

Abstract
Oestradiol and the selective oestrogen receptor modulator (SERM) raloxifene have been shown to ameliorate collagen-induced arthritis (CIA) in rats and in mice. One aim was to investigate if raloxifene exerts its anti-arthritic and anti-osteoporotic effects during the induction or effector phase of arthritis. A second aim was to analyse if raloxifene activates the oestrogen response element (ERE) to produce its immune-modulator effects. CIA or collagen–antibody-induced arthritis (CAIA) was induced in ovariectomized DBA/1-mice. CIA was used for evaluation of treatment during the induction, and CAIA for the effector phase of arthritis and osteoporosis development. Raloxifene, oestradiol or vehicle was administered 5 days/week. The clinical disease was evaluated continuously. Bone marrow density (BMD) was analysed with peripheral quantitative computer tomography, paws were collected for histological examination, and sera were analysed for markers of bone and cartilage turnover and proinflammatory cytokines. Transgenic luciferase (Luc)-ERE mice were immunized with collagen (CII), and after 10 days injected once with raloxifene, oestradiol or vehicle before termination. Spleens were analysed for luciferase activity to measure ERE activation. Treatment with oestradiol or raloxifene during the induction phase of CIA failed to affect arthritis. Raloxifene did not hamper disease activity in CAIA, whereas oestradiol delayed the onset and ameliorated the severity. Both raloxifene and oestradiol preserved BMD in CAIA. CII-immunization increased the oestradiol-induced ERE activation in spleen, and raloxifene activated the ERE at about 25% the intensity of oestradiol. Further experiments are needed to elucidate the exact mechanisms behind this finding.
Keywords: BMD, CAIA, CIA, oestradiol, luciferase-ERE, ovariectomy, raloxifene
Introduction
Rheumatoid arthritis (RA) is a progressive systemic autoimmune disease, causing great morbidity. Both focal joint erosions and generalized osteoporosis result in a disabling disease. The prevalence is 0·5–1% worldwide [1], with a female to male ratio of 3:1, and the prevalence of concurrent osteoporosis is 50% [2,3]. The female sex steroid oestradiol has been shown to be beneficial in postmenopausal osteoporosis, and also to influence the incidence and progression of RA. We have previously reported decreased joint destruction and disease progression in postmenopausal RA patients treated with oestrogen-containing hormone replacement therapy (HRT) [4]. Unfortunately, HRT has been associated with severe side effects [5], and is no longer recommended for long-term therapy. Therefore, there is a need to find alternative oestrogen-like substances with the beneficial properties, and lacking the side effects.
We and others have shown previously that administration of both oestradiol and raloxifene, a selective oestrogen receptor modulator (SERM) approved for the treatment of postmenopausal osteoporosis, can ameliorate collagen-induced arthritis (CIA), a murine model of human RA [6,7]. Even when treatment was initiated in mice with severe, established disease, these effects were substantial [7]. Also, when oestradiol was administered (at doses equivalent to estrus, resulting in serum levels of 400 pg/ml, or 50% of pregnancy levels, with serum levels of 4000 pg/ml) from 7 days prior to immunization until termination, three different mouse models failed to develop arthritis [8]. In addition to the anti-arthritic properties, treatment with raloxifene also prevented arthritis-induced osteoporosis development [6,7]. CIA and the loss of endogenous oestrogen after ovariectomy (OVX) have been shown to contribute to osteoporosis development in an additive way [9].
In the present study we wanted to investigate whether raloxifene would display anti-arthritic effects with treatment only during the induction phase of CIA, or during the effector phase of the disease. For treatment during the induction phase we used the CIA model, and treated the mice from 2 days pre- to 10 days postimmunization. Treatment during the effector phase was evaluated using the collagen–antibody-induced arthritis (CAIA) model [10]. In CAIA, the introduction of preformed antibodies induces arthritis. Antibodies to collagen II (CII) have been shown previously to be involved in both human and experimental RA [11], and oestradiol has been shown to hamper the disease in CAIA [12].
Oestrogens activate target genes via various signalling pathways, including the classical pathway, in which oestrogen receptors (ER) α and β bind to oestrogen response elements (ERE) on DNA, and thereby promote gene transcription. We therefore wanted to investigate whether the ERE is activated by CII immunization, and whether raloxifene can activate this pathway. To this end, we used a transgenic ERE-luciferase (Luc) reporter mouse model [13].
One recent 10-week Phase II clinical trial investigated an agonist of ERα (Org 37663) in postmenopausal RA, and found no clinical benefit despite induction of oestrogenic responses in several organ systems [14]. In another recent 12-week Phase II trial an agonist of ERβ (ERB-041) was investigated, and similar results were found [15]. One explanation may be that the duration of the trials was not long enough to induce clinical benefit, as a previous trial with HRT showed benefit only after 2 years. As animal studies with both compounds had shown anti-arthritic properties, it is important to investigate further the mechanisms for these effects, and to evaluate whether there is a difference between different species [16,17]. Indeed, we have shown previously that stimulation of ERα ameliorated CIA in mice, whereas an agonist of ERβ did not, while the specific ERβ agonist ERB-041 improved arthritis in rats [18].
Based on the current study, we conclude that neither the SERM raloxifene nor oestradiol had any effect on the induction phase of CIA. Oestradiol, but not raloxifene, modified the effector phase of the disease in CAIA. Raloxifene activated the classical signalling pathway to promote gene transcription, although not to the same extent as oestradiol. However, both compounds displayed potent anti-osteoporotic properties in lipopolysaccharide (LPS)-induced bone loss seen in CAIA.
