

Abstract
Currently, the genome sequencing community is producing shotgun sequence data at a very high rate, but finishing (collecting additional directed sequence data to close gaps and improve the quality of the data) is not matching that rate. One reason for the difference is that shotgun sequencing is highly automated but finishing is not: Most finishing decisions, such as which directed reads to obtain and which specialized sequencing techniques to use, are made by people. If finishing rates are to increase to match shotgun sequencing rates, most finishing decisions also must be automated. The Autofinish computer program (which is part of the Consed computer software package) does this by automatically choosing finishing reads. Autofinish is able to suggest most finishing reads required for completion of each sequencing project, greatly reducing the amount of human attention needed. Autofinish sometimes completely finishes the project, with no human decisions required. It cannot solve the most complex problems, so we recommend that Autofinish be allowed to suggest reads for the first three rounds of finishing, and if the project still is not finished completely, a human finisher complete the work. We compared this Autofinish-Hybrid method of finishing against a human finisher in five different projects with a variety of shotgun depths by finishing each project twice—once with each method. This comparison shows that the Autofinish-Hybrid method saves many hours over a human finisher alone, while using roughly the same number and type of reads and closing gaps at roughly the same rate. Autofinish currently is in production use at several large sequencing centers. It is designed to be adaptable to the finishing strategy of the lab—it can finish using some or all of the following: resequencing reads, reverses, custom primer walks on either subclone templates or whole clone templates, PCR, or minilibraries. Autofinish has been used for finishing cDNA, genomic clones, and whole bacterial genomes (see http://www.phrap.org).
When the shotgun phase of a sequencing project has been completed, the assembly typically consists of a number of contigs (groups of overlapping reads). The contigs are separated by gaps, for which the sequence is unknown. Within each contig, there are regions of high error rate in which the correct sequence is uncertain. The goal of the finishing phase is then to get a single contiguous contig with low error rate. Prior to the availability of consensus error probabilities, finishers invented various ad hoc rules (such as requiring that each consensus base agree with at least one read on each strand) to determine whether additional data were needed. With the availability of consensus error probabilities, there is an objective way to decide when a finisher is done: He or she decides what error rate is acceptable and acquires additional data until that error rate is achieved. Because the error probabilities do not take into account the possibility of subclone rearrangements, a reasonable additional requirement is that there should be no single subclone regions (regions supported by aligned reads from only one template).
Autofinish is designed to achieve these objectives, suggesting reads to close gaps (using a variety of methods), improve sequence quality in regions of high error rate, and eliminate any single subclone regions. The user can configure Autofinish to perform all or any combination of these tasks.
Autofinish will suggest the following types of experiments: Universal primer reads (reads primed from the subclone vector at one side of the insert will be referred to as “forward universal primer reads” and those primed from the vector at the other side of the insert “reverse universal primer reads”); custom primer reads with subclone templates (e.g., plasmid or M13); custom primer reads with a whole clone template [e.g., bacterial artificial chromosome (BAC), cosmid, or bacterial genome]; minilibraries (shotgun libraries prepared from a subclone insert) (McMurray et al. 1998); PCR to close gaps. The user can configure Autofinish to suggest any combination of these types of reads. For example, by default custom primer reads with a whole clone template are turned off because most labs do not wish to create such reads.
Autofinish has been used in production since mid-1998. Labs that currently use Autofinish include the Genome Center at the University of Washington (Seattle), the Genome Sequencing Center at Washington University (St. Louis), the Department of Energy Joint Genome Microbial Project, the Berkeley Drosophila Genome Project at Lawrence Berkeley National Laboratory, the Lita Annenberg Hazen Genome Center at Cold Spring Harbor Laboratory, and other private and public labs.
RESULTS
Autofinish is sometimes unable to completely finish a project. Thus it typically is used immediately after shotgun and then, if there are still remaining problems, a human finisher takes over (what we refer to as Autofinish-Hybrid finishing). Table 1 shows a list of BACs, along with one complete bacterial genome, (Pseudomonas aeruginosa) that were finished at the University of Washington Genome Center from 1998–1999 using Autofinish followed by a human finisher. Notice that Autofinish suggests a substantial fraction, ranging from 60% to 100%, of the finishing reads. For example, the finisher who completed PA01, the 6Mb bacterial genome, was spared having to pick all but 342 of the 1637 finishing reads. For five of the BACs, Autofinish completely finished the projects with no human decisions. For more recently finished BACs (Table 2) Autofinish suggested all finishing reads for nearly all of the projects, reflecting improvements in Autofinish such as the ability to specify PCR reactions and minilibraries for closing gaps, the ability to choose redundant reads in case some fail, and improved positioning of gap-closing reads. The present paper includes both tables, to show that Autofinish– unlike Athena–did not spring into the world fully armed and developed, but benefited from years of production use.
Table 1.
Use of Autofinish on University of Washington Projects during 1998 and 1999
| Project | No. of finishing reads | No. of Autofinish reads (% Autofinish reads) |
|---|---|---|
| djs201 | 267 | 161 (60%) |
| rg391h05 | 390 | 236 (61%) |
| djs104 | 140 | 90 (64%) |
| djs259 | 148 | 103 (70%) |
| PAO1 | 1637 | 1195 (73%) |
| djs333 | 141 | 112 (79%) |
| djs119 | 82 | 66 (80%) |
| djs228 | 128 | 104 (81%) |
| djs362 | 99 | 82 (83%) |
| djs146 | 141 | 121 (86%) |
| djs94 | 127 | 111 (87%) |
| djs327 | 173 | 156 (90%) |
| djs77 | 113 | 107 (95%) |
| djs306 | 192 | 183 (95%) |
| djs124 | 149 | 145 (97%) |
| djs301 | 224 | 219 (98%) |
| djs14 | 91 | 91 (100%) |
| djs40 | 11 | 11 (100%) |
| djs45 | 66 | 66 (100%) |
| djs58 | 72 | 72 (100%) |
| djs292 | 86 | 86 (100%) |
Autofinish (versions 5.0 to 9.0) was used on these projects at the University of Washington during 1998 and 1999. All are 150-kb to 200-kb BACs except for PA01, the six-megabase Pseudomonas aeruginosa bacterial genome. Immediately after shotgun, Autofinish was run for three rounds on each project: Each time, the lab made the reads suggested by Autofinish (without adding or removing any suggested reads) and reassembled the project. After this, if the project was still not finished, a human finisher manually completed the project. All of these projects have been submitted to GenBank as Phase 3 Complete sequence. (No. of finishing reads) The sum of Autofinish-selected reads and human-selected reads. (No. of Autofinish reads [% Autofinish Reads] The number and percentage of finishing reads that were suggested by Autofinish.
