereas there was no reduction in anti-IL-25 treated animals (P < 0·01 compared to naive mice). CD4+ T cells recovered from control antibody-treated mice exhibited increased expression of the key Th2 cytokines, including IL-4 (P < 0·001), IL-5 (P < 0·01) and IL-13 (P < 0·05), compared to untreated, naive animals (Fig. 3b–d). Both antibodies, and in particular anti-IL-25, inhibited the Th2 response, by abrogating up-regulation of IL-4 (P < 0·001 for both), IL-5 (at least P < 0·05) and IL-13 (not significant).
Fig. 3.
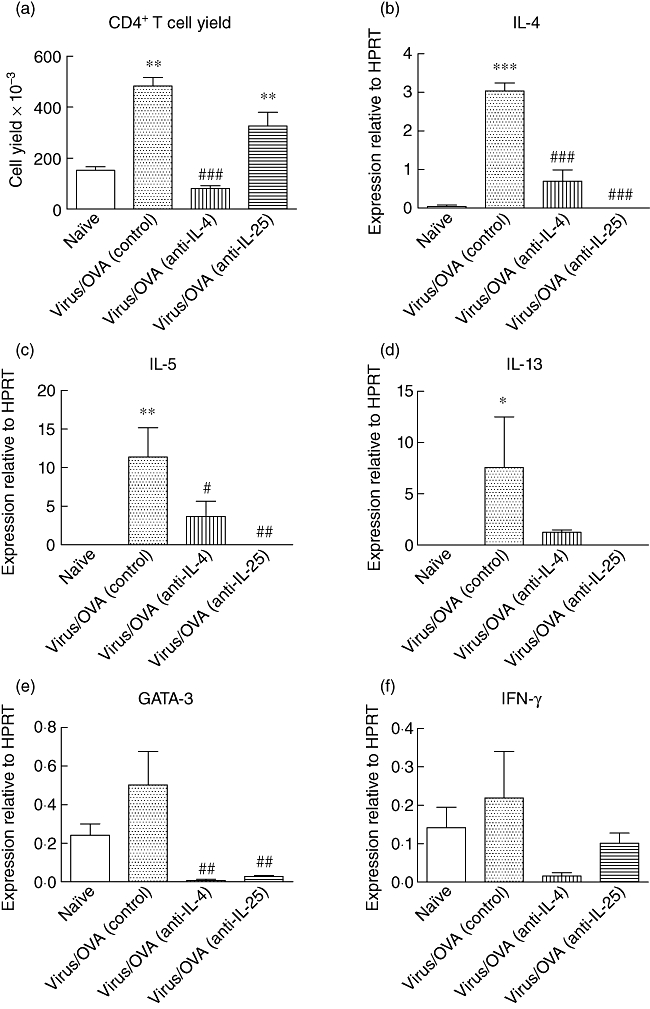
Effect of antibody treatment during the chronic challenge period on T helper type 2 (Th2)-biased immunological response. (a) Numbers of CD4+ T cells recovered from disaggregated lung tissue. (b–f) Levels of expression of mRNA for interleukin (IL)-4, IL-5, IL-13, GATA-3 and interferon (IFN)-γ by CD4+ T cells, relative to hypoxanthine phosphoribosyltransferase (HPRT). Significant differences compared to naive animals shown as *P < 0·05, **P < 0·01 and ***P < 0·001, compared to control animals shown as #P < 0·05, ##P < 0·01 and ###P < 0·001.
Levels of GATA-3, the Th2-associated transcription factor, mimicked the pattern of the major Th2 cytokines, with control mice exhibiting a modest increase, whereas there was a significant decrease in both anti-IL-4 and anti-IL-25 treatment groups (P < 0·01) (Fig. 3e). No evidence of increased IFN-γ expression was detected in any experimental group (Fig. 3f), nor was there any increase in the expression of T-bet, the Th1-associated transcription factor (not shown).
A striking finding was that CD4+ T cells recovered from anti-IL-25-treated mice exhibited borderline increased expression of IL-17A (not significant) as well as markedly increased expression of IL-17F (P < 0·01) and IL-6 (P < 0·01) compared to control mice (Fig. 4a–c). In the context of the increase in total number of CD3+ T cells in lung tissue, these findings are consistent with expansion of the population of pulmonary Th17 cells.
Fig. 4.

