

Abstract
We examined dinucleotide relative abundances and their biases in recent sequences of eukaryotic genomes and chromosomes, including human chromosomes 21 and 22, Saccharomyces cerevisiae, Arabidopsis thaliana, and Drosophila melanogaster. We found that dinucleotide relative abundances are remarkably constant across human chromosomes and within the DNA of a particular species. The dinucleotide biases differ between species, providing a genome signature that is characteristic of the bulk properties of an organism's DNA. We detail the relations between species genome signatures and suggest possible mechanisms for their origin and maintenance.
The recent sequencing of the complete genomes of Saccharomyces cerevisiae, Caenorhabditis elegans and Drosophila melanogaster, along with human chromosomes 21 and 22 and chromosomes 2 and 4 of Arabidopsis thaliana, provides new opportunities for studying higher eukaryote genome organization (C. elegans Sequencing Consortium 1998; Dunham et al. 1999; Lin et al. 1999; Mayer et al. 1999; Adams et al. 2000; Hattori et al. 2000). Every genome has a unique signature based on dinucleotide relative abundances (Karlin and Ladunga 1994). This genome signature is a characteristic of the genome as a whole and does not depend on knowledge of individual genes or alignment of homologous sequences. Instead, it reflects the response of the whole genome to overall selective pressures, operating through limits on compositional and/or structural variations in DNA. It is essentially constant in both coding and noncoding sequences and is independent of renaturation fraction (G + C isochores) and of base compositional fractions (Russell et al. 1976; Russell and Subak-Sharpe 1977). The mechanisms that determine and maintain the signature are not understood, but they could involve DNA replication and repair mechanisms and biases in DNA modification processes. They can operate on the whole genome through DNA structure (e.g., base-step stacking energies and DNA conformational tendencies), context dependent mutation, and DNA methylation patterns (for review, see Karlin 1998).
Dinucleotide Relative Abundances
The dinucleotide relative abundance is defined as
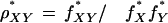 |
where f*X is the frequency of the nucleotide X and f*XY is the frequency of the dinucleotide XY, calculated over a sequence concatenated with its inverted complement. (Throughout we refer to the dinucleotide pair XpY as XY.) ρ* measures the abundance of dinucleotides relative to what would be expected from the component base frequencies. Hence, ρ* (actually ρ* − 1) can also be referred to as the dinucleotide bias.
The vector of ρ* values constitutes the genome signature. In practice, a given sequence is split into equal (typically 50-kb) segments and the signature is calculated for each. Distributions of ρ* values for the 50-kb segments can be compared with each other within a species or between different species. Thus, it can be judged which dinucleotide pairs are relatively over- or underrepresented in the genome. Theoretical and empirical studies indicate that if the dinucleotide XY has a mean ρ*XY ≤ 0.78, then XY is significantly underrepresented (suppressed), whereas ρ*XY ≥ 1.23 indicates over-representation. Corresponding expressions can be constructed for tri- and tetranucleotide relative abundances but add little additional information, suggesting that DNA conformational stacking arrangements are determined mainly through the dinucleotide base-step configurations.
The genome signature is highly invariant across the DNA of an organism and is similar for closely related species. Strong support for the invariance of the signature within species comes from both sequence analysis and experimental studies of nearest-neighbor frequencies, which have shown that the set of dinucleotide relative abundance values for 50-kb DNA contigs is a characteristic of an organism's DNA and distinguishes it from other species (Russell et al. 1976; Russell and Subak-Sharpe 1977; Karlin and Burge 1995; Karlin 1998).
ρ*XY Distributions Across Species
Each available data set (see Methods) was divided into nonoverlapping 50-kb samples and the ρ*XY values determined for each sample. For every organism, one obtains a list of ρ* values for each 50-kb sample for all dinucleotides XY. These are plotted as histograms of ρ* values for each dinucleotide in Figure 1, which compares the distributions for human, S. cerevisiae, D. melanogaster, C. elegans, and A. thaliana. The distributions are all homogeneous within species and distinctly different between species. Histograms are superior to simple variance statistics. Individual p* values do not discriminate between, for example, yeast and Arabidopsis, between mouse and human, betwee4n the protists Plasmodium falciparum and Trypanosoma brucei, or among most prokaryotes. The whole genome signature vector (10 components) does discriminate these cases.