Table 2.
Use of Autofinish on University of Washington Projects During 2000
| Project | No. of finishing reads | No. of Autofinish reads (% Autofinish reads) |
|---|---|---|
| djs13 | 237 | 237 (100%) |
| djs267 | 267 | 267 (100%) |
| djs510 | 350 | 326 (93%) |
| djs696 | 294 | 294 (100%) |
| djs702 | 206 | 206 (100%) |
| djs712 | 265 | 265 (100%) |
| djs701 | 234 | 234 (100%) |
| djs718 | 381 | 381 (100%) |
| djs713 | 170 | 170 (100%) |
| djs714 | 243 | 243 (100%) |
| djs715 | 428 | 428 (100%) |
| djs721 | 76 | 76 (100%) |
| djs513 | 128 | 128 (100%) |
| djs704 | 500 | 500 (100%) |
| djs709 | 421 | 421 (100%) |
BACs that the University of Washington started and completed finishing between August and December 2000, using Autofinish version 10.0 and better. Seven additional BACs, not listed, were started but not completed during this period.
One might, however, question these results: Were these projects particularly easy to finish? In the cases in which Autofinish did not completely finish the project and a human finisher needed to suggest reads, did Autofinish help very much or were most of its suggested reads superfluous, and the reads suggested by the human finisher the reads that really helped? Did Autofinish suggest an excessive number of reads to finish the project? Could a human finisher have completed the project much more cheaply?
To address these questions we held a competition between Autofinish-Hybrid finishing and Human-only finishing. The human finisher was C.D., who had over two years experience in finishing. We selected five BACs for this competition, each with a different degree of shotgun coverage (Table 3). For each BAC, the finisher started with an assembly of just the shotgun reads, and finished this Human-only project without Autofinish. Simultaneously, the finisher started with an identical but separate assembly of just the shotgun reads and did three rounds of Autofinish finishing (Fig. 1). If the Autofinish copy of the project wasn't completely finished at this point, she finished the project manually using a combination of resequencing, subclone and BAC template walking, PCR product sequencing, and minilibraries, just as she did with the Human-only copy of the project.
Table 3.
Shotgun Statistics of the Five BACs Selected for the Autofinish-Hybrid versus Human-Only Competition
| Project | GenBank accession no. | Q20 coverage | Size in kB | #Shotgun reads | # Contigs after shotgun phase | Avg. # Q20 bases per read |
|---|---|---|---|---|---|---|
| BAC218 | AC009362 | 4× | 155 | 1934 | 24 | 325 |
| BAC383 | AC010656 | 5× | 139 | 1979 | 12 | 353 |
| BAC270 | AC009517 | 7× | 141 | 2766 | 7 | 359 |
| BAC366 | AC009358 | 11× | 185 | 5455 | 5 | 377 |
| BAC422 | AC009332 | 14× | 148 | 5379 | 8 | 380 |
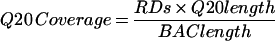 where RDs is the number of shotgun reads, Q20length is the average number of bases in a read which are PHRED quality 20 or greater, and BAClength is the size of the BAC in base pairs.
where RDs is the number of shotgun reads, Q20length is the average number of bases in a read which are PHRED quality 20 or greater, and BAClength is the size of the BAC in base pairs.
Figure 1.

Finishing procedures with Autofinish and with a human finisher.
Figure 2 compares how quickly the two methods close gaps. Rounds 1–3 in each panel are particularly relevant because they represent a race between the finisher and Autofinish (without any human help). As is evident in Figure 2, by the third round, the finisher and Autofinish have closed the same number of gaps in the 7x and 14x projects, Autofinish is ahead by one gap in the 5x project, and is behind by one gap in the 4x and 11x projects. This shows that Autofinish is effective at closing gaps. The reason Autofinish-Hybrid took an extra round to close the final gap in the 11x project was the laboratory failure of a single gap-closing Autofinish read. The finisher had in fact independently chosen precisely the same read to close the corresponding gap in the Human-only version of the project, but in that case the read was successful and closed the gap.
Figure 2.





Gap Closing: Autofinish-Hybrid vs. Human-only for five different BACs.
The Autofinish-Hybrid method required the same number of rounds to close all gaps as the Human-only method, except in two cases: the 5x project where the Autofinish-Hybrid method took two fewer rounds, and the 11x project where it took one more round. In the 14x project, Phrap (P. Green, unpubl.) assembled the Human-only version into a single contig after round 1, while the Autofinish-Hybrid version initially consisted of two contigs as a result of a simple misassembly, not a gap. This misassembly was resolved by Phrap after round 2 Autofinish reads were added.
For the 4x project, notice that in round 4 the Autofinish-Hybrid method pulls ahead of the Human-only method. How can this occur when round 4 is human versus human, because the finisher has taken over after the three rounds of Autofinish? The reason is that Autofinish has reduced the size of a gap more than the finisher did in the Human-only project, and thus it is easier for the finisher in round 4 to close gaps in the Autofinish project.
Also notice that the 4x project was initially in 24 contigs, which is a higher number than what most centers would consider desirable to finish. Autofinish and the finisher each closed half of those gaps after a single round, indicating that Autofinish's gap-closing capabilities match those of a human finisher even for projects where shotgun coverage is very low. At this writing, the 4x project still is not completed. Both the Autofinish-Hybrid and Human-only projects have the same two stubborn gaps. However, finishing them would have no bearing on this competition because a human finisher has taken over.
But how many reads is Autofinish using? Could Autofinish be competing so successfully with the human finisher by suggesting many more reads? No. As is evident from Figure 3, the number of reads used by the Autofinish-hybrid method of finishing and the Human-only method are very similar. Nor is Autofinish using more expensive reads. Figure 4 shows that the Autofinish-Hybrid method used slightly fewer PCR reads overall than the Human-only method. Figure 5 shows that the number of custom primers required was slightly larger for the Autofinish-Hybrid method. As discussed below, a more recent version of Autofinish (10.0) uses fewer custom primers.