Effect of antibody treatment during the chronic challenge period on expression of T helper type 17 (Th17) cytokines. Levels of expression of mRNA for (a) interleukin (IL)-17A, (b) IL-17F and (c) IL-6 by CD4+ T cells, relative to hypoxanthine phosphoribosyltransferase (HPRT). Significant differences compared to naive animals shown as **P < 0·01 and ***P < 0·001, compared to control animals shown as ##P < 0·01.
Immunostaining for TGF-β1
Limited immunoreactivity for active transforming growth factor-β1 (TGF-β1) was detected in the airway epithelium and the subepithelial connective tissue of naive animals. In mice treated with control antibody, the median grade of immunoreactivity was increased significantly, both in the epithelium and in the subepithelial region (Fig. 5). Treatment with anti-IL-4 did not decrease the median grade, whereas treatment with anti-IL-25 decreased staining for active TGF-β1 in both the epithelium and subepithelial zone, although these changes were not statistically significant (Fig. 5).
Fig. 5.
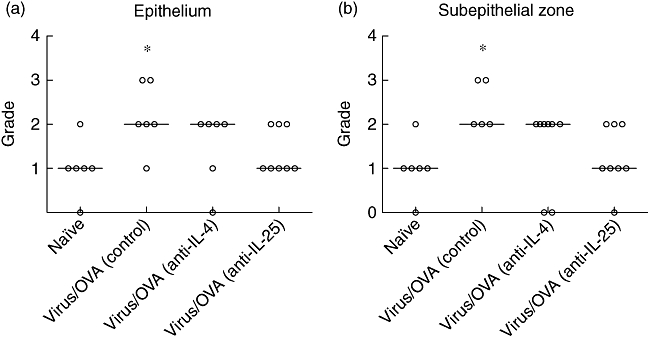
Effect of antibody treatment during the chronic challenge period on immunoreactivity for active transforming growth factor (TGF)-β1. Intensity of staining (a) in the epithelium and (b) in the subepithelial zone, in animals treated with anti-interleukin (IL)-4 or anti-IL-25 during the chronic challenge period. Significant differences compared to naive animals shown as *P < 0·05.
Effects of treatment with anti-IL-25 in early life alone
Based on the above findings that treatment with anti-IL-25 inhibited some aspects of allergic inflammation as well as preventing airway remodelling, we tested the effect of administering this antibody immediately following neonatal infection with PVM. Mice were injected with an appropriate dose of antibody on days 10, 17 and 24 of life.
Inflammatory response
Following early-life treatment with anti-IL-25, animals exhibited an increased yield of BAL cells (Table 3). Although there were decreased proportions of lymphocytes (P < 0·001 compared to the control group) and eosinophils (P < 0·05), the percentage of neutrophils was increased dramatically compared to other groups (P < 0·001) (Table 3). Again, however, no changes suggestive of pulmonary infection or pneumonia were found on histopathological examination of these animals.
Table 3.
Bronchoalveolar lavage (BAL) cellular response following anti-IL-25 treatment in early life
| Groups | Naive | Control | Anti-IL-25 |
|---|---|---|---|
| Total | 176 (19·6) | 320 (27·3) | 900 (319·2) |
| Macrophages | 90·0% (2·0) | 87·2% (1·6) | 65·9% (6·3)** ### |
| Lymphocytes | 9·8% (2·0) | 10·5% (1·8) | 1·4% (0·5)** ### |
| Neutrophil | 0·2% (0·1) | 3·3% (0·9) | 32·6% (6·4)*** ### |
| Eosinophils | 0·0% (0·0) | 0·3% (0·1)** | 0·1% (0·0)# |
Data are presented as mean numbers of cells (thousands) ± standard error of the mean and as percentages for individual cell types (n = 8). Significant differences compared to naive animals shown as **P < 0·01 and ***P < 0·001, compared to control animals shown as #P < 0·05 and ###P < 0·001. IL: interleukin.
In the lung tissue, the modest relative increase in eosinophils observed in the control group was again not diminished in animals treated with anti-IL-25 in early life (P < 0·05 compared to naive mice) (Table 4). As expected, increased numbers of intrapulmonary CD3+ T cells were observed in the lungs of mice treated with control antibody (P < 0·001 compared to the naive group) (Table 4). Similar to the experiment involving treatment during chronic challenge, administration of anti-IL-25 in early life did not diminish the numbers of intrapulmonary T cells, which remained comparable to the control group (P < 0·01 compared to naive animals). Chronic inflammatory cells in the lamina propria of conducting airways also remained increased significantly in anti-IL-25 treated animals (P < 0·05 compared to the naive group) (Table 4).