Materials and methods
Animals and experimental procedures
The ethical committee for animal experiments at Gothenburg University approved this study. Female DBA/1 mice were purchased from Taconic M&B A/S (Ry, Denmark). Male transgenic 3 × ERE-TAT-Luc (ERE-luciferase) mice, on a mixed CBA × C56Bl/6 J background, were generated as described previously [13]. Mice were electronically tagged and kept, five to 10 animals per cage, under standard environmental conditions, and fed standard laboratory chow and tap water ad libitum.
OVX and sham operations were performed at 10 weeks of age. Ovaries were removed through a midline incision of the skin, and flank incisions of the peritoneum. The skin incision was then closed with metallic clips. Sham-operated animals had their ovaries exposed but not removed. Orchiectomy was performed at 20 weeks of age. Testes were removed through an incision of the scrotum, and the incision was closed with a metallic clip. Surgery was performed after ketamine (PfizerAB, Täby, Sweden) and medetomidin (OrionPharma, Espoo, Finland) anaesthesia. Carprofen (OrionPharma) was used postoperatively as a painkiller.
Experimental design
Induction phase
CIA was induced 2 weeks after OVX in female DBA/1 mice. Treatment with raloxifene, oestradiol or vehicle 5 days per week was started 2 days prior to immunization and continued for 12 days. A booster injection of collagen II with incomplete Freund's adjuvant was given 3 weeks after immunization, and arthritic score and severity were monitored. Mice were terminated 2 weeks later.
Effector phase
CAIA: Treatment with raloxifene, oestradiol or vehicle 5 days per week started 10 days after OVX or sham operation, and continued until termination of the experiment. The mice received a four-collagen–antibodies cocktail intravenously (i.v.) 10 days later (20 days after surgery, day 0). One week later, they received an intraperitoneal (i.p.) injection of LPS to enhance arthritis incidence and severity, and the experiment was terminated on day 14. Control mice were injected with phosphate-buffered saline (PBS) i.v. and LPS i.p.
Male transgenic ERE-luciferase mice were castrated and 11 days later immunized with chicken CII and adjuvant. After 9 days they received one subcutaneous injection of raloxifene, oestradiol or vehicle, and were then terminated 10 h later (day 10 after immunization).
Treatment
Mice were given subcutaneous injections 5 days per week of the raloxifene analogue LY117018 (generous gift from Eli Lilly, Indianapolis, IN, USA) (60 µg/mouse/day) or 17β-oestradiol-3-benzoate (E2) (Sigma, St Louis, MO, USA) (1·0 µg/mouse/day) dissolved in Miglyol812 (OmyaPeralta GmbH, Hamburg, Germany). Control mice received Miglyol812 (100 µl/mouse/day). The dosages of Ral and E2 have been shown previously to prevent osteoporosis equally well in mice [19–21]. LY117018 differs from raloxifene at only one site on the molecule, with a pyrrolidine ring on the basic side chain instead of a piperidine ring. This small difference does not affect its biological properties. Thus, Ral and LY117018 can be regarded as replaceable with respect to their biological properties.
Induction of arthritis
Experiment 1
Two weeks after ovariectomy DBA/1 mice were immunized with 100 µg of chicken CII (Sigma, St Louis, MO, USA) dissolved in 0·1 m acetic acid and emulsified with an equal volume of incomplete Freund's adjuvant (Sigma) supplemented with 0·5 mg/ml Mycobacterium tuberculosis (Sigma). A total volume of 100 µl was injected intradermally at the base of the tail. After 21 days, mice received a booster injection with CII emulsified in incomplete Freund's adjuvant. Arthritis developed shortly thereafter, and was evaluated continuously for frequency and severity.
Experiment 2
Twenty days after OVX or sham-operation, DBA/1 mice received an intravenous shot of a four-antibody cocktail [monoclonal immunoglobulin (Ig)G antibodies specific for the C1, J1, D3 and U1 epitopes on the collagen type II molecule], according to the protocol of Nandakumar and Holmdahl [10]. Non-arthritic controls received equal volumes of PBS. One week later, all mice received an intraperitoneal injection of 25 µg LPS (Escherichia coli 055 : B5; Difco Laboratories, Detroit, MI, USA).
Experiment 3
ERE-luciferase mice were immunized with 100 µg of chicken CII (Sigma) dissolved in 0·1 m acetic acid and emulsified with an equal volume of incomplete Freund's adjuvant (Sigma) supplemented with 0·5 mg/ml M. tuberculosis (Sigma). A total volume of 100 µl was injected intradermally at the base of the tail. Control mice were injected with 100 µl PBS.
Evaluation of arthritis
In experiments 1 and 2, the animals were evaluated every other day for frequency and severity of arthritis. Scoring was performed in a blinded manner without knowledge of the treatment groups and previous scores. Severity was graded as described previously [22], scoring 1–3 in each paw (maximum of 12 points per mouse) as follows: (i) swelling or erythema in one joint; (ii) swelling or erythema in two joints; or (iii) severe swelling of the entire paw or ankylosis.
Tissue collection and histological examination
At termination of the experiments, mice were anaesthetized for blood withdrawal, and then killed by cervical dislocation. Sera were collected individually and stored at −20°C until used. Successful removal of the ovaries was confirmed by weighing the uteri. For experiment 2, one femur was placed in formaldehyde for analysis of bone mineral density. The paws (experiments 1 and 2) were placed in formaldehyde, decalcified and embedded in paraffin. Sections were stained with haematoxylin and eosin and encoded before examination. In sections from each animal, the distal and proximal areas of all four paws were graded separately on a scale of 0–4 and the score was then divided by 2, which yielded a maximum histological destruction score of 16 points per mouse, assessed as follows: 1 = synovial hypertrophy; 2 = pannus, discrete erosions of cartilage and bone; 3 = severe erosions of cartilage and bone; and 4 = complete ankylosis. In experiment 3, spleens were collected and frozen individually in liquid nitrogen, and kept at −20°C until use.