Figure 3.

Finishing reads required: Autofinish-Hybrid vs. Human-only.
Figure 4.

PCR reads required: Autofinish-Hybrid vs. Human-only.
Figure 5.

Custom primers required: Autofinish-Hybrid vs. Human-only.
After finishing the Autofinish-Hybrid and Human-only versions of a BAC, we compared the consensus sequences and examined discrepancies to identify the number of errors in each version, as shown in Table 4. Although all projects attained the target error rate of one error per 10,000 bases or better, the Human-only consensus was more accurate than the Autofinish-Hybrid consensus in three out of four projects, indicating that the human finisher “overshot” the target error rate by a greater amount. This indicates that for a comparable number of finishing reads, the human finisher was able to attain a somewhat lower error rate (albeit lower than the competition specified). By performing additional Autofinish runs and examining the location of suggested reads, we attempted to estimate how many additional reads Autofinish would have had to call to attain the same final error rate as the human finisher. For the four projects combined, we estimate an additional 64 reads would have been required, for a total of 608 reads, about 12% more than in the Human-only projects. Because of improvements in Autofinish and Phred (Ewing et al. 1998) since this competition, we believe Autofinish now would use fewer reads to reach the same level of accuracy (see Discussion).
Table 4.
Predicted and Observed Errors in the Completed Autofinish-Hybrid and Human-Only Projects
| Project | Autofinish-Hybrid | Human-Only | ||
|---|---|---|---|---|
| predicted errors (errors/10kb) | observed errors (errors/10kb) | predicted errors (errors/10kb) | observed errors (errors/10kb) | |
| BAC383 (5×) | 8.7 (0.68) | 12 (0.93) | 5.9 (0.46) | 2 (0.16) |
| BAC270 (7×) | 5.7 (0.41) | 12 (0.85) | 4.9 (0.35) | 15 (1.07) |
| BAC366 (11×) | 8.9 (0.48) | 19 (1.03) | 2.2 (0.12) | 3 (0.16) |
| BAC422 (14×) | 1.5 (0.10) | 3 (0.20) | 1.0 (0.07) | 1 (0.07) |
| Total | 24.8 (0.41) | 46 (0.76) | 14.0 (0.23) | 21 (0.35) |
| Observed errors/predicted errors | 1.85 | 1.5 | ||
| Total predicted errors | 38.8 | |||
| Total observed errors | 67 | |||
| Observed errors/Predicted errors | 1.72 | |||
The true number of errors is probably higher than the observed counts, because some errors may be the same in both versions and would not be detected by the comparison. For this analysis we used the more recent PHRED version 0.000925.c because it gives more accurate quality values for MegaBACE data than the older versions of PHRED available during the competition. Parts of the BACs containing misassembled or misplaced reads were excluded.
It also is apparent from Table 4 that there are ∼70% more errors than predicted. We believe this excess is partly because of errors introduced by PCR amplification of the subclones, which are not reflected in the Phred quality values, and partly to a slight overestimation of the Phred quality values on MegaBACE data.
The main difference between the two methods is shown in Figure 6: The Human-only method required two- to six-fold as much time picking reads as the Autofinish-Hybrid method. There can be a large difference in the length of time it takes different finishers to finish a project. If we had used a less-experienced finisher than C.D., this figure likely would have shown an even more dramatic difference.
Figure 6.

Human hours required for choosing reads: Autofinish-Hybrid vs. Human-only.
Autofinish's ability to discern which contigs in an assembly to finish is an important and useful feature, because most assemblies contain small “junk” contigs that normally would be ignored by a human finisher. In our experiment, usually Autofinish and the human finisher agreed on which contigs were worthy of additional finishing and which should not be pursued further. Every contig that Autofinish chose to finish eventually integrated into the final assembly.
DISCUSSION
The competition described above shows the Autofinish-Hybrid method saves a finisher a substantial amount of time sitting in front of a computer picking reads, which should enable finishing at a greater rate. Autofinish also relieves finishers of much of the routine, tedious work and allows them to concentrate their time on the most interesting and challenging problems.
According to the traditional finishing model, when a clone has completed the shotgun stage the clone is assigned to a particular person to finish. That person could choose finishing reads manually, or use Autofinish. The competition above shows the advantages of using Autofinish for the first three rounds. However, a lab may gain efficiency by not assigning the clone to a finisher until after Autofinish has been run. Instead, all clones that have completed the shotgun stage can be assigned to a production group having the skills to do the reactions suggested by Autofinish, but not necessarily the skills to choose finishing reads or edit the assembly. This group has no need to even view the assembly because Autofinish can be run automatically; they would instead focus on processing multiple finishing reactions at high throughput, just as the shotgun production group does. This is the model used by the University of Washington Genome Center where it has yielded an increase in finishing rate (R. Kaul, pers. comm.) We believe this model is more scaleable than the Human-only finishing model. Each time more people are hired, there are training costs, the new hires are initially not as productive as existing employees, and the management burden increases in degree and complexity. Increasing Autofinish's workload does not create these problems, and may even have decreasing marginal cost.
Autofinish does not yet completely automate the finishing process: There is a need before submission for a human finisher to correct any misassemblies and misplaced reads. Consed (Gordon et al. 1998) has the tools to detect these by locating high-quality discrepant bases and reads with unaligned, high-quality regions, and to correct them.
The competition shows that the Autofinish-Hybrid method requires roughly the same number and type of reads and closes gaps at roughly the same rate as a human finisher alone. Because laboratory costs depend on number and type of reads, these results show that Autofinish probably does not increase lab costs. The project with the greatest difference between the two methods in number of reads is the 7x coverage project, BAC270, in which the Autofinish-Hybrid method used 155 reads to the Human-only method's 198 reads. We found that there were very few reads in common and thus we could not identify any reads as being “extra”. Instead there were three sources of this discrepancy: (1) The Human-only method had 22 more laboratory failures that had to be redone than the Autofinish-Hybrid method; (2) the Human-only method suggested a larger portion of universal primers than the Autofinish-Hybrid method did, and this also may have increased the number of reads required; and (3) there were seven cases in which the human finisher suggested reads that would not fix more than the 0.05 minimum number of errors threshold that the finisher set for Autofinish and thus do not help much in reducing the error rate. There also were four regions that the human finisher missed entirely that exceeded this threshold; Autofinish correctly suggested reads for these regions.