Table 4.
Inflammatory and epithelial changes in the airways and lungs following anti-IL-25 treatment in early life
| Experimental groups | |||
|---|---|---|---|
| Naive | Control | Anti-IL-25 | |
| Intrapulmonary eosinophils (OD450) | 0·06 (0·01) | 0·16 (0·04) | 0·31 (0·09)* |
| Intrapulmonary CD3+ T cells/mm2 | 3·6 (0·9) | 28·1 (3·2)*** | 28·7 (6·6)** |
| Lamina propria cells/100 µm basement membrane | 25·0 (1·3) | 38·4 (1·7)*** | 30·7 (1·5)* ## |
| Thickness of subepithelial collagen zone (µm) | 1·7 (0·1) | 4·3 (0·3)*** | 2·9 (0·1)* ## |
| Thickness of epithelial cell layer (µm) | 12·0 (0·4) | 16·8 (0·7)*** | 13·7 (0·8)## |
| Grade of goblet cell hyperplasia/metaplasia† | 0·0 (0·0) | 4·0 (2·0)*** | 2·0 (4·0)* |
Data are presented as mean ± standard error of the mean, except †which are median (interquartile range). Significant differences compared to naive animals shown as *P < 0·05, **P < 0·01 and ***P < 0·001, compared to control animals shown as ##P < 0·01. IL: interleukin.
Airway wall remodelling
Similar to the experiment involving treatment during chronic challenge, early-life treatment with anti-IL-25 prevented the subsequent development of subepithelial fibrosis or epithelial hypertrophy (P < 0·01 compared to control animals for both) (Table 4). Also, as for treatment during chronic challenge, anti-IL-25 treatment in early life had no effect on mucin-secreting goblet cells, with numbers equivalent to control animals (P < 0·05 compared to the naive group).
Pulmonary CD4+ T cell response and cytokine profile
Analogous to results in the chronic challenge experiment, significantly increased numbers of CD4+ T cells were recovered from animals administered anti-IL-25 in early life (P < 0·001 compared to naive mice). These cells exhibited decreased expression of the Th2 cytokines including IL-4 (P < 0·001), IL-5 and IL-13, as well as of the Th2 transcription factor, GATA-3 (not significant). Again, there was no evidence of a Th1 response but increased levels of IL-17A, IL-17F and IL-6 were detected (data not shown).
Discussion
We have demonstrated recently that in a model of the induction phase of childhood asthma, the interaction between early-life viral infections and subsequent allergen sensitization/challenge is crucial to the development of key features of asthma [5]. Furthermore, we showed that the low-level allergic airway inflammation that is observed during this induction phase is dependent upon the development of a Th2-biased immunological response. This is consistent with clinical evidence of the relevance of Th2-driven inflammation in childhood asthma [20]. To investigate further the mechanisms underlying development of childhood asthma, we assessed the effects on airway inflammation and remodelling of suppressing the induction of a Th2 response, by neutralizing IL-4 or IL-25.
IL-4 was of interest as it is the prototypic Th2-promoting cytokine, which drives Th2 differentiation and expansion of this population of cells [8]. In addition, because increasing evidence implicates AECs as having a crucial role in orchestrating allergic inflammation [21,22], we were also interested in IL-25, which is expressed in the airway epithelium following allergen exposure [9] and is also induced strongly in AECs following respiratory viral infection [23]. To assess whether development of an asthmatic phenotype could be inhibited, neutralizing antibodies were administered during the chronic challenge period. Furthermore, as we had previously demonstrated increased expression of IL-25 immediately following an early-life infection with PVM [5], we also tested the effect of administering neutralizing antibodies against IL-25 during early life alone.
