

Abstract
Large-scale genomic sequencing projects have provided DNA sequence information for many genes, but the biological functions for most of them will only be known through functional studies. Bacterial artificial chromosomes (BACs) and P1-derived artificial chromosomes (PACs) are large genomic clones stably maintained in bacteria and are very important in functional studies through transfection because of their large size and stability. Because most BAC or PAC vectors do not have a mammalian selection marker, transfecting mammalian cells with genes cloned in BACs or PACs requires the insertion into the BAC/PAC of a mammalian selectable marker. However, currently available procedures are not satisfactory in efficiency and fidelity. We describe a very simple and efficient procedure that allows one to retrofit dozens of BACs in a day with no detectable deletions or unwanted recombination. We use a BAC/PAC retrofitting vector that, on transformation into competent BAC or PAC strains, will catalyze the specific insertion of itself into BAC/PAC vectors through in vivo cre/loxP site-specific recombination.
Bacterial artificial chromosomes (BACs) and P1-derived artificial chromosomes (PACs), with their capacity for large inserts, stability, and lack of chimerism, play important roles in genome mapping and sequencing (Osoegawa et al. 2000). For these reasons and the fact that virtually every gene will have one or more corresponding BAC/PAC clones, they are the first choice for functional complementation or dose-effect studies in differentiated or embryonic stem (ES) cells. Mouse ES cells are of special interest for gene function studies because of their capacity for in vitro differentiation into different lineages (O'Shea 1999) and the ability to generate transgenic animals through germline transmission. The lack of a universal selection marker for mammalian cell transfection makes it difficult to include any particular marker in the BAC/PAC vectors. Therefore, the vectors for most current BAC or PAC libraries do not contain any mammalian selection marker for transfection studies. This necessitates the development of efficient ways to insert such selection markers into individual clones chosen from BAC/PAC libraries, a “retrofitting” operation.
Several strategies have been developed to retrofit BACs/PACs on the basis of restriction digestion, transposition, homologous recombination, or site-specific recombination. Certain rare-cutter restriction enzymes have been used to linearize both the BAC clone and a retrofitting cassette. These sticky ends were then ligated to form retrofitted BACs with the selectable marker inserted (Mejia and Monaco 1997; Hejna et al. 1998). For this strategy to work, it has to be certain that there is no such restriction site in the genomic insert, which often is not the case with large inserts. It also requires subsequent transformation of the retrofitted BAC/PAC into Escherichia coli, which is both very inefficient and deleterious to the genomic insert. Recombination-based approaches allow the direct integration of a selectable marker cassette into BAC/PAC clones without the need for linearization and retransformation. Several homologous recombination-based strategies have been described, including RecA- (Yang et al. 1997) or RecE/RecT-promoted (Muyrers et al. 1999; Narayanan et al. 1999), χ-stimulated (Jessen et al. 1998), and alu-repeat-based (Chatterjee and Sternberg 1996) strategies. These methods are very useful for introducing changes to the genomic insert. However, for the simple insertion of a selectable marker into BAC vector and when many BAC clones need to be modified and transfected, these methods are inadequate and pose the risk of unwanted recombination because of the expression of Rec genes. Another method for retrofitting BACs is by transposition of a retrofitting cassette to the BAC/PAC, either randomly (Chatterjee and Sternberg 1996) or at a predefined site (Frengen et al. 1999), but this method also requires multiple manipulations. In summary, although all of these methods work to some extent in BAC/PAC retrofitting, they are relatively complicated and most of them impose the potential risk of causing deletions and unwanted recombination to the genomic insert, hindering the process of large-scale BAC retrofitting.