Figure 1.

Distribution of ρ* values for all 50-kb samples from human (red), Drosophila melanogaster (black), Saccharomyces cerevisiae (green), Caenorhabditis elegans (blue), and Arabidopsis thaliana (orange).
The most striking feature is the CG underrepresentation in human DNA. GC relative abundances tend to be in the normal range across eukaryotic species, except for Drosophila which has high ρ*GC. Human DNA has higher relative abundances of CC/GG, AG/CT, and CA/TG dinucleotides than the other species, but neither dinucleotide pair is significantly biased. ρ*CA/TG is slightly high in human but normal in Drosophila, yeast, Arabidopsis, and C. elegans. TA is modestly suppressed in all organisms, with human and C. elegans showing the lowest ρ*TA. Yeast and Arabidopsis have very similar ρ* values for all dinucleotides, with generally sharply peaked distributions and low variance, the exception being CG in Arabidopsis. In contrast human, C. elegans, and, to a lesser extent, Drosophila all exhibit a moderate spread in ρ* values. AC/GT, AA/TT, and AT relative abundances do not differ much between species and are all in the normal (unbiased) range of ρ* values.
Human Chromosomes 21 and 22
The recent completion of human chromosomes 21 and 22 makes them particularly interesting sequences to study. Both were partitioned into contiguous 50-kb windows and the ρ*XY values for each window are plotted across the chromosomes in Figure 2. One can see immediately that, with minor exceptions, all dinucleotide biases are clearly invariant both across and between chromosomes. This is conspicuous in the ρ* values for CG, GC, GA/TC, AC/GT, and AT. From around position 10 Mb to 25 Mb on chromosome 21, the AG/CT dinucleotide bias is slightly reduced compared to the rest of the chromosome and to chromosome 22. In addition, the chromosome-21 TA bias is slightly elevated over this region. The only other notable variation is around position 13.4 Mb of chromosome 22 in the 406-kb long contig NT002447. Closer inspection reveals that a large portion of this contig (GenBank accession no. AP000536) is dominated by a 47-kb tandem repeat of an ∼ 50-bp subunit.
Figure 2.

Variation of ρ* across human chromosomes 21 (red) and 22 (black) for each unique dinucleotide. The horizontal scale is identical in all graphs. Each vertical scale graduation is 0.1 in ρ*.
It is noteworthy that the genome signature does not change according to the predicted gene density on either chromosome; nor does it change as one approaches the centromeric heterochromatin or the telomeres. For example, there is a 7-Mb region of chromosome 21 from position 5 Mb to 12 Mb that has a low (G + C) content, no CG islands, a few Alu repeats, and low gene numbers relative to the rest of the chromosome (Hattori et al. 2000). Yet the signature does not vary across this region or relative to distant regions of chromosome 21.
{ρ*XY} Comparisons
Table 1 shows the mean ρ*XY values of nonoverlapping 50-kb samples for each dinucleotide pair in several eukaryotes and for each human chromosome. Mean ρ*XY values are strongly conserved across all human chromosomes. The ranges are in CG, 0.18 to 0.31; GC, 0.96 to 1.02; TA, 0.66 to 0.75; AT, 0.84 to 0.89; CC/GG, 1.22 to 1.24; TT/AA, 1.11 to 1.13; TG/CA, 1.20 to 1.24; AG/CT, 1.15 to 1.24; AC/GT, 0.82 to 0.86; and GA/TC 0.98 to 1.00. The largest variation is in ρ*CG, where the highest value is 0.31 for chromosome 19, followed by chromosomes 16 and 22 at 0.28. The lowest values occur for chromosomes Y (0.18), X (0.20), and 18 (0.21). There is a positive correlation between ρ*XY values and the CG-island densities implied by in situ fluorescence hybridization of human chromosomes during metaphase (Cross and Bird 1995). Chromosomes 19 and 22 are rich in CG islands, whereas chromosomes 18, X, and Y are CG-island poor, in agreement with the ρ*CG values noted above.