In the first part of this investigation, we confirmed that animals which had recovered from neonatal PVM infection and then been sensitized and chronically challenged with antigen developed key features of childhood asthma, including low-level allergic inflammation of the airways with modest eosinophil recruitment, a Th2-biased immunological response, and changes of airway wall remodelling. In animals treated with the control antibody βGl-113, we observed inflammation and changes of remodelling, essentially identical to those described previously [5]. Treatment with anti-IL-4 or anti-IL-25 had divergent effects, with suppression of different elements of inflammation and/or airway wall remodelling by each antibody. Specifically, following treatment with anti-IL-4, chronic lymphocytic inflammation and both acute eosinophilic and neutrophilic inflammation were inhibited. Goblet cell metaplasia/hyperplasia was also abrogated, but other changes of remodelling were not suppressed. In contrast, neutralizing IL-25 did not suppress recruitment of lymphocytes into lung tissue – indeed, numbers of pulmonary CD3+ T cells increased – but had some effects on the numbers of lymphocytes and eosinophils in BAL fluid. Treatment with anti-IL-25 did not suppress the neutrophil response, instead being associated with increased recruitment of these cells into BAL fluid. Strikingly, anti-IL-25 suppressed subepithelial fibrosis and epithelial cell hypertrophy completely. Both antibodies inhibited the Th2 response, with abrogated up-regulation of IL-4, IL-5, IL-13 and GATA-3 in isolated pulmonary CD4+ T cells.
A prominent effect of neutralizing IL-25 was enhanced expression of IL-17 family cytokines by CD4+ T cells, consistent with development of a Th17 phenotype. Both IL-17A and IL-17F are secreted by Th17 cells [24], while IL-6 is important for the development of these cells [25,26] and may also be secreted by them. The evidence of amplification of a Th17 response following treatment with anti-IL-25 is consistent with earlier published data on regulation of Th17 differentiation by IL-25 [27,28]. This effect could explain how treatment with anti-IL-25, in contrast to anti-IL-4, increased the numbers of neutrophils in BAL significantly, because Th17 cells have a strong association with neutrophilic inflammation in asthma [29–31], which may be steroid-resistant [31].
It was noteworthy that the effects of the two antibodies on changes of airway remodelling were markedly different. Neutralizing IL-4 inhibited goblet cell changes, but had no effect on subepithelial fibrosis or epithelial hypertrophy. Neutralizing IL-25 produced the converse outcomes. To date, limited data are available regarding the relationship between IL-25 and remodelling, because this is not a significant feature of conventional short-term models of allergic bronchopulmonary inflammation. In this study, we have not only demonstrated such a link but also obtained preliminary evidence of a possible mechanism, because reduced remodelling correlated with reduced immunoreactivity for active TGF-β1 in the airways. This observation is consistent with our previous demonstration of the relationship between expression of TGF-β1 and changes of airway wall remodelling in a murine model of chronic asthma [16]. However, in the present study of the induction phase of asthma, the increase in TGF-β1 staining in control mice was relatively modest and changes in median staining intensity following treatment with anti-IL-25 were not statistically significant.
In the second part of this investigation, we went on to assess the possibility of suppressing the initial induction of Th2 response by treatment in early life alone, i.e. at the time of up-regulation by viral infection, but without later treatment during chronic challenge. Blocking of IL-4 in childhood would have too many systemic adverse effects to have significant potential in this regard, whereas blocking of locally produced IL-25 appeared to be much more attractive. We found that treatment on days 10, 17 and 24 of life was able to suppress the subsequent induction of asthmatic inflammation and remodelling, with a long-lasting effect equivalent to later administration of anti-IL-25. Our results imply that up-regulated expression of IL-25 following early-life infection with PVM, which we have demonstrated previously [5], is likely to be biologically important. Other cytokines produced by AECs, such as IL-33 and thymic stromal lymphopoietin (TSLP), may also promote an asthmatic response by biasing CD4+ T cell differentiation towards a Th2 phenotype [32–34]. However, our findings suggest that IL-25 plays a crucial role in the pathogenesis of asthma following early-life viral infection. Whether this observation is applicable to childhood asthma in general will require further investigation in other relevant models. Nevertheless, the apparent induction of a Th17 response by treatment with anti-IL-25 is likely to limit its usefulness as a therapeutic intervention.
In conclusion, we have shown that in our novel murine model of childhood asthma, blockade of cytokines associated with the generation of a Th2-biased immunological response inhibits the development of key features of an asthmatic response, including allergic inflammation and/or airway wall remodelling. This work highlights the potential of early intervention to suppress induction of a Th2-biased response in the primary prevention of childhood asthma.
Acknowledgments
Supported by grants from the National Health and Medical Research Council of Australia.
Disclosure
The authors declare that they have no conflict of interest related to the publication of this manuscript.