Assessment of bone mineral density (BMD)
One femur was subjected to a peripheral quantitative computed tomography (pQCT) scan with a Stratec pQCT XCT Research M, software version 5·4B (Norland, Fort Atkinson, WI, USA) at a resolution of 70 µm, as described previously [23]. Trabecular BMD was determined with a metaphyseal scan at a point 3% of the length of the femur from the growth plate. The inner 45% of the area was defined as the trabecular bone compartment. Cortical BMD was determined with a mid-diaphyseal scan.
Identification of serological markers of bone and cartilage remodelling
For measurement of bone resorption, serum levels of fragments of type I collagen were assessed using a RatLaps enzyme-linked immunosorbent assay (ELISA) kit (Nordic Bioscience Diagnostics A/S, Herlev, Denmark). Serum levels of osteocalcin, a marker of bone formation, were determined with a mouse osteocalcin immunoradiometric assay (IRMA) kit (Immutopics, Inc., San Clemente, CA, USA). As a marker of cartilage destruction, serum levels of cartilage oligomeric matrix protein (COMP) were determined with an animal COMP® ELISA kit (AnaMar Medical AB, Uppsala, Sweden).
Serum anti-CII antibody ELISA and serum interleukin (IL)-6 bioassay
By use of a previously described ELISA, serum levels of anti-CII antibodies were determined [24]. A bioassay with cell line B13·29, subclone B9 (which is dependent on IL-6 for growth), was used to measure serum levels of IL-6, as described previously [25,26].
Protein preparation and luciferase analysis
The frozen spleen was homogenized in lysis buffer [25 mm Tris pH 7·8, 1·5 mm ethylenediamine tetraacetic acid (EDTA), 10% glycerol, 1% Triton X-100, 2 mm dithiothreitol (DTT) and complete protease inhibitors, #1535101; Roche Diagnostics, Mannheim, Germany] and separated by centrifugation at 10 650 g for 30 min. The supernatant was stored at −20°C until further analysis. The protein content was measured using Bio-Rad DC protein assay (#500–0116; Promega, Madison, WI, USA). The luciferase activity was performed using a standard luciferase assay (#E4030; Promega) according to the manufacturer's instructions and measured on a GloMax™ 20/20 luminometer (#E5311; Promega).
Statistical analysis
For statistical evaluation, the Kruskall–Wallis test followed by a post hoc test was used for comparisons between all groups in each experiment. A P-value ≤ 0·05 was considered significant.
Results
Raloxifene or oestradiol treatment did not influence the induction phase of collagen-induced arthritis
To investigate whether raloxifene can influence the induction phase of CIA, OVX DBA/1 mice were treated from 2 days pre-immunization until 10 days postimmunization with either raloxifene (60 µg/day), oestradiol (1 µg/day) or the Miglyol812 vehicle control (100 µl/day), as described in Materials and methods. Arthritis scores were evaluated every other day after administration of the booster injection of CII on day 21. In this experiment raloxifene or oestradiol did not hamper the development of arthritis significantly, as measured by frequency (Fig. 1) and severity (data not shown) of arthritis. In addition, we found no differences in the serum levels of anti-CII antibodies, IL-6 or the cartilage degradation marker COMP (Fig. 1).
Fig. 1.
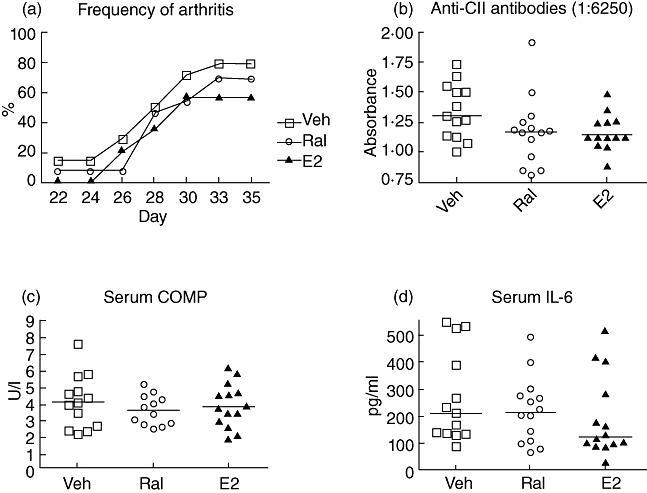
Treatment with raloxifene or oestradiol during the induction phase of collagen-induced arthritis (CIA). Female ovariectomized (OVX) DBA/1 mice were treated with raloxifene (60 µg/day; n = 13) (open circles), oestradiol (1 µg/day; n = 14) (triangles) or vehicle control (Miglyol812; n = 14) (open squares) from 2 days before until 10 days after immunization. The development of arthritis was evaluated every other day until termination on day 35. Lines represent medians. Kruskall–Wallis test with post hoc comparison was used. There was no statistical difference regarding frequency of arthritis (a), anti-collagen II (CII) antibodies (b), serum interleukin (IL)-6 (c) or cartilage oligomeric matrix protein (COMP) (d) between the treatment groups.
Oestradiol delayed the onset and reduced the severity of collagen-antibody induced arthritis, but raloxifene did not
To investigate the anti-arthritic properties of raloxifene, female DBA/1-mice were ovariectomized or sham-operated, and CAIA was induced. Ten days prior to receiving the antibody cocktail, administration of raloxifene (60 µg/day), oestradiol (1 µg/day) or vehicle (Miglyol812, 100 µl/day) was started, and continued 5 days per week until termination of the experiment. Figure 2a shows that treatment with oestradiol resulted in a significantly later onset of disease compared to vehicle-treated OVX controls (P < 0·001 on day 7 and P < 0·01 on day 9). The presence of endogenous hormones (sham-operated mice) also delayed the onset of arthritis (P < 0·01 on day 7), but this effect was not sustained. Raloxifene treatment did not result in delayed onset compared to vehicle controls. Figure 2b shows that oestradiol treatment resulted in less severe arthritic disease, and this effect was sustained throughout the experiment (P < 0·001 compared to vehicle-treated controls). There was no maintained difference in arthritic severity between the OVX and sham vehicle-treated groups, although the groups differed significantly (P < 0·05) on day 7. Raloxifene treatment did not alter disease frequency or severity significantly compared to OVX vehicle controls at any time-point.