In the case of the 14x project, Autofinish suggested 14 more reads, or 28% more reads than the human finisher. Here, the Autofinish-Hybrid project required redoing 10 more reads than the Human-only method, which accounts for much of the difference.
In the four finished BACs, the human finisher attained higher accuracy with the identical number of reads; we estimate that Autofinish would have required 12% more reads to reach the same level of accuracy. However, improvements have been made since the competition was held that should reduce Autofinish's number of reads. These include fixing a bug that occasionally caused it to resuggest a read that failed in a previous round, identifying particular problems (e.g., stops and low complexity regions) that require alternate chemistry, and a newer version of Phred (0.000925.c) which, for MegaBACE data, gives more accurate (and lower) quality values in locations that likely contain errors.
Different labs and even different finishers within a lab have different finishing styles. In particular, Washington University Genome Sequencing Center (Wash-U) finishers routinely choose more reads than C.D. for each problem region. A comparison of results from Autofinish version 9.66 (the version used in this competition) with their finishers' results would likely result in the Autofinish-Hybrid method finishing the project with far fewer total reads, although possibly more rounds and more custom primer reads. Wash-U requested that Autofinish suggest redundant universal primer reads, in the event that some reads failed. The Baylor Human Genome Sequencing Center requested that Autofinish suggest all reverses that might help in closing gaps. Many users requested that Autofinish prefer multiple universal primers to a single custom primer because the universal primers are cheaper and this method would save calendar time waiting for primers. These changes all have been incorporated in Autofinish version 10.0, which by default calls more universal primer reads than version 9.66. The user can make Autofinish 10.0 suggest reads in the parsimonious manner of Autofinish 9.66 by setting the parameters appropriately.
For this experiment, we chose projects with a wide range of shotgun depths of coverage to see whether Autofinish's effectiveness is restricted to a particular shotgun depth. Anecdotally, we observed that the 5x project had 787 fewer shotgun reads than the 7x project, but required only 111 more finishing reads for the hybrid method and 43 more using the Human-only method. This suggests that using a lower shotgun depth may be more efficient. (Albeit these are different projects.) So what is the optimal shotgun coverage? This question could be addressed by the following experiment: Several high-depth-of-coverage (e.g., 14x) shotgun projects could have reads randomly removed, to create projects of successively lower depths of coverage. Then each of these projects could be finished independently, including making the reads in the lab, and one could compare the total (shotgun + finishing) cost of sequencing each of these projects. Because Autofinish decreases the cost of finishing reads by saving human labor, Autofinish should lower the optimal shotgun coverage.
In our experience, when a lab is considering the use of Autofinish, one of its skilled finishers looks at the Autofinish output and sometimes disagrees with some of the suggestions and dismisses Autofinish. We think that the above-described controlled competition is a better way of evaluating Autofinish, and it indicates that Autofinish is roughly as good as a human finisher for the first three rounds of finishing after shotgun.
METHODS
Competition Between Autofinish-Hybrid and Human-Only
C.D. did the lab work for both her manually chosen reads and for the Autofinish reads using the same techniques and equipment, to attempt to eliminate any potential laboratory bias.
Template DNA for shotgun reads was prepared mainly by PCR although a small number were prepared using an alkaline lysis method. Finishing reads template DNA consisted of subclones prepared with a Qiagen prep robot, or BAC DNA prepared using an alkaline lysis method. ABI dye terminator chemistry and an ABI 3700 capillary sequencer were used for all finishing reads, and Amersham dye terminator chemistry and a MegaBACE capillary sequencer were used for most shotgun reads (with a small number run on an ABI 373 or 377). Bases were called with Phred version 0.980904.a for shotgun reads and version 0.990722.g for finishing reads.
If a read failed (meaning that when viewed by the eye, the chromatogram was blank or had fewer than 100 discernable bases), the finisher would attempt a second reaction from the same DNA template stock and would count both reads. We counted all samples run, whether successes or failures, with one exception: There were three 96-well plates that failed to produce data because of a clogged pipette tip on an overnight run on the 3700. The finisher simply restarted the sequencer the next day after unclogging the tip and reran the three plates without redoing the sequencing reactions. We did not count the failed batch in the total number of reads because such a machine failure has no bearing on how Autofinish compares to a human finisher.
During the first three rounds of manual finishing for each project, the finisher only picked resequencing reads (reads redone using dye terminator chemistry) and custom primer walks on subclone or clone (BAC) templates. The finisher did not use PCR or minilibraries until after the first three rounds. (In retrospect, she thinks it would have been more efficient to use some PCR in the third round in areas of low subclone coverage, but she thinks this would have made little difference in the competition results because she eventually did the PCR in a subsequent round.) The finisher did not view Autofinish's read choices prior to manually selecting reads, in order to prevent Autofinish from influencing her choices. She measured the time spent sitting at the computer choosing reads and preparing primer orders.
During the first three rounds using Autofinish on the Autofinish-Hybrid projects, the finisher made all reads Autofinish suggested. There was one partial exception: She initially ran Autofinish on a very low-coverage project (BAC218) with Autofinish's redundancy parameter (a parameter that determines how few or many reads Autofinish suggests) set high, but after seeing the huge number of primers suggested, she decided that redundancy 1 was more appropriate for the initial stages of a project shotgunned to such a low depth of coverage. She reran Autofinish with redundancy 1 and used all of Autofinish's suggested reads without change.
The finisher did not use data from overlapping BACs, GenBank, or any other outside sources in either the Autofinish-Hybrid method or in the Human-only method. To do so would be to effectively decrease the size of the region to be finished but otherwise be irrelevant to the competition.
How to Use Autofinish
To use Autofinish, begin by running the phredPhrap script that, among other things, runs Phred and then Phrap. Autofinish requires precisely the same input files that Consed needs: the ace file (output from Phrap) and the PHD files (output from Phred).