References
- 1.Holt PG, Sly PD. Prevention of allergic respiratory disease in infants: current aspects and future perspectives. Curr Opin Allergy Clin Immunol. 2007;7:547–55. doi: 10.1097/ACI.0b013e3282f14a17. [DOI] [PubMed] [Google Scholar]
- 2.Kristjansson S, Bjarnarson SP, Wennergren G, et al. Respiratory syncytial virus and other respiratory viruses during the first 3 months of life promote a local TH2-like response. J Allergy Clin Immunol. 2005;116:805–11. doi: 10.1016/j.jaci.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 3.Kusel MM, de Klerk NH, Kebadze T, et al. Early-life respiratory viral infections, atopic sensitization, and risk of subsequent development of persistent asthma. J Allergy Clin Immunol. 2007;119:1105–10. doi: 10.1016/j.jaci.2006.12.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jackson DJ, Gangnon RE, Evans MD, et al. Wheezing rhinovirus illnesses in early life predict asthma development in high-risk children. Am J Respir Crit Care Med. 2008;178:667–72. doi: 10.1164/rccm.200802-309OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Siegle JS, Hansbro N, Herbert C, et al. Early-life viral infection and allergen exposure interact to induce an asthmatic phenotype in mice. Respir Res. 2010;11:14. doi: 10.1186/1465-9921-11-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Domachowske JB, Bonville CA, Rosenberg HF. Animal models for studying respiratory syncytial virus infection and its long term effects on lung function. Pediatr Infect Dis J. 2004;23:S228–34. doi: 10.1097/01.inf.0000144672.81955.a4. [DOI] [PubMed] [Google Scholar]
- 7.Rosenberg HF, Domachowske JB. Pneumonia virus of mice: severe respiratory infection in a natural host. Immunol Lett. 2008;118:6–12. doi: 10.1016/j.imlet.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paul WE, Zhu J. How are T(H)2-type immune responses initiated and amplified? Nat Rev Immunol. 2010;10:225–35. doi: 10.1038/nri2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Angkasekwinai P, Park H, Wang YH, et al. Interleukin 25 promotes the initiation of proallergic type 2 responses. J Exp Med. 2007;204:1509–17. doi: 10.1084/jem.20061675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Temelkovski J, Hogan SP, Shepherd DP, Foster PS, Kumar RK. An improved murine model of asthma: selective airway inflammation, epithelial lesions and increased methacholine responsiveness following chronic exposure to aerosolised allergen. Thorax. 1998;53:849–56. doi: 10.1136/thx.53.10.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Siegle JS, Hansbro N, Herbert C, Yang M, Foster PS, Kumar RK. Airway hyperreactivity in exacerbation of chronic asthma is independent of eosinophilic inflammation. Am J Respir Cell Mol Biol. 2006;35:565–70. doi: 10.1165/rcmb.2006-0135OC. [DOI] [PubMed] [Google Scholar]
- 12.Ohara J, Paul WE. Production of a monoclonal antibody to and molecular characterization of B-cell stimulatory factor-1. Nature. 1985;315:333–6. doi: 10.1038/315333a0. [DOI] [PubMed] [Google Scholar]
- 13.Schneider T, Issekutz AC. Quantitation of eosinophil and neutrophil infiltration into rat lung by specific assays for eosinophil peroxidase and myeloperoxidase. Application in a Brown Norway rat model of allergic pulmonary inflammation. J Immunol Methods. 1996;198:1–14. doi: 10.1016/0022-1759(96)00143-3. [DOI] [PubMed] [Google Scholar]
- 14.Foster PS, Webb DC, Yang M, Herbert C, Kumar RK. Dissociation of T helper type 2 cytokine-dependent airway lesions from signal transducer and activator of transcription 6 signalling in experimental chronic asthma. Clin Exp Allergy. 2003;33:688–95. doi: 10.1046/j.1365-2222.2003.01647.x. [DOI] [PubMed] [Google Scholar]
- 15.Herbert C, Hettiaratchi A, Webb DC, Thomas PS, Foster PS, Kumar RK. Suppression of cytokine expression by roflumilast and dexamethasone in a mouse model of chronic asthma. Clin Exp Allergy. 2008;38:847–56. doi: 10.1111/j.1365-2222.2008.02950.x. [DOI] [PubMed] [Google Scholar]