Site-specific recombination, on the other hand, involves a specific sequence element on the BAC/PAC vector and its partner in the retrofitting construct. Therefore, the expression of Rec genes is not needed, and unwanted homologous recombination can be avoided. One commonly used site-specific recombination scheme is P1 phage-derived cre/loxP recombination. The cre recombinase recognizes and recombines loxP sites, sequences of 34 bp in length with two inverted repeats. Current PAC and BAC vectors all contain a loxP site and successful retrofitting reactions have been performed in vitro with a loxP-containing construct, pRetroBAC, and commercial cre recombinase (Kim et al. 1998). However, when modifying BACs in vitro by using pRetroBAC and a retrofitting vector that we made, we observed problems of low transformation efficiency of large BACs and deletions to the genomic inserts. Presumably, nucleases contaminating the cre recombinase preparations caused deletions, which were further enriched through BAC retransformation because of much higher transformation efficiency with smaller DNA molecules (Sheng et al. 1995). Therefore, a large number of recombinant clones have to be screened for the integrity of the genomic insert.
Here we report a single-step, in vivo cre/loxP-based approach in which competent BAC strains are transformed with a novel retrofitting vector, pRetroES. We believe it is by far the simplest and most efficient way of inserting a mammalian selectable marker into a BAC or PAC clone without changing the genomic insert. With this method, one can easily retrofit dozens or even hundreds of BACs in 1 d with limited hands-on time. Furthermore, because of the high fidelity of this method, the screening of retrofitted clones is usually not necessary and problems of deletions and unwanted recombination are prevented.
RESULTS AND DISCUSSION
Rationale and the Structure of pRetroES
In our effort to transfect candidate BACs into mouse ES cells for the functional complementation screen of Ssm1, a mouse gene responsible for strain-specific transgene methylation (Engler et al. 1991), we explored various strategies of BAC retrofitting. We decided to pursue strategies based on cre/loxP site-specific recombination because of its precise specificity. pRetroES-t, constructed for in vitro BAC retrofitting, contains a loxP site, a PGKneo gene, and a conditional replication origin oriR6Kγ, which needs the λpir gene product to function (Metcalf et al. 1996). We tested pRetroES-t, along with a similar BAC retrofitting vector, pRetroBAC (Kim et al. 1998), on retrofitting our candidate BACs in vitro with either commercial (Novagen) or, later, self-made (Liu et al. 1998) cre recombinase. The in vitro modified BACs were then transformed into DH10B competent cells (Stratagene) by electroporation.
The results (not shown) indicate that both pRetroES-t and pRetroBAC worked properly in retrofitting BACs with relatively smaller inserts (<150 kb). However, with BACs having inserts larger than 150 kb, the transformation efficiency decreased dramatically. We could only get a few recombinant colonies for a 246-kb test BAC after many retrofitting and transformation experiments. After restriction digestion and electrophoresis of the retrofitted clones, we found that many clones had deletions to the genomic insert (data not shown). Because transformation efficiency drops dramatically as the size increases (Sheng et al. 1995), we postulate that cre recombinase or, more likely, contaminating endonucleases might have caused deletions to some DNA molecules and those molecules with deletions may have been selectively transformed because of their smaller size, resulting in frequent deletions observed in the retrofitted clones.
The problems of low transformation efficiency with large insert BACs and deletions to the inserts prompted us to explore better strategies for BAC retrofitting. The ideal way would be in vivo retrofitting, where the BAC molecules will remain in bacteria which then are transformed with the retrofitting construct to introduce the selectable marker by recombination. With this strategy, it is essential to have a transient and limited supply of cre recombinase, enough to promote the integration reaction, but not excessive so as to cause unwanted recombination events, including looping out of the inserted mammalian selection marker. We therefore designed a retrofitting vector, pRetroES, for such in vivo BAC/PAC retrofitting (Fig. 1A).
Figure 1.