Fig. 2.

Exogenous and endogenous oestradiol hampered the development of collagen II-antibody induced arthritis (CAIA), but raloxifene did not. Female ovariectomized (OVX) DBA/1 mice were treated with raloxifene (60 µg/day; n = 11) (open circles), oestradiol (1 µg/day; n = 8) (triangles) or vehicle control (Miglyol812; n = 10) (open squares) and CAIA was induced. Sham-operated controls received vehicle control (n = 11) (open triangles), as did OVX non-arthritic mice (n = 11) (solid squares). Treatment was started 10 days prior to injection of the four-antibody cocktail to induce CAIA. Frequency (a) and severity (b) were evaluated continuously. Severity was graded 1–3 in each paw (maximum score of 12 per mouse), expressed as the mean in each group. Asterisks show the difference between oestradiol and vehicle treatment, and the pound signs show the difference between sham-operated and OVX controls. (c) Histological destruction scores of paw sections. Scatter-plots show the scores of individual mice, and bars show the median in each group. (d) Representative images of paw tissue sections, revealing treatment effects on histological features in each group. The Kruskall–Wallis test with post hoc comparison was used; *P < 0·05; ***P < 0·001.
Histological examination of the paw sections (Fig. 2c and d) revealed the same degree of destruction in joints from OVX and sham-operated controls (median destruction scores of 5·2 and 6·0 of a maximum of 16, respectively). Oestradiol treatment resulted in preserved joint architecture (P < 0·001) compared to vehicle-treated OVX controls. Raloxifene did not affect the degree of joint destruction significantly.
Treatment with raloxifene or oestradiol protected mice with CAIA from osteoporosis
Non-arthritic OVX controls and both OVX and sham-operated mice with CAIA had low trabecular bone mineral density (BMD), with median values of 184, 170 and 185 mg/cm3, respectively (Fig. 3). In contrast, treatment with raloxifene increased the BMD (median 271 mg/cm3) compared to controls (P < 0·01), although raloxifene did not hamper arthritis development. Oestradiol treatment resulted in a trabecular BMD of 469 mg/cm3. The cortical thickness was higher in sham-operated than in OVX mice (P < 0·01), and was increased by treatment with both oestradiol and raloxifene (P < 0·001).
Fig. 3.
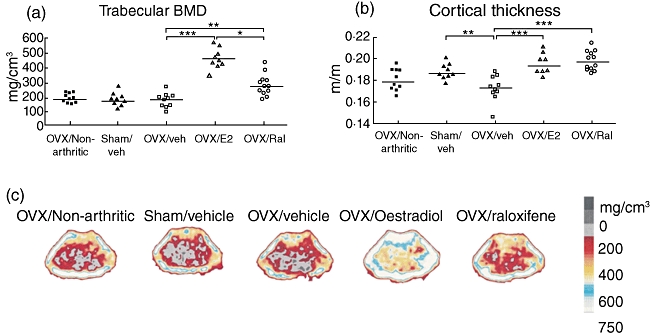
Treatment with raloxifene or oestradiol protected arthritic mice from osteoporosis. Scatter-plots showing the trabecular bone mineral density (BMD) (a) and the cortical thickness (b), measured with peripheral quantitative computer tomography (pQCT) in mice with collagen II-antibody induced arthritis (CAIA) treated with raloxifene (60 µg/day; n = 11) (open circles), oestradiol (1 µg/day; n = 8) (triangles) or vehicle control (Miglyol812; n = 10) (open squares). Sham-operated controls received vehicle control (n = 11) (open triangles), as did ovariectomized (OVX) non-arthritic mice (n = 11) (solid squares). Lines represent medians. Kruskall–Wallis test with post hoc comparison was used; *P < 0·05; **P < 0·01; ***P < 0·001. (c) Representative pQCT scans of cross-sections of the femur, showing the bone mineral density (BMD). The pictures show one representative mouse in each treatment group. The bar shows the density of the bone, from 0 (black) to > 750 mg/cm3 (white).
Serological markers of bone and cartilage remodelling in mice with collagen-antibody induced arthritis
Bone formation, as measured by serum levels of osteocalcin, was significantly higher in non-immunized mice versus arthritic OVX mice (Table 1). Raloxifene increased the osteocalcin levels compared to both oestradiol treatment and vehicle controls. In contrast, the levels of RatLaps (indicating bone resorption) did not differ between the raloxifene, oestradiol and vehicle groups, whereas sham-operated mice had lower levels than OVX mice.
Table 1.
Serum levels of bone and cartilage turnover markers and interleukin (IL)-6 in mice with collagen–antibody-induced arthritis (CAIA) treated with raloxifene, oestradiol or vehicle control.*
| Non-immunized OVX vehicle healthy controls | Immunized sham vehicle | Immunized OVX vehicle | Immunized OVX E2 | Immunized OVX raloxifene | |
|---|---|---|---|---|---|
| Bone formation | 178††† | 118 | 137 | 138 | 170††# |
| Osteocalcin, ng/ml | (145–213) | (103–135) | (114–151) | (121–172) | (155–185) |
| Bone resorption | 14·1 | 13·2††† | 15·9 | 14·5 | 15·6 |
| RatLaps, ng/ml | (13·4–15·8) | (12·7–13·5) | (13·4–17·9) | (14·2–16·1) | (14·6–17·0) |
| Cartilage destruction | 3·0††† | 4·2† | 8·3 | 3·9†† | 5·8# |
| COMP, U/l | (2·6–4·6) | (3·4–6·8) | (3·8–10·1) | (3·4–4·6) | (4·0–7·0) |
| IL-6 (pg/ml) | 105††† | 240 | 230 | 203 | 199 |
| (85–125) | (217–290) | (145–397) | (130–301) | (154–297) |
Values are the median (interquartile range) of eight to 12 animals per group.