To start Autofinish, from the command line type: consed -ace (ace filename) -autofinish
(This is the same way one starts Consed, except for the “-autofinish”.)
Autofinish will create the following files:
A file of forward universal primer reads to make
A file of reverse universal primer reads to make
A file of custom primer reads to make
A custom navigation file that allows one to use Consed to easily and quickly examine each read suggested by Autofinish by successively clicking “Next”
A combination of files 1, 2, and 3 sorted by contig and contig position so one can see at a glance what Autofinish has suggested for a particular region of the assembly
A verbose “.out” file in which Autofinish explains its reasons for the decisions it makes
A file of minilibraries to make
autofinish.fof which contains the names of the files listed above
Files 1, 2, 3, and 7 (above) are in a delimited text file format, making it easy to integrate Autofinish into existing work-flow setups incorporating robotics or LIMS systems.
Customizing Autofinish
The user can customize Autofinish by creating a file named .consedrc containing some parameters and their customized values. A complete list of the customizable parameters is in the beginning of the Autofinish “.out” file. This list is quite long and the user must understand a parameter in order to correctly modify it, as this is the price of Autofinish's flexibility.
How Autofinish Works
Autofinish has the following phases:
Evaluates the success of previous rounds of Autofinish on this project
Evaluates the error rate of all the existing reads in the project and creates “model reads”, which include estimates of the length and position-specific quality values of each finishing read
Determines the location of subclone templates that are acceptable for creating additional reads
Decides which contigs to try to finish
- For each contig:
- checks if either end of the contig is the clone end
- orders and orients the contig with respect to other contigs
- suggests minilibraries for closing gaps
- suggests universal primer reads for closing gaps and improving the error rate in regions of high error rate
- chooses redundant universal primer reads
- chooses custom primer walking reads for any region left unfixed by universal primer reads
- suggests custom primer reads to eliminate single subclone regions
Suggests PCR for closing gaps
Prints output files and, if Autofinish was run with the option -doExperiments, adds tags to the ace file recording which reads and primers Autofinish suggested
Let us examine these phases in detail:
Evaluates the Success of Previous Runs of Autofinish on This Project
If Autofinish was previously run with the -doExperiments parameter, as in consed -ace (acefilename) -autofinish -doExperiments, Autofinish will have a record in the ace file of reads it suggested for this project in the past. For informational purposes only, Autofinish reports, for each suggested read, whether there now exists a corresponding actual read and how well the actual read achieved its objective—whether the read is aligned where Autofinish intended and how well the read improved the predicted error rate of the region.
Creates Model Finishing Reads
Autofinish assumes that any read it suggests (except for fixing single subclone regions) will have a particular length and its bases particular qualities. Autofinish creates this imaginary “model read” by examining all existing reads in the project and compiling a histogram indicating at each base position what fraction of reads have quality 0, what fraction of reads have quality 1 or less, what fraction of reads have quality 2 or less, etc. Phred and Phrap quality values are defined by
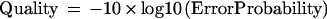 |
Because Phred quality values are not valid in the unaligned part of a read, only bases that align against the consensus or vector are counted in this histogram.
Autofinish then constructs a model read whose quality value at a particular base position is the median quality value of the histogram at that base position. The length of the model read is the 90th percentile length (customizable) of existing reads.
Autofinish examines all existing reads and calculates the probability that a read is alignable against the consensus at any particular base position in the read. Autofinish finds the read positions at which 90% or more (customizable) of existing reads are aligned against the consensus and constructs a second model read, which is aligned at these positions. This model read (which does not utilize quality information) is used for fixing single subclone regions.
The user can view in Consed these model reads for a particular project by clicking on Info/Show Model Read on the Main Consed Window.
Determines the Location of Subclone Templates that are Acceptable to Use for Additional Reads
Autofinish does not use a template if it has any of the following properties: the template has a chimeric read detected by Phrap; the template is listed in the badTemplates.txt file, a file created by the lab specifically to exclude particular templates from finishing; the library of the template is listed in the badLibraries.txt file, a file created by the lab specifically to exclude particular libraries from finishing, e.g., libraries used in the shotgun that are unavailable for finishing; the template's insert is shorter than 300 bases (customizable); the existing reads from the template have any indication of being misassembled. In such a case, Autofinish cannot be sure of the location of the template. Indications of a read being misassembled include high-quality discrepancies with the consensus, high-quality regions that are unaligned with the consensus, and inconsistencies between existing reads made from the template. Such inconsistencies may take the following forms: (1) The reads are further apart than the maximum subclone size; (2) The reads are in different contigs and not both close enough to the ends of the contigs to be consistent with the maximum subclone size even if the contigs were joined; (3) The reads are in different contigs, there are 0 (customizable) other forward-reverse pairs between the same two contig ends, and these reads are inconsistent with the contig order and orientation as deduced from a majority of linking forward-reverse pairs; and (4) The reads are in the same contig, but the orientations of forward/reverse pair universal primer reads are inconsistent, e.g., they are pointing away from each other (← →) or in the same direction (→ →) instead of towards each other (→ ←).
It is vital that Autofinish determine the location of each template's insert with respect to a contig in the assembly, both for choosing which templates are available for making a particular read with a particular custom primer and also for determining the location of each “de novo universal primer read” (a forward universal primer read in the case in which the template has an existing reverse but no forward, and a reverse universal primer read in the case in which the template has an existing forward but no reverse).
To find the location of the insert of a particular template, Autofinish examines each existing universal primer read from that template and finds the vector/insert junction. Ideally, the template would have an existing forward universal primer read and a reverse universal primer read, which together would determine the precise location of the insert of the template. Frequently, however, there is only a single universal primer read from a subclone template, so Autofinish can determine precisely one end of the template insert, but must guess at the other end. It makes this guess in two ways: (1) If there are at least 100 (customizable) existing forward/reverse pairs from the same library as this subclone template, Autofinish calculates the mean and standard deviation of the insert sizes of templates of these forward/reverse pairs, and guesses that the insert size of this particular template will be the mean minus 0.2 standard deviations (customizable). (2) If there are not enough forward/reverse pairs, then Autofinish will assume the insert size is 1500 bases (customizable). If there are several subclone libraries in the same project, Autofinish can make these estimates in a library-specific manner. Autofinish gets the library name and subclone template name of a read from the PHD file. Similarly, Autofinish reads from the PHD file whether a read is a universal primer read (and thus delineates the end of a subclone template) or a walk. It is the lab's responsibility to put this information into the PHD file by customizing the perl script determineReadTypes.perl. Typically, labs have the library name, template name, and template type as part of the read name, but different labs have different naming conventions. Hence the need for this script.