Table 1.
Mean Eukaryotic ρ* Values for All Available DNA Contigs at Least 50kb in Size
 |
In common with all mammalian genomes, Mus musculus and the human chromosomes exhibit extreme CG underrepresentation, with ρ*CG = 0.21 in mouse and 0.18–0.31 for human chromosomes. CG suppression is usually explained through the methylation-deamination-mutation hypothesis, whereby methylation of CG to 5-methylcytosine and subsequent deamination to thymine results, if unrepaired, in conversion of CG to TG/CA. The methylation hypothesis is supported by the fact that invertebrates that do not possess a methylase, such as Drosophila and C. elegans, do not exhibit significant CG dinucleotide bias (ρ*CG = 0.92 for Drosophila and 0.96 for C.elegans). ρ*CG is significantly low in A. thaliana (0.72) but not in yeast (0.80), concurring with the occurence of methylation in dicots such as Arabidopsis but with its absence from monocots. However, in human and mouse, TG/CA is only marginally overrepresented (ρ*TG/CA = 1.20–1.24 and 1.20–1.23, respectively), in marked contrast to the extreme underrepresentation of CG. Moreover, CG is underrepresented in all animal mitochondria despite the lack of methylase activity in mitochondria. There is also no significant bias in TG/CA in animal mitochondria. This indicates that although methlyation may contribute to vertebrate CG suppression, it does not fully account for it.
All of the eukaryotes except Plasmodium falciparum (0.99) show low ρ*TA, ranging from 0.56 in Leishmania major and 0.62 in C. elegans to 0.75 in Arabidopsis and Drosophila. TA is the least stable dinucleotide stacking pair and is prominent in some regulatory signals, such as the TATA box and 3′ polyadenylation signal. Avoidance of spurious signal sequences and considerations of DNA stability could both act to suppress overall levels of TA. In coding regions, TA may be low because UA is disfavored in mRNAs, where it is relatively susceptible to cleavage by ribonucleases (Beutler et al. 1989). ρ*GC is high in Drosophila (1.27), whereas C.elegans has high ρ*TT/AA = 1.28. Mouse shows high ρ*AG/CT (1.25), with human (range 1.15–1.22) hardly biased. The other most biased dinucleotide abundances are in Leishmania (ρ*TG/CA = 1.25) and Plasmodium (ρ*CC/GG = 1.51). Yeast is unusual among these eukaryotes in having no significantly biased dinucleotide relative abundances. All yeast ρ*s are in the range 0.8–1.13 except for ρ*TA, which qualifies as marginally underrepresented at 0.77.
Table 2 shows the unsymmetrized (single-strand) ρ values for CG and TA at different codon positions, introns, and intergenic regions in human DNA. Both CG and TA are suppressed in coding and noncoding regions, with TA being less biased in all cases. Introns and intergenic DNA exhibit stronger CG suppression than coding sequences but are less biased in TA. This is consonant with higher substitution rates in noncoding regions, which do not have the constraints on amino acid and codon usage, which affect coding sequences. The higher CG usage at codon positions 1,2—compared to 2,3 or 3,1—probably reflects the fact that in human proteins, arginine is more frequently coded for by CGN (3.2% of the time) than by an AGR codon (2.2%). Paradoxically, G is highest at codon position 1 (32%) and C is highest at position 3 (29%), yet CG is highly suppressed at positions 3,1.
Table 2.