P < 0·05 versus ovariectomy (OVX) vehicle
P < 0·01 versus OVX vehicle
P < 0·001 versus OVX vehicle
P < 0·05 versus OVX E2. COMP: cartilage oligomeric matrix protein.
Measurement of cartilage degradation in CAIA
The serum level of COMP is a marker of the degree of cartilage destruction, and has been shown to increase both in human RA [27,28] and in murine CIA [29]. In the CAIA experiment, COMP was increased in OVX mice compared to non-arthritic OVX mice, and arthritic OVX mice had significantly higher levels than the sham-operated controls. Oestradiol lowered the COMP level significantly, compared to arthritic OVX controls, whereas raloxifene did not. These findings are consistent with the degree of cartilage destruction seen in histological sections (Table 1).
IL-6 levels in serum
The serum levels of the proinflammatory cytokine IL-6 were measured using a bioassay. Data from the CAIA experiment are depicted in (Table 1). Mice immunized with CAIA had significantly higher serum levels of IL-6 compared with non-immunized healthy controls (P < 0·001). All CAIA mice had similar levels of IL-6, regardless of treatment, at the time of termination when sera were collected.
Raloxifene activated the oestrogen response element to a lesser degree than oestradiol
Transgenic Luc-ERE mice were orchiectomized and 11 days later they were immunized with CII and Freund's adjuvant, as described in Materials and methods. Ten days after immunization they were terminated after having received one subcutaneous injection of raloxifene (60 µg), oestradiol (1 µg) or vehicle (Miglyol812, 100 µl) 10 h previously. The amount of luciferase activity in spleen was measured and related to the amount of protein present (Fig. 4). Compared to non-immunized oestradiol controls, there was a 10-fold increase in luciferase activity in the spleen of immunized oestradiol-treated mice, demonstrating increased ERE activation after CII immunization. The luciferase activity was enhanced more than 100-fold in immunized oestradiol-treated mice compared to vehicle controls, with median values of 2400 and 12 units/mg protein, respectively. Raloxifene treatment resulted in a median value of 570 units/mg protein, displaying an activation of the ERE, but not to the same extent as oestradiol.
Fig. 4.
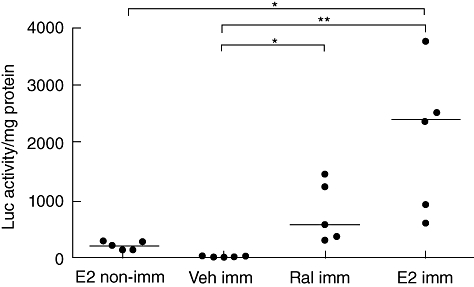
Raloxifene activated the oestrogen response element to a lesser degree than oestradiol. Transgenic luciferase-oestrogen response element (ERE) mice were orchiectomized and 2 weeks later immunized with type II collagen and Freund's adjuvant, as described in Materials and methods. Ten days after immunization they were terminated after having received one subcutaneous injection of raloxifene (60 µg), oestradiol (1 µg) or vehicle (Miglyol812, 100 µl) 10 h previously. The amount of luciferase activity in spleen was measured and related to the amount of protein in the tissue. Bars show the median in each group; n = 5 per group, *P < 0·05; **P < 0·01.
Discussion
It has been shown previously that both exogenous and endogenous oestradiol hamper CAIA [12], as well as CIA [30–32]. We have shown potent anti-arthritic effects of raloxifene previously in the CIA model [6,7], with protection against both erosivity and generalized osteoporosis even when treatment was started in established disease. The present study is the first to show that despite these anti-arthritic properties, raloxifene did not affect CAIA. The CAIA model does not involve the induction phase, but instead only the antibody-mediated effector phase of arthritic disease. Our results suggest therefore that raloxifene does not exert its effects during the effector phase, in contrast to oestradiol, which has an effect at this stage of disease development [12].
It has also been shown that oestradiol treatment during the induction phase of CIA delays the onset of the disease by approximately 10 days [33]. Therefore, in an additional study, mice were treated with raloxifene, oestradiol or vehicle during the induction phase, and were then evaluated continuously for arthritis. However, in this study treatment with oestradiol or raloxifene daily for 12 days, starting 2 days before immunization, did not influence the appearance of arthritis significantly.
Recent studies have proposed that the anti-inflammatory mechanisms may be different during raloxifene treatment compared to oestradiol treatment. Oestradiol down-regulated T lymphocyte-dependent and granulocyte-mediated inflammation, but raloxifene did not [19]. Raloxifene lowered the levels of tumour necrosis factor (TNF)-α and receptor activator of nuclear factor kappa-B ligand (RANKL) mRNA in spleen from arthritic mice, whereas oestradiol did not affect these mediators of inflammation [6].
To elucidate further the differences between these two compounds, we investigated the activation of the ERE in spleen from ERE-Luc reporter mice immunized with CII and Freund's complete adjuvant. As expected, exposure to oestradiol resulted in increased luciferase activity in the spleen, whereas vehicle controls displayed a total lack of luciferase activity. Immunization with CII greatly enhanced the luciferase activity (indicating oestradiol-induced ERE activation). One previous in vitro study shows that raloxifene acts ERE-dependently in osteoblasts as an oestradiol agonist, and in breast cancer cells as an antagonist [34]. In addition, both oestradiol and raloxifene can act via the raloxifene response element [35] and at an AP1 enhancer element [36,37] (non-classical pathway), suggesting different pathways of activation in different cells. Interestingly, exposure to raloxifene increased the ERE-induced luciferase activity in spleen, but to a lesser degree than oestradiol. These results indicate that although raloxifene affected tissues via the classical oestrogen receptors and ERE in spleen, it did not have the same impact as oestradiol. The doses of raloxifene and oestradiol were chosen for their equipotent effects on BMD, and therefore it is possible that a higher dose of raloxifene could have activated the ERE to the same extent as oestradiol.