Decides Which Contigs to Try to Finish
Autofinish tries to distinguish real contigs from spurious contigs (such as those deriving from contaminant reads), and does not finish the latter. If a contig has <10 reads in it (customizable) or is shorter than 1 kb (customizable), Autofinish does not try to finish it. If any contig has a depth of coverage (sum of aligned read lengths divided by the consensus length) exceeding six times (customizable) the depth of coverage of the longest contig, Autofinish does not finish the shorter contig, because it is likely to be contaminant or misassembled.
Checks if Either End of the Contig is the Clone End
Autofinish checks each contig end to see if it contains the clone end. The user can specify that Autofinish do this in either of two ways: one applying when the clone vector is included in the shotgun (see Fig. 7), and the other when there are two reads that are known to lie at the ends of the clone. The latter case has been used for finishing cDNA clones.
Figure 7.

Autofinish checks each contig to see if either end is the clone end. Masked clone vector and subclone vector both appear as Xs, and Bs are high quality bases that do not match subclone or clone vector. Notice that vector-insert junctions of reads 1–4 are aligned. If read 5 were not present, this figure would suggest a typical clone end with the Xs clone vector. If only read 1 and read 2 were present, the Xs could instead be subclone vector, which just happens to align; but the presence of two additional reads (read 3 and read 4), or additional vector bases in one read, would make this less likely. However, if read 5 were present (all high quality bases surrounding the putative vector/insert junction), it would be unlikely that this is the clone end.
Orders and Orients the Contig
Autofinish tries to order and orient contigs by using forward/reverse pair information. If it finds two (customizable) forward/reverse pairs linking the same two ends of contigs, then Autofinish considers the orientation to be certain and prints it to the “.out” file. If there are not two forward/reverse pairs linking the end of one contig to an end of another contig, then Autofinish will try to suggest additional reads so there will be at least two such pairs.
Optionally, Autofinish will suggest all reverse universal primer reads that have a chance of either extending the contig or closing the gap; these must be within the maximum insert size of the library from the contig end.
Suggests Minilibraries For Closing Gaps
If a subclone spans a gap, Autofinish can suggest a minilibrary be made from that subclone in order to close the gap. Autofinish can optionally suggest minilibraries for all such gaps, or just for such gaps that are predicted to be larger than a user-settable size. Autofinish estimates the size of the gap and the insert size of each template spanning the gap by assuming that the mean of the insert sizes of the templates that span the gap equals the mean insert size of the library of these templates. When choosing a subclone for the minilibrary, Autofinish will prefer subclone templates whose size is within two (customizable) standard deviations of the mean size of subclones in the library over templates that are further from the mean. Within each of these two groups, Autofinish will prefer templates that will fix more errors, i.e., which cover more of the low quality bases near the ends of the contigs.
Suggests Universal Primer Reads for Closing Gaps and Improving the Error Rate in Regions of High Error Rate
This and the subsequent steps are the heart of Autofinish, in which it chooses reads based on error probabilities.
Autofinish calculates the Phrap error probability of each consensus base from the Phrap-generated consensus quality values using the formula:
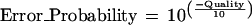 |
An error probability of .01 means that out of 100 different bases each having an error probability of .01, roughly 99 of them will be correct and 1 will be incorrect, so the “expected” (or “predicted”) number of errors of each base is .01. The expected number of errors in a region, E, is the sum of the error probabilities of the bases in the region. When Autofinish is considering how many errors a particular finishing read will fix, it uses this method to estimate the expected number of errors in the region of the consensus covered by the read. Because a finishing read is never perfect, it will on average fix fewer errors than the expected number in the consensus. We explain below how we estimate the number of errors a finishing read will fix.
Consensus error probabilities are stored in the elements of an array, with one element of the array for each base in the consensus. Autofinish extends this array to the left and to the right with elements representing the gap bases between contigs, and assigns error probability 1.0 to these bases since they are unknown. If either contig end is a clone end, Autofinish assigns 0.0 instead of 1.0 to those gap bases so it will not try to extend the contig in that direction. Similarly, if any base has a doNotFinish tag covering it, Autofinish assigns an error probability of 0.0 to that base. This is particularly useful if the clone being finished is overlapped by another finished clone and the lab does not want to finish the overlapping region again. (The user can optionally specify that Autofinish not finish regions covered by other types of tags, such as repeat tags.)
Autofinish now creates a list of possible candidate universal primer reads (both resequencing universal primer reads and de novo universal primer reads) for this contig.
Autofinish calculates the expected number of errors in the consensus fixed by each candidate read (described later). Each such read has to fix at least 0.02 expected errors (customizable) in order to be added to the list. This prevents Autofinish from suggesting reads that will not reduce the error rate significantly. Labs frequently customize this parameter because it allows them to control whether Autofinish tries to fix more or fewer minor problems: consed.AutofinishMinNumberOfErrorsFixedByAnExp: 0.02
For each read candidate, Autofinish divides the expected number of errors fixed by the (user-estimated) cost of the read to yield the number of errors fixed per dollar. Autofinish looks through this list of candidate reads and picks the single read that fixes the most errors per dollar. (If a problem can be fixed either by a resequencing read or a de novo universal primer read, the user can determine which one Autofinish will choose by varying the costs of these two types of reads.)
Autofinish will reject a read if it already had been chosen in a previous round, in order to keep from suggesting the same failing read over and over again. If a read passes this check, it is transferred to a list of chosen reads. If the chosen read crosses a “stop” (a region that dye terminator chemistry has been unable to sequence through) or low complexity region, Autofinish suggests that special chemistry (e.g., dGTP) be used. Otherwise it suggests dye terminator chemistry.