CG and TA Dinucleotide Biases in Human Coding and Noncoding DNA
| Codon positions | Introns | Intergenic | ||||
|---|---|---|---|---|---|---|
| (1,2) | (2,3) | (3,1) | ||||
| ρCG | 0.70 | 0.36 | 0.44 | 0.24 | 0.29 | |
| ρTA | 0.54 | 0.56 | 0.61 | 0.72 | ||
δ* Comparisons
It is useful to have a measure of the difference between the signatures of DNA sequences. For this purpose, we use the dinucleotide relative abundance distance, which for sequences p and q is defined as
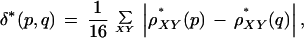 |
where the sum is over all dinucleotides XY. The value of δ* is quoted after multiplying by 1000. The average distance δ* between random sequences of length 50 kb is then ∼ 10–20. In comparing DNA sequences, the mean δ* value is found for all pairwise comparisons of 50-kb contigs. This can be done within a species and between different species. Thus, a matrix of distances is built up, which is the mean δ* distance between 50-kb segments from each species or sequence. Extensive testing has shown that the δ* distance is not distorted by extreme biases in a single dinucleotide (Karlin and Ladunga 1994).
δ* Comparisons within Species
Human within-chromosome δ* scores range from 30 in chromosome 7 to 48 in chromosome 11. The range of δ* between chromosomes is from 30 (chromosome 18 vs. 13) to 54 (19 vs. Y), with 35–45 being typical. The δ* distance between chromosomes, therefore, is approximately the same as within chromosomes, despite the differences in base composition, gene density, and repeat frequencies between them. The δ*distances between and within Drosophila chromosomes range from 42 to 68. As shown in Figure 3, the left (L) and right (R) arms of chromosomes 2 and 3 are a close group, with δ* between 42 and 57. The Drosophila X chromosome is slightly more variable both within itself (δ* = 68) and in comparison to the other chromosomes, with a distance of 57 from 2L, 3L, and 3R and 65 from 2R. With the exception of the X chromosome, these values are similar to human within- and between-chromosome δ* values. Finally, in C. elegans the six chromosomes exhibit a range of δ* within themselves from 49 (chromosome 4) to 70 (chromosome 2). Between-chromosome distances are from 51 (chromosome X vs. 4 and 5) to 70 (3 vs. 2 and X). The δ* values thus exhibit the same invariance within a species as the dinucleotide ρ* biases.
Figure 3.

Drosophila melanogaster δ−distances within and between chromosomes X, 2 (right arm, R, and left arm, L), and 3 (R, L).
δ* Comparisons between Species
Figure 4 shows the mean δ* distances between the eukaryotes discussed above. P. falciparum chromosomes 2 and 3, L. major chromosome 1, and the complete E. coli genome are included for comparison.
Figure 4.

δ* Distances between Homo sapiens, homsa; Mus musculus, musmu; Drosophila melanogaster, drome; Caenorhabditis elegans, caeel; Saccharomyces cerevisiae, sacce; Arabidopsis thaliana, arath; Escherichia coli, ecoli; Plasmodium falciparum, plafa; and Leishmania major, leima.
Human and mouse show moderate similarity (δ* = 58), as one would expect. Arabidopsis and yeast are close (δ* = 45); surprisingly, their δ* distance from each other is nearly as low as their mean within-species distances. E. coli is very distant from human (210), mouse (241), and both protoctists (196, 174). It is also distant from C. elegans (128), Arabidopsis (148), and yeast (122). Mysteriously, however, there is moderate similarity between the signatures of E. coli and D. melanogaster (δ* =74).
DISCUSSION
We have confirmed, through our analysis of the current complete eukaryotic genomes and chromosomes 21 and 22 of human, the constancy and validity of the genome signature for each species. Signature comparisons have revealed a number of intriguing relations between organisms. For example, bacterial phage genome signatures are strongly correlated with the nature of the host and the extent to which the phage uses the host-cell machinery (Blaisdell et al. 1996). Both broad-range and specialized plasmids in prokaryotes share moderate to close genome signature with their host (Campbell et al. 1999). Although mammalian mitochondria are close to each other in signature and reflect relationships parallel to those derived from nuclear DNA, they are not close to their host nuclear DNA, with typical δ* differences between 140 and 200 (Karlin and Mrázek 1997).