The present study is the first to analyse the effects of CAIA on BMD and cartilage and bone remodelling. Sham-operated mice with CAIA, non-arthritic OVX mice and OVX mice with CAIA displayed the same trabecular BMD. These results were unexpected, as both OVX and CIA have been shown to induce bone loss separately and additively [9]. All mice had received an intraperitoneal injection of LPS 1 week prior to termination. LPS is well known to induce osteoporosis quickly [38,39]. Because we did not find any difference in BMD between the vehicle-treated mice that had received collagen-antibodies and the non-arthritic controls, osteoporosis may have been induced by the administration of LPS. Also, the duration of the experiment was 2 weeks after administration of antibodies, and this short observation time may conceal pro-osteoporotic properties of CAIA. This issue needs to be studied further. Interestingly, raloxifene treatment resulted in increased BMD, although it did not affect the severity of the arthritic disease, suggesting anti-osteoporotic properties by raloxifene during LPS-induced inflammation. In addition, raloxifene increased bone formation as measured by serum levels of osteocalcin. This is in accordance with our previous results [6].
The histological destruction found in paw sections was not as severe as in some previous studies [10,12], and this was due most probably to the short experiment protocol (2 weeks of disease).
Serum levels of COMP reflect the degree of cartilage destruction during arthritic disease [27–29]. To our knowledge, this has not been investigated previously in CAIA. The arthritic disease resulted in a significant increase in COMP levels in OVX mice compared to non-arthritic controls (P < 0·001). As both groups had received an injection of LPS, administration of anti-CII antibodies contributed to the cartilage destruction. Indeed, it has been shown previously in vitro that anti-collagen II antibodies are pathogenic to chondrocytes, affecting both cartilage formation [40] and cartilage explants [41]. Administration of oestradiol and sham operation lowered the COMP levels compared to arthritic OVX controls, indicating protection of cartilage by both exogenous and endogenous oestradiol. In contrast, raloxifene did not influence the serum levels of COMP or the destruction of cartilage. It has been reported previously that raloxifene does not hamper granulocyte-mediated inflammation, whereas oestradiol does [19]. This could explain the difference between raloxifene and oestradiol treatment, as CII antibodies have been shown to mediate cartilage destruction even in the absence of inflammation [42,43].
In conclusion, we found that raloxifene and oestradiol, in doses that have been shown previously to have anti-arthritic and anti-osteoporotic properties, both activated the ERE but did not affect the induction phase of CIA. In contrast to oestradiol, raloxifene did not have the capacity to ameliorate the effector phase of arthritis. We also report that the induction of CAIA, by itself, did not induce osteoporosis. Interestingly, both raloxifene and oestradiol prevented LPS-induced trabecular bone loss. Additional experiments are needed to elucidate the mechanisms whereby oestradiol and raloxifene exert their beneficial effects on arthritis and inflammation-triggered osteoporosis.
Acknowledgments
We thank Margareta Rosenkvist, Berit Eriksson, Anette Hansevi and Maud Petersson for excellent technical assistance. This study was supported by grants from the Medical Faculty of Göteborg University (ALF), Göteborg Medical Society, King Gustav V's 80 years' foundation, the Sahlgrenska Foundation, the NovoNordic Foundation, the Börje Dahlin foundation, the Association against Rheumatism, Reumaforskningsfond Margareta and the Swedish Research Council.
Disclosure
The authors declare that they have no competing interests.
References
- 1.Doran MF, Pond GR, Crowson CS, O'Fallon WM, Gabriel SE. Trends in incidence and mortality in rheumatoid arthritis in Rochester, Minnesota, over a forty-year period. Arthritis Rheum. 2002;46:625–31. doi: 10.1002/art.509. [DOI] [PubMed] [Google Scholar]
- 2.Forsblad D'Elia H, Larsen A, Waltbrand E, et al. Radiographic joint destruction in postmenopausal rheumatoid arthritis is strongly associated with generalised osteoporosis. Ann Rheum Dis. 2003;62:617–23. doi: 10.1136/ard.62.7.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sinigaglia L, Nervetti A, Mela Q, et al. A multicenter cross sectional study on bone mineral density in rheumatoid arthritis. Italian Study Group on Bone Mass in Rheumatoid Arthritis. J Rheumatol. 2000;27:2582–9. [PubMed] [Google Scholar]
- 4.Forsblad D'Elia H, Larsen A, Mattsson LA, et al. Influence of hormone replacement therapy on disease progression and bone mineral density in rheumatoid arthritis. J Rheumatol. 2003;30:1456–63. [PubMed] [Google Scholar]
- 5.Stefanick ML. Estrogens and progestins: background and history, trends in use, and guidelines and regimens approved by the US Food and Drug Administration. Am J Med. 2005;118:64–73. doi: 10.1016/j.amjmed.2005.09.059. [DOI] [PubMed] [Google Scholar]