Autofinish then recalculates the error probabilities of consensus bases predicted to be overlapped by the chosen read. Autofinish scans its list of candidate reads for those that overlap the chosen read, and recalculates how many errors each such candidate read would now fix (and errors fixed per dollar), because some of the errors are expected to be fixed by the chosen read.
Autofinish repeats this process of picking the read that fixes the most errors per dollar until there are no more candidate reads left that fix at least 0.02 errors (see above).
Chooses Redundant Universal Primer Reads
Autofinish optionally suggests redundant universal primer reads so that if one fails, the problem may still be fixed by another read. To do this, Autofinish restores the consensus error probabilities to what they originally were (with all the expected errors unfixed). It then chooses universal primer reads again using the same criteria as in the first pass, but excluding the reads chosen in the first pass.
Chooses Custom Primer Walking Reads
If problems still remain after choosing universal primer reads, Autofinish tries to choose custom primer walking reads. Autofinish considers every possible custom primer that can prime within this contig and screens these primers using the same criteria as Consed's primer picker. (The number of possible primers is the same order of magnitude as the number of bases in the contig.) For each primer, Autofinish picks three templates (a customizable number) that are located such that the primer is within the template and the vector-insert junction is at least 500 bases away (customizable). If more than three templates are acceptable for a particular primer, Autofinish picks three templates based either on which are the “best quality” or which are in the “best position” (the default, but customizable).
Best quality: The quality of the template is determined by finding the read from the template that has the highest predicted error rate from base 80 to base 300 (customizable). The best quality template has the lowest such value.
Best position: Autofinish prefers templates that have a forward/reverse pair (because the beginning and end of such templates are known with relative certainty). Next best are templates that have either a forward universal primer read or a reverse universal primer read. In each case Autofinish chooses among templates based on which template extends furthest in the direction of the candidate read, where size and location of the insert are determined as described previously.
For each custom primer and its templates, Autofinish calculates the number of errors fixed and the number of errors fixed per dollar. Just as with universal primer reads, the custom primer reads must exceed the 0.02 expected error threshold to be considered.
Autofinish looks through this list of candidate reads and picks the single read (or primer and its templates) that fixes the most errors per dollar.
This read must pass these additional checks:
A previous run of Autofinish must not have suggested this primer or any other primer within 50 bases on the same strand (customizable).
Autofinish must not have already suggested a custom primer read on the same strand within 200 bases (customizable) in the same run of Autofinish.
If the potential read extends into the gap, it still must overlap the “high quality segment” of the consensus by at least 70 (customizable) bases. (The high quality segment is calculated as follows: Each base is assigned a score by subtracting the Phrap error probability from 0.05. The high quality segment is the region of contiguous bases for which the sum of the corresponding scores is a maximum. Usually this corresponds to nearly all of the consensus except at the extreme left and right ends where the quality drops below 13.) This is to prevent Autofinish from picking a read that overlaps the contig so little that the read fails to assemble with the contig, and thus does not extend it.
The custom primer pass ends precisely the same way that the universal primer passes end.
Fixes Single Subclone Regions
Autofinish determines all existing single subclone regions. Then Autofinish examines whether any existing single subclone regions will be fixed by reads that Autofinish has suggested in this round for other purposes, assuming that each suggested read will succeed, assemble where it is expected to, and be aligned as specified by the second model read (described earlier).
If there are still any single subclone regions remaining, Autofinish will try to suggest custom primer reads to fix these regions. Autofinish chooses templates for these custom primers based on which reads fix the most single subclone bases. Autofinish divides the number of single subclone bases fixed by a read by the user-estimated cost of the read and selects the read that fixes the greatest number of single subclone bases per dollar. If this custom primer read is too close (within 50 bases by default) to a custom primer read that Autofinish chose in a previous run, or too close (within 200 bases by default) to a custom primer read that Autofinish chose in this same run, Autofinish rejects this primer. Otherwise Autofinish chooses it. Autofinish then must adjust which templates cover which bases and also adjust the number of single subclone bases improved by any candidate read that overlaps the candidate read just picked. Autofinish stops choosing reads to improve single subclone bases when it can find no more reads that will fix any single subclone bases.
Suggests PCR for Closing Gaps
PCR is considered a last resort: If Autofinish can suggest a single non-PCR read to extend any contig in the assembly, Autofinish will not suggest PCR. If there is no such read, Autofinish will choose one PCR primer for the end of each contig (except for clone ends). Autofinish PCR primers must pass all of the Consed primer picking checks, although with modified settings for melting temperatures, primer size, and matchElsewhere score (a measure of the possibility of priming to the wrong location). In addition, each PCR primer must be at least 100 (customizable) bases within the high quality segment of the contig so that there is enough overlap to ensure that Phrap will assemble the resulting reads with the contig. Autofinish will choose this set of PCR primers so that they all have melting temperatures within two degrees (customizable) of each other and that no two of them anneal to each other. Autofinish will print a list of pairs of PCR primers to use.
How Many Errors Will A Read Fix?
Each suggested read is represented by a model read that has a quality histogram indicating, for each position and quality level Q, the probability that a read will have quality ≤Q at that position.
At a particular base, if Autofinish is suggesting several reads from different subclone templates but the same strand, Autofinish assumes errors in these reads are independent. Autofinish combines the reads' histograms at that base to get a histogram indicating the probability that all reads have quality ≤Q. It does this by multiplying the values at each quality level, because the probability that two reads will both have a quality value ≤Q equals the product of the probability that the first read will have quality value ≤Q times the probability that the second read will have quality value ≤Q.
At a particular base, if Autofinish is suggesting several reads from the same subclone template and the same strand, Autofinish assumes errors in these reads are completely dependent. The quality histograms of these reads at this base are combined by taking the minimum of the values at each quality level.
After combining histograms in this way, Autofinish ends up with at most two histograms at each consensus base, one for top-strand suggested reads and one for bottom-strand suggested reads. If there are any top-strand suggested reads at this position, Autofinish creates a single-top strand “combined quality” equal to the median quality at this position of this histogram (i.e., the value Q such that there is a 50% probability that the reads will have quality ≤Q). Similarly, if there are any bottom-strand reads, Autofinish creates a single bottom strand combined quality.