Among bacteria, there are signature similarities between closely related species (such as E. coli vs. Salmonella typhimirium and Streptococcus pyogenes vs. Lactococcus lactis) but no groupings that can be attributed to obvious causes such as the environment in which the bacteria live. Likewise, archaea do not form a coherent clade in terms of their signature; for example, halobacteria sp. and methanogens have extremely different genome signatures. Anomalies in the signature have been used to detect bacterial pathogenicity islands and laterally transferred operons in Helicobacter pylori and Mycobacterium tuberculosis (Karlin 1998) and in Neisseria meningitidis, Vibrio cholerae, Campylobacter jejuni, and E. coli (data not shown). Unmethylated CG shows normal dinucleotide bias in most proteobacteria and can provoke an immune response in mammals (Krieg et al. 1998). CG is also suppressed in most small (< 30 kb length) vertebrate viral genomes, except for a few togaviruses (Karlin et al. 1994). Another intriguing result is that the signature of mammalian retroviruses shows moderate similarity to the nuclear DNA into which they integrate with a range δ* = 70–90 (data not shown). This might have resulted from the processing of the viral genetic program by the host-cell machinery or a selective shift in the viral genome toward a genome signature that is more compatible with the host.
There are a number of unanswered questions concerning the nature of the genome signature. The homogeneity of the signature is clearly maintained by processes that operate at the scale of the whole genome. However, it is not known if the signature corresponds to a frozen event or if it is a dynamical feature of a genome that changes over time, albeit slowly. How did the signature arise for a given genome and how fast can it change? Many DNA repair enzymes recognize the shape of the DNA molecule rather than specific sequences (Echols and Goodman 1991; Kunkel 1992). Stacking energies, charge interactions, and conformational tendencies all bear on local DNA structure and thus influence the intrinsic curvature of DNA (Bolshoy 1995). In addition, the efficiency of DNA repair is affected by neighboring-base context.
METHODS
Data
The human, mouse, A. thaliana, P. falciparum, L. major, C. elegans, S. cerevisiae, and E. coli sequences were acquired from GenBank. Except for chromosomes 21 and 22, sequence sets for human chromosomes were produced using the lists of contigs maintained by the Computational Biosciences Section at Oak Ridge National Laboratory. Only contigs ≥50 kb in length were used. The complete D. melanogaster genome was obtained from the Gadfly database maintained by the Berkeley Drosophila Genome Project.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
NOTE ADDED IN PROOF
The p* values for human chromosomes are essentially unchanged when calculated across the recently released draft sequence of the complete human genome (International Human Genome Sequencing Consortium 2001).
Footnotes
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.163101.
REFERENCES
- Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, et al. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- Beutler E, Gelbart T, Han JH, Koziol JA, Beutler B. Evolution of the genome and the genetic code: Selection at the dinucleotide level by methylation and polyribonucleotide cleavage. Proc Natl Acad Sci. 1989;86:192–196. doi: 10.1073/pnas.86.1.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaisdell BE, Campbell AM, Karlin S. Similarities and dissimilarities of phage genomes. Proc Natl Acad Sci. 1996;93:5854–5859. doi: 10.1073/pnas.93.12.5854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolshoy A. Dinucleotides contribute to the bending of DNA in chromatin. Nat Struct Biol. 1995;2:446–448. doi: 10.1038/nsb0695-446. [DOI] [PubMed] [Google Scholar]