- 6.Jochems C, Islander U, Kallkopf A, Lagerquist M, Ohlsson C, Carlsten H. Role of raloxifene as a potent inhibitor of experimental postmenopausal polyarthritis and osteoporosis. Arthritis Rheum. 2007;56:3261–70. doi: 10.1002/art.22873. [DOI] [PubMed] [Google Scholar]
- 7.Jochems C, Lagerquist M, Hakansson C, Ohlsson C, Carlsten H. Long-term anti-arthritic and anti-osteoporotic effects of raloxifene in established experimental postmenopausal polyarthritis. Clin Exp Immunol. 2008;152:593–7. doi: 10.1111/j.1365-2249.2008.03660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Latham KA, Zamora A, Drought H, et al. Estradiol treatment redirects the isotype of the autoantibody response and prevents the development of autoimmune arthritis. J Immunol. 2003;171:5820–7. doi: 10.4049/jimmunol.171.11.5820. [DOI] [PubMed] [Google Scholar]
- 9.Jochems C, Islander U, Erlandsson M, Verdrengh M, Ohlsson C, Carlsten H. Osteoporosis in experimental postmenopausal polyarthritis: the relative contributions of estrogen deficiency and inflammation. Arthritis Res Ther. 2005;7:R837–43. doi: 10.1186/ar1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nandakumar KS, Holmdahl R. Efficient promotion of collagen antibody induced arthritis (CAIA) using four monoclonal antibodies specific for the major epitopes recognized in both collagen induced arthritis and rheumatoid arthritis. J Immunol Methods. 2005;304:126–36. doi: 10.1016/j.jim.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 11.Burkhardt H, Koller T, Engstrom A, et al. Epitope-specific recognition of type II collagen by rheumatoid arthritis antibodies is shared with recognition by antibodies that are arthritogenic in collagen-induced arthritis in the mouse. Arthritis Rheum. 2002;46:2339–48. doi: 10.1002/art.10472. [DOI] [PubMed] [Google Scholar]
- 12.Nandakumar KS, Svensson L, Holmdahl R. Collagen type II-specific monoclonal antibody-induced arthritis in mice: description of the disease and the influence of age, sex, and genes. Am J Pathol. 2003;163:1827–37. doi: 10.1016/S0002-9440(10)63542-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lemmen JG, Arends RJ, van Boxtel AL, van der Saag PT, van der Burg B. Tissue- and time-dependent estrogen receptor activation in estrogen reporter mice. J Mol Endocrinol. 2004;32:689–701. doi: 10.1677/jme.0.0320689. [DOI] [PubMed] [Google Scholar]
- 14.van Vollenhoven RF, Houbiers JG, Buttgereit F, et al. The selective estrogen receptor alpha agonist Org 37663 induces estrogenic effects but lacks antirheumatic activity: a phase IIa trial investigating efficacy and safety of Org 37663 in postmenopausal female rheumatoid arthritis patients receiving stable background methotrexate or sulfasalazine. Arthritis Rheum. 2010;62:3832–3. doi: 10.1002/art.27196. [DOI] [PubMed] [Google Scholar]
- 15.Roman-Blas JA, Castaneda S, Cutolo M, Herrero-Beaumont G. Efficacy and safety of a selective estrogen receptor-beta agonist, ERB-041, in patients with rheumatoid arthritis. Arthritis Care Res. 2010;62:1588–93. doi: 10.1002/acr.20275. [DOI] [PubMed] [Google Scholar]
- 16.Dulos J, Vijn P, van Doorn C, et al. Suppression of the inflammatory response in experimental arthritis is mediated via estrogen receptor alpha but not estrogen receptor beta. Arthritis Res Ther. 2010;12:R101. doi: 10.1186/ar3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harris HA, Albert LM, Leathurby Y, et al. Evaluation of an estrogen receptor-beta agonist in animal models of human disease. Endocrinology. 2003;144:4241–9. doi: 10.1210/en.2003-0550. [DOI] [PubMed] [Google Scholar]
- 18.Engdahl C, Jochems C, Windahl SH, et al. Amelioration of collagen-induced arthritis and immune-associated bone loss through signaling via estrogen receptor alpha, and not estrogen receptor beta or G protein-coupled receptor 30. Arthritis Rheum. 2010;62:524–33. doi: 10.1002/art.25055. [DOI] [PubMed] [Google Scholar]
- 19.Erlandsson MC, Gomori E, Taube M, Carlsten H. Effects of raloxifene, a selective estrogen receptor modulator, on thymus, T cell reactivity, and inflammation in mice. Cell Immunol. 2000;205:103–9. doi: 10.1006/cimm.2000.1719. [DOI] [PubMed] [Google Scholar]
- 20.Erlandsson MC, Jonsson CA, Lindberg MK, Ohlsson C, Carlsten H. Raloxifene- and estradiol-mediated effects on uterus, bone and B lymphocytes in mice. J Endocrinol. 2002;175:319–27. doi: 10.1677/joe.0.1750319. [DOI] [PubMed] [Google Scholar]
- 21.Onoe Y, Miyaura C, Ito M, Ohta H, Nozawa S, Suda T. Comparative effects of estrogen and raloxifene on B lymphopoiesis and bone loss induced by sex steroid deficiency in mice. J Bone Miner Res. 2000;15:541–9. doi: 10.1359/jbmr.2000.15.3.541. [DOI] [PubMed] [Google Scholar]
- 22.Holmdahl R, Jansson L, Larsson E, Rubin K, Klareskog L. Homologous type II collagen induces chronic and progressive arthritis in mice. Arthritis Rheum. 1986;29:106–13. doi: 10.1002/art.1780290114. [DOI] [PubMed] [Google Scholar]
- 23.Windahl SH, Vidal O, Andersson G, Gustafsson JA, Ohlsson C. Increased cortical bone mineral content but unchanged trabecular bone mineral density in female ERbeta(–/–) mice. J Clin Invest. 1999;104:895–901. doi: 10.1172/JCI6730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Verdrengh M, Jonsson IM, Holmdahl R, Tarkowski A. Genistein as an anti-inflammatory agent. Inflamm Res. 2003;52:341–6. doi: 10.1007/s00011-003-1182-8. [DOI] [PubMed] [Google Scholar]