At each base, Autofinish applies a heuristic formula (see below) to combine the top strand combined quality with the consensus quality, and then, with the resulting value, applies the formula again with the bottom strand combined quality to obtain a new predicted consensus quality. The difference between the original consensus error probability and the error probability corresponding to the new predicted consensus quality is the predicted number of errors fixed by all suggested reads at that base position. The errors fixed by a single read is simply the difference between the errors fixed by this group of reads with and without the single read.
The formula for the number of errors fixed at a base is derived as follows:
Let r be the error probability of the combined model read base at the particular base position and let c be the error probability of the existing consensus base. Let e = MIN( r, c), f = MAX( r, c), and A and B be the base calls for the predicted more accurate of the two sequences and the predicted less accurate sequence, respectively.
Case 1: The probability, a1, that A and B agree and are correct = (1-e)(1-f) if the errors in the sequences are independent and (1-e)1 if the errors are completely dependent. It is likely that the sequencing errors will be somewhere in the continuum between independence and complete dependence. If 0 < = t < = 1 represents this continuum with complete dependence at t = 0 and independence at t = 1, then a1 is taken to be (1−e)( t(1−f) + (1−t)1), a convex combination of the independent and dependent cases.
Case 2: The probability, d1, that A is correct and B is incorrect (hence A and B disagree) is (1−e)f under independence and 0 under complete dependence, hence d1 is taken to be t(1−e)f + (1−t)0
Case 3: The probability, d2, that A is incorrect and B is correct (hence A and B disagree) is e(1−f) under independence and 0 under complete dependence hence d2 is taken to be te(1−f)
Case 4: The probability, a2, that A is incorrect and B is incorrect and they agree is ef under independence and e under complete dependence hence a2 is taken to be tef + (1−t)e. [We are assuming (conservatively) that if they are both incorrect, they always agree.]
The probability the bases agree is pa = a1 +a 2. The probability the bases disagree is pd = d1 + d2. If they agree, the probability there is an error (given that they agree) is ea = a2/(a1 + a2 ). If they disagree, and base A is chosen because it has the lowest error probability, the probability of an error (given that they disagree) is ed = d2/( d1 + d2 ). Let QA = −10*log10(ea) and QD = −10*log10(ed). Then the predicted quality of the resulting new consensus base is taken to be QC = pa*QA + pd*QD.
We experimented with values of t and chose t = 1 when there is no existing aligned read with the same chemistry and strand for which the combined quality was computed, and t = 0.8 otherwise. Table 5 shows the result of using this formula in the simple case in which a particular base is covered by a single Autofinish read. In the case in which the Autofinish read is the same strand and chemistry as an existing read, the new consensus quality is not much better than the maximum of the old consensus quality and the new read quality. However, if the new read is a different strand or chemistry than any existing read, the new consensus quality is close to the sum of the old consensus quality and the new read quality.
Table 5.
Example Values of the Errors Fixed Formula
| Old cons | New read | New unique | New redundant |
|---|---|---|---|
| 5 | 10 | 11.0 | 10.6 |
| 5 | 20 | 21.2 | 20.8 |
| 10 | 10 | 16.2 | 13.1 |
| 10 | 20 | 27.5 | 23.9 |
| 20 | 10 | 27.5 | 23.9 |
| 20 | 20 | 39.2 | 26.4 |
| 30 | 10 | 37.6 | 34.0 |
| 30 | 20 | 49.5 | 36.5 |
| 40 | 10 | 47.6 | 44.0 |
| 40 | 20 | 59.6 | 46.6 |
Old cons: Quality of the consensus base. New read: Quality of the corresponding base of the Autofinish candidate read. New unique: The resulting consensus quality value if the candidate read has a different chemistry or strand from any existing read aligned at that base position. New redundant: the resulting consensus quality value if the candidate read has the same strand and chemistry as some existing read aligned at that base position.
New Consed Features
Some projects cannot be completely finished using Autofinish and human finishers must finish the job. To make this interactive process easier using Consed, we have added the following features: tear a contig in two; remove a single read from a contig; join two contigs together; add new reads to an assembly without reassembling with Phrap; pick PCR primers; search for string with differences (Wu and Manber 1992); translate to amino acids and detect open reading frames; rapidly review putative polymorphic sites by integration with Polyphred (Nickerson et al. 1997); and many user-friendly improvements.
We continue to improve both Consed and Autofinish.
How to Get Consed and Autofinish
Autofinish and Consed are available on most UNIX platforms (Compaq, HP, SGI, Sun) and on Intel Linux. Executables are available at no charge to academic users and by commercial license from the University of Washington for other users. Currently there are ∼900 licensed sites in 36 countries, at least 230 of which are actively using the software. See http://www.phrap.org for more information.
Acknowledgments
We thank P. Minx for numerous suggestions and hard work over a long period of time. R. Maupin put in many hours testing Autofinish. In addition, the following people found problems or suggested improvements: D. Grafham, P. Havlak, B. Kronmiller, D. Hutchison, Y. Zhou, S. Dugan, B. Tubby, D. Johnson, and C. Nusbaum. E. Sims helped with lab work. L. Rowen inspired this competition by her friendly skepticism. Compaq, Hewlett-Packard, Silicon Graphics, and Sun Microsystems provided computers used in the development of Consed and Autofinish. Work partially supported by NIH Grant R01 HG 00774, and the Howard Hughes Medical Institute.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL gordon@genome.washington.edu.
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.171401.
REFERENCES
- Ewing B, Hillier L, Wendl M, Green P. Basecalling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998;8:175–185. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- Ewing B, Green P. Basecalling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998;8:186–194. [PubMed] [Google Scholar]
- Gordon D, Abajian C, Green P. Consed: A graphical tool for sequence finishing. Genome Res. 1998;8:195–202. doi: 10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- McMurray A, Sulston J, Quail M. Short-Insert Libraries as a Method of Problem Solving in Genome Sequencing. Genome Res. 1998;8:562–566. doi: 10.1101/gr.8.5.562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickerson D, Tobe V, Taylor S. PolyPhred: automating the detection and genotyping of single nucleotide substitutions using fluorescence-based resequencing. Nucleic Acids Res. 1997;25:2745–2751. doi: 10.1093/nar/25.14.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Manber U. Fast text searching allowing errors. Communications of the ACM. 1992;35:83–91. [Google Scholar]