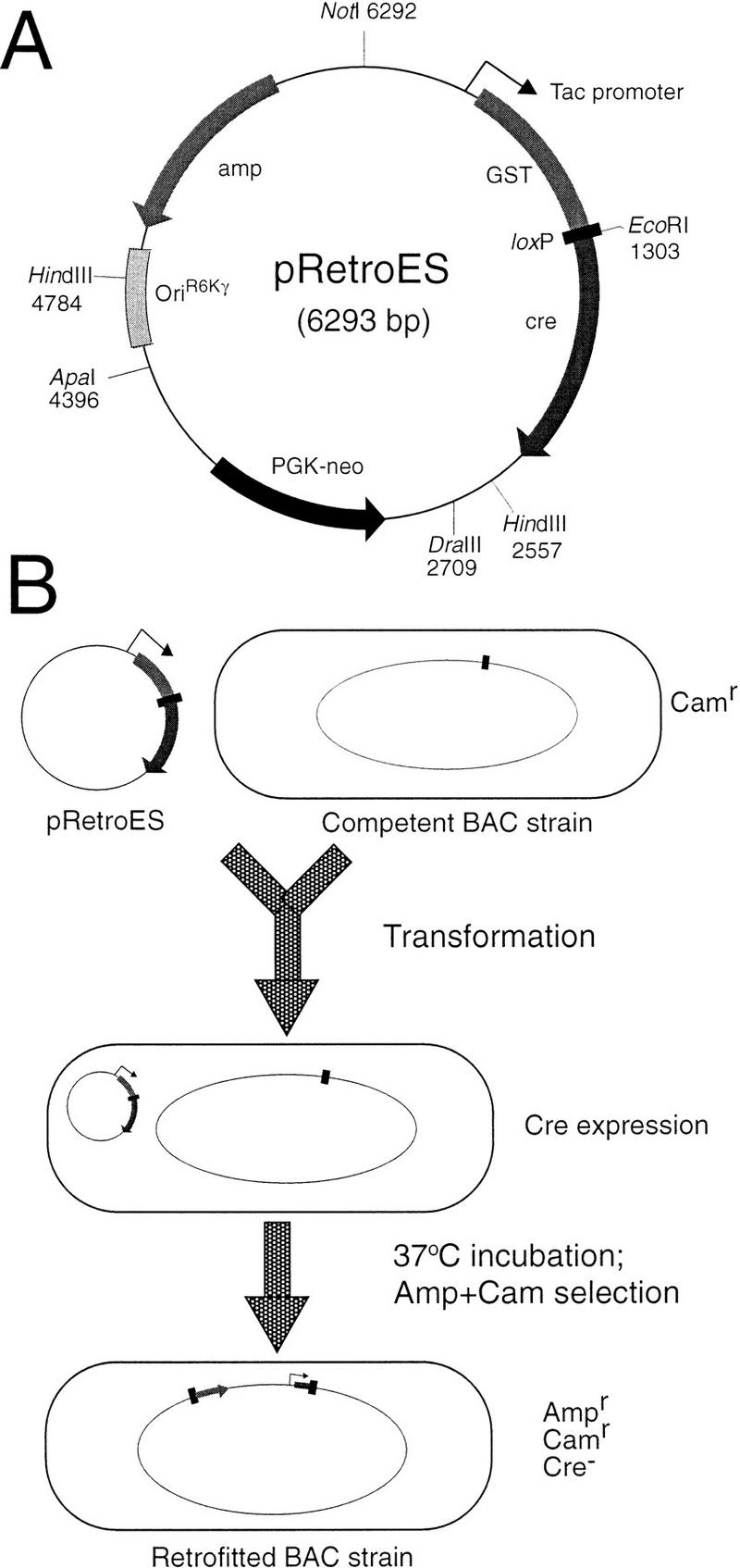
Map of the retrofitting vector pRetroES and schematic illustration of the retrofitting procedure. (A) Map of pRetroES. Essentially, it has a GST-loxP-cre fusion gene driven by a tac promoter that encodes a fusion cre recombinase for promoting cre/loxP recombination. The conditional replication origin, oriR6Kγ, will not function in the usual BAC host. The PGKneo is a selectable marker useful for embryonic stem cell transfection. (B) Schematic representation of the retrofitting procedure. In brief, a small volume of competent BAC-bearing Escherichia coli is prepared and transformed with pRetroES by electroporation. During the posttransformation incubation, the fusion cre recombinase will be expressed and will promote the recombination between the loxP sites on the retrofitting vector and the BAC vector, leading to the integration of pRetroES into the BAC at the loxP site. After integration, the tac promoter is separated from the fusion gene and no more cre recombinase will be produced. For detailed protocol, see Methods.