- 25.Bremell T, Abdelnour A, Tarkowski A. Histopathological and serological progression of experimental Staphylococcus aureus arthritis. Infect Immun. 1992;60:2976–85. doi: 10.1128/iai.60.7.2976-2985.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Helle M, Boeije L, Aarden LA. Functional discrimination between interleukin 6 and interleukin 1. Eur J Immunol. 1988;18:1535–40. doi: 10.1002/eji.1830181010. [DOI] [PubMed] [Google Scholar]
- 27.Saxne T, Heinegard D. Cartilage oligomeric matrix protein: a novel marker of cartilage turnover detectable in synovial fluid and blood. Br J Rheumatol. 1992;31:583–91. doi: 10.1093/rheumatology/31.9.583. [DOI] [PubMed] [Google Scholar]
- 28.Mansson B, Carey D, Alini M, et al. Cartilage and bone metabolism in rheumatoid arthritis. Differences between rapid and slow progression of disease identified by serum markers of cartilage metabolism. J Clin Invest. 1995;95:1071–7. doi: 10.1172/JCI117753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Joosten LA, Lubberts E, Helsen MM, et al. Protection against cartilage and bone destruction by systemic interleukin-4 treatment in established murine type II collagen-induced arthritis. Arthritis Res. 1999;1:81–91. doi: 10.1186/ar14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holmdahl R, Jansson L, Andersson M. Female sex hormones suppress development of collagen-induced arthritis in mice. Arthritis Rheum. 1986;29:1501–9. doi: 10.1002/art.1780291212. [DOI] [PubMed] [Google Scholar]
- 31.Jansson L, Holmdahl R. Oestrogen induced suppression of collagen arthritis. IV: progesterone alone does not affect the course of arthritis but enhances the oestrogen-mediated therapeutic effect. J Reprod Immunol. 1989;15:141–50. doi: 10.1016/0165-0378(89)90033-8. [DOI] [PubMed] [Google Scholar]
- 32.Yamasaki D, Enokida M, Okano T, Hagino H, Teshima R. Effects of ovariectomy and estrogen replacement therapy on arthritis and bone mineral density in rats with collagen-induced arthritis. Bone. 2001;28:634–40. doi: 10.1016/s8756-3282(01)00426-4. [DOI] [PubMed] [Google Scholar]
- 33.Jansson L, Mattsson A, Mattsson R, Holmdahl R. Estrogen induced suppression of collagen arthritis. V: physiological level of estrogen in DBA/1 mice is therapeutic on established arthritis, suppresses anti-type II collagen T-cell dependent immunity and stimulates polyclonal B-cell activity. J Autoimmun. 1990;3:257–70. doi: 10.1016/0896-8411(90)90145-i. [DOI] [PubMed] [Google Scholar]
- 34.Nuttall ME, Fisher PW, Suva LJ, Gowen M. The selective oestrogen receptor modulators idoxifene and raloxifene have fundamentally different cell-specific oestrogen-response element (ERE)-dependent/independent mechanisms in vitro. Eur J Cancer. 2000;36(Suppl 4):S63–4. doi: 10.1016/s0959-8049(00)00230-6. [DOI] [PubMed] [Google Scholar]
- 35.Yang NN, Venugopalan M, Hardikar S, Glasebrook A. Identification of an estrogen response element activated by metabolites of 17beta-estradiol and raloxifene. Science. 1996;273:1222–5. doi: 10.1126/science.273.5279.1222. [DOI] [PubMed] [Google Scholar]
- 36.Paech K, Webb P, Kuiper GG, et al. Differential ligand activation of estrogen receptors ERalpha and ERbeta at AP1 sites. Science. 1997;277:1508–10. doi: 10.1126/science.277.5331.1508. [DOI] [PubMed] [Google Scholar]
- 37.Weisz A, Rosales R. Identification of an estrogen response element upstream of the human c-fos gene that binds the estrogen receptor and the AP-1 transcription factor. Nucleic Acids Res. 1990;18:5097–106. doi: 10.1093/nar/18.17.5097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blanque R, Cottereaux C, Gardner CR. Increases in osteocalcin after ovariectomy are amplified by LPS injection: strain differences in bone remodelling. Gen Pharmacol. 1998;30:51–6. doi: 10.1016/s0306-3623(97)00067-0. [DOI] [PubMed] [Google Scholar]
- 39.Gao Y, Grassi F, Ryan MR, et al. IFN-gamma stimulates osteoclast formation and bone loss in vivo via antigen-driven T cell activation. J Clin Invest. 2007;117:122–32. doi: 10.1172/JCI30074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Amirahmadi SF, Pho MH, Gray RE, et al. An arthritogenic monoclonal antibody to type II collagen, CII-C1, impairs cartilage formation by cultured chondrocytes. Immunol Cell Biol. 2004;82:427–34. doi: 10.1111/j.0818-9641.2004.01267.x. [DOI] [PubMed] [Google Scholar]
- 41.Crombie DE, Turer M, Zuasti BB, et al. Destructive effects of murine arthritogenic antibodies to type II collagen on cartilage explants in vitro. Arthritis Res Ther. 2005;7:R927–37. doi: 10.1186/ar1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Amirahmadi SF, Whittingham S, Crombie DE, et al. Arthritogenic anti-type II collagen antibodies are pathogenic for cartilage-derived chondrocytes independent of inflammatory cells. Arthritis Rheum. 2005;52:1897–906. doi: 10.1002/art.21097. [DOI] [PubMed] [Google Scholar]
- 43.Nandakumar KS, Bajtner E, Hill L, et al. Arthritogenic antibodies specific for a major type II collagen triple-helical epitope bind and destabilize cartilage independent of inflammation. Arthritis Rheum. 2008;58:184–96. doi: 10.1002/art.23049. [DOI] [PubMed] [Google Scholar]