- 16.Kumar RK, Herbert C, Foster PS. Expression of growth factors by airway epithelial cells in a model of chronic asthma: regulation and relationship to subepithelial fibrosis. Clin Exp Allergy. 2004;34:567–75. doi: 10.1111/j.1365-2222.2004.1917.x. [DOI] [PubMed] [Google Scholar]
- 17.Herbert C, Scott MM, Scruton KH, et al. Alveolar macrophages stimulate enhanced cytokine production by pulmonary CD4+ T-lymphocytes in an exacerbation of murine chronic asthma. Am J Pathol. 2010;177:1657–64. doi: 10.2353/ajpath.2010.100019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yi X, Feng F, Xiang Z, Ge L. The effects of allitridin on the expression of transcription factors T-bet and GATA-3 in mice infected by murine cytomegalovirus. J Med Food. 2005;8:332–6. doi: 10.1089/jmf.2005.8.332. [DOI] [PubMed] [Google Scholar]
- 19.Norzila MZ, Fakes K, Henry RL, Simpson J, Gibson PG. Interleukin-8 secretion and neutrophil recruitment accompanies induced sputum eosinophil activation in children with acute asthma. Am J Respir Crit Care Med. 2000;161:769–74. doi: 10.1164/ajrccm.161.3.9809071. [DOI] [PubMed] [Google Scholar]
- 20.Anderson GP. Endotyping asthma: new insights into key pathogenic mechanisms in a complex, heterogeneous disease. Lancet. 2008;372:1107–19. doi: 10.1016/S0140-6736(08)61452-X. [DOI] [PubMed] [Google Scholar]
- 21.Barrett NA, Austen KF. Innate cells and T helper 2 cell immunity in airway inflammation. Immunity. 2009;31:425–37. doi: 10.1016/j.immuni.2009.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang YH, Liu YJ. Thymic stromal lymphopoietin, OX40-ligand, and interleukin-25 in allergic responses. Clin Exp Allergy. 2009;39:798–806. doi: 10.1111/j.1365-2222.2009.03241.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaiko GE, Phipps S, Angkasekwinai P, Dong C, Foster PS. NK cell deficiency predisposes to viral-induced Th2-type allergic inflammation via epithelial-derived IL-25. J Immunol. 2010;185:4681–90. doi: 10.4049/jimmunol.1001758. [DOI] [PubMed] [Google Scholar]
- 24.Alcorn JF, Crowe CR, Kolls JK. TH17 cells in asthma and COPD. Annu Rev Physiol. 2010;72:495–516. doi: 10.1146/annurev-physiol-021909-135926. [DOI] [PubMed] [Google Scholar]
- 25.Furuzawa-Carballeda J, Vargas-Rojas MI, Cabral AR. Autoimmune inflammation from the Th17 perspective. Autoimmun Rev. 2007;6:169–75. doi: 10.1016/j.autrev.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 26.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 27.Zaph C, Du Y, Saenz SA, et al. Commensal-dependent expression of IL-25 regulates the IL-23-IL-17 axis in the intestine. J Exp Med. 2008;205:2191–8. doi: 10.1084/jem.20080720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kleinschek MA, Owyang AM, Joyce-Shaikh B, et al. IL-25 regulates Th17 function in autoimmune inflammation. J Exp Med. 2007;204:161–70. doi: 10.1084/jem.20061738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laan M, Palmberg L, Larsson K, Linden A. Free, soluble interleukin-17 protein during severe inflammation in human airways. Eur Respir J. 2002;19:534–7. doi: 10.1183/09031936.02.00280902. [DOI] [PubMed] [Google Scholar]
- 30.Bullens DM, Truyen E, Coteur L, et al. IL-17 mRNA in sputum of asthmatic patients: linking T cell driven inflammation and granulocytic influx? Respir Res. 2006;7:135. doi: 10.1186/1465-9921-7-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McKinley L, Alcorn JF, Peterson A, et al. TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J Immunol. 2008;181:4089–97. doi: 10.4049/jimmunol.181.6.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmitz J, Owyang A, Oldham E, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–90. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 33.Al-Shami A, Spolski R, Kelly J, Keane-Myers A, Leonard WJ. A role for TSLP in the development of inflammation in an asthma model. J Exp Med. 2005;202:829–39. doi: 10.1084/jem.20050199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lambrecht BN, Hammad H. Biology of lung dendritic cells at the origin of asthma. Immunity. 2009;31:412–24. doi: 10.1016/j.immuni.2009.08.008. [DOI] [PubMed] [Google Scholar]