- C. elegans Sequencing Consortium. Genome sequence of the nematode C. elegans: A platform for investigating biology. Science. 1998;282:2012–2018. doi: 10.1126/science.282.5396.2012. [DOI] [PubMed] [Google Scholar]
- Campbell A, Mrázek J, Karlin S. Genome signature comparisons among prokaryote, plasmid and mitochondrial DNA. Proc Natl Acad Sci. 1999;96:9184–9189. doi: 10.1073/pnas.96.16.9184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross SH, Bird AP. CpG islands and genes. Curr Opin Genet Dev. 1995;5:309–314. doi: 10.1016/0959-437x(95)80044-1. [DOI] [PubMed] [Google Scholar]
- Dunham I, Shimizu N, Roe BA, Chissoe S, Hunt AR, Collins JE, Bruskiewich R, Beare DM, Clamp M, Smink LJ, et al. The DNA sequence of human chromosome 22. Nature. 1999;402:489–495. doi: 10.1038/990031. [DOI] [PubMed] [Google Scholar]
- Echols H, Goodman MF. Fidelity mechanisms in DNA replication. Annu Rev Biochem. 1991;60:477–511. doi: 10.1146/annurev.bi.60.070191.002401. [DOI] [PubMed] [Google Scholar]
- Hattori M, Fujiyama A, Taylor TD, Watanabe H, Yada T, Park HS, Toyoda A, Ishii K, Totoki Y, Choi DK, et al. The DNA sequence of human chromosome 21. Nature. 2000;405:311–319. doi: 10.1038/35012518. [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Karlin S. Global dinucleotide signatures and analysis of genomic heterogeneity. Curr Opin Microbiol. 1998;1:598–610. doi: 10.1016/s1369-5274(98)80095-7. [DOI] [PubMed] [Google Scholar]
- Karlin S, Burge C. Dinucleotide relative abundance extremes: A genomic signature. Trends Genet. 1995;11:283–290. doi: 10.1016/s0168-9525(00)89076-9. [DOI] [PubMed] [Google Scholar]
- Karlin S, Ladunga I. Comparisons of eukaryotic genomic sequences. Proc Natl Acad Sci. 1994;91:12832–12836. doi: 10.1073/pnas.91.26.12832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin S, Mrázek J. Compositional differences within and between eukaryotic genomes. Proc Natl Acad Sci. 1997;94:10227–10232. doi: 10.1073/pnas.94.19.10227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin S, Doerfler W, Cardon LR. Why is CpG suppressed in the genomes of virtually all small eukaryotic viruses but not in those of large eukaryotic viruses? J Virol. 1994;68:2889–2897. doi: 10.1128/jvi.68.5.2889-2897.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieg AM, Yi AK, Schorr J, Davis HL. The role of CpG dinucleotides in DNA vaccines. Trends Microbiol. 1998;6:23–27. doi: 10.1016/S0966-842X(97)01145-1. [DOI] [PubMed] [Google Scholar]
- Kunkel TA. Biological asymmetries and the fidelity of eukaryotic DNA replication. Bioessays. 1992;14:303–308. doi: 10.1002/bies.950140503. [DOI] [PubMed] [Google Scholar]
- Lin XY, Kaul S, Rounsley S, Shea TP, Benito MI, Town CD, Fujii CY, Mason T, Bowman CL, Barnstead M, et al. Sequence and analysis of chromosome 2 of the plant Arabidopsis thaliana. Nature. 1999;402:761–768. doi: 10.1038/45471. [DOI] [PubMed] [Google Scholar]
- Mayer K, Schuller C, Wambutt R, Murphy G, Volckaert G, Pohl T, Dusterhoft A, Stiekema W, Entian KD, Terryn N, et al. Sequence and analysis of chromosome 4 of the plant Arabidopsis thaliana. Nature. 1999;402:769–777. doi: 10.1038/47134. [DOI] [PubMed] [Google Scholar]
- Russell GJ, Subak-Sharpe JH. Similarity of the general designs of protochordates and invertebrates. Nature. 1977;266:533–536. doi: 10.1038/266533a0. [DOI] [PubMed] [Google Scholar]
- Russell GJ, Walker PM, Elton RA, Subak-Sharpe JH. Doublet frequency analysis of fractionated vertebrate nuclear DNA. J Mol Biol. 1976;108:1–23. doi: 10.1016/s0022-2836(76)80090-3. [DOI] [PubMed] [Google Scholar]