The essential part of pRetroES is the tac-GST-loxP-cre fusion gene, which is both self-catalytic and suicidal. Liu et al. (1998) showed that loxP sequence can be transcribed and translated into a fusion protein, and we found that the fusion cre recombinase described earlier is functional (data not shown). On transformation into the BAC-bearing E. coli, the fusion gene provides enough cre recombinase to promote site-specific recombination for integration of the construct into the BAC vector. Once integrated, the cre fusion gene is broken at the loxP site and becomes nonfunctional, preventing the production of any excess cre recombinase (Fig. 1B). Similar to our in vitro retrofitting vector pRetroES-t, pRetroES contains a conditional replication origin oriR6Kγ, which functions only in the presence of the λpir gene product. It maintains the stability of the recombinant BAC molecule and eliminates the need to delete the normal replication origin through restriction digestion, fragment purification, and ligation (Kim et al. 1998). The latter process often produces nonrecombinant, cotransformed, double resistant colonies in our hands (data not shown).
Optimization of the Retrofitting Procedure
To transform the retrofitting vector, one must make competent cells for each BAC strain. We found that electroporation of competent cells prepared from small-volume culture is the most convenient and efficient approach. Overnight cultures of BAC strains are diluted 1∶100 into 5 mL LB media and grown with shaking at 37°C for 3 h followed by several washes in the cold and then electroporation. It is not difficult in this way to prepare hundreds of competent BAC strains at the same time, paving the way for large-scale BAC/PAC retrofitting. Even with relatively low competence, one can still obtain enough retrofitted clones. We routinely are able to generate a few hundred to a few thousand recombinant colonies from each transformation. The retrofitting efficiency is independent of BAC insert size, as demonstrated by roughly equal numbers of recombinant colonies obtained from retrofitting three sample BACs with insert sizes of 104, 196, and 246 kb, respectively.
After electroporation, the cells are incubated in LB media at 37°C with shaking for a period of time to allow cre expression, cre/loxP recombination, and the expression of antibiotic resistance genes. Although longer incubation might increase retrofitting efficiency, we found that 75–90-min postelectroporation incubation is long enough to generate sufficient recombinant colonies.
Integration at loxP Versus lox511 Sites
RPCI-23 and other BAC libraries are constructed with the pBACe3.6 vector (Frengen et al. 1999), which has two loxP sites in opposite orientation initially designed as a means for vector replacement: a wild-type loxP and a mutant form, called lox511. It was reported that lox511, having a single base substitution in the loop, recombines inefficiently with wild-type loxP (Hartung and Kisters-Woike 1998). To characterize the nature and location of the pRetroES integrations, we performed two sets of colony PCR reactions on the chloramphenicol/ampicillin double-resistant (retrofitted BAC-containing) colonies: one to amplify recombinants integrated at the loxP site and the other at lox511 (Fig. 2A). Surprisingly, the results, some of which are shown in Figure 2B, indicate that there is no preference for recombining at the loxP site over the lox511 site. Whether the fusion cre recombinase, which contains a GST tag and amino acids coded by the loxP sequence, has altered specificity and has lost the preference of loxP over lox511 is not known. Integration at either site should make no difference for most applications, including ES cell transfection. Some of the recombinants have retrofitting vector integrated at both loci (see Fig. 2B clone #31), a phenomenon more frequently observed when more retrofitting vector DNA was used in transformation (data not shown). Such double-integrated recombinants seem to be stable in E. coli because restriction analysis of DNA prepared from such clones shows expected vector fragments and no deletion to the insert. We have not tested if the added homology resulting from such double integrations would cause unwanted recombination during transfection. If it is a concern for particular applications, double integrated recombinants can be easily detected and eliminated through colony PCR (Fig. 2B).
Figure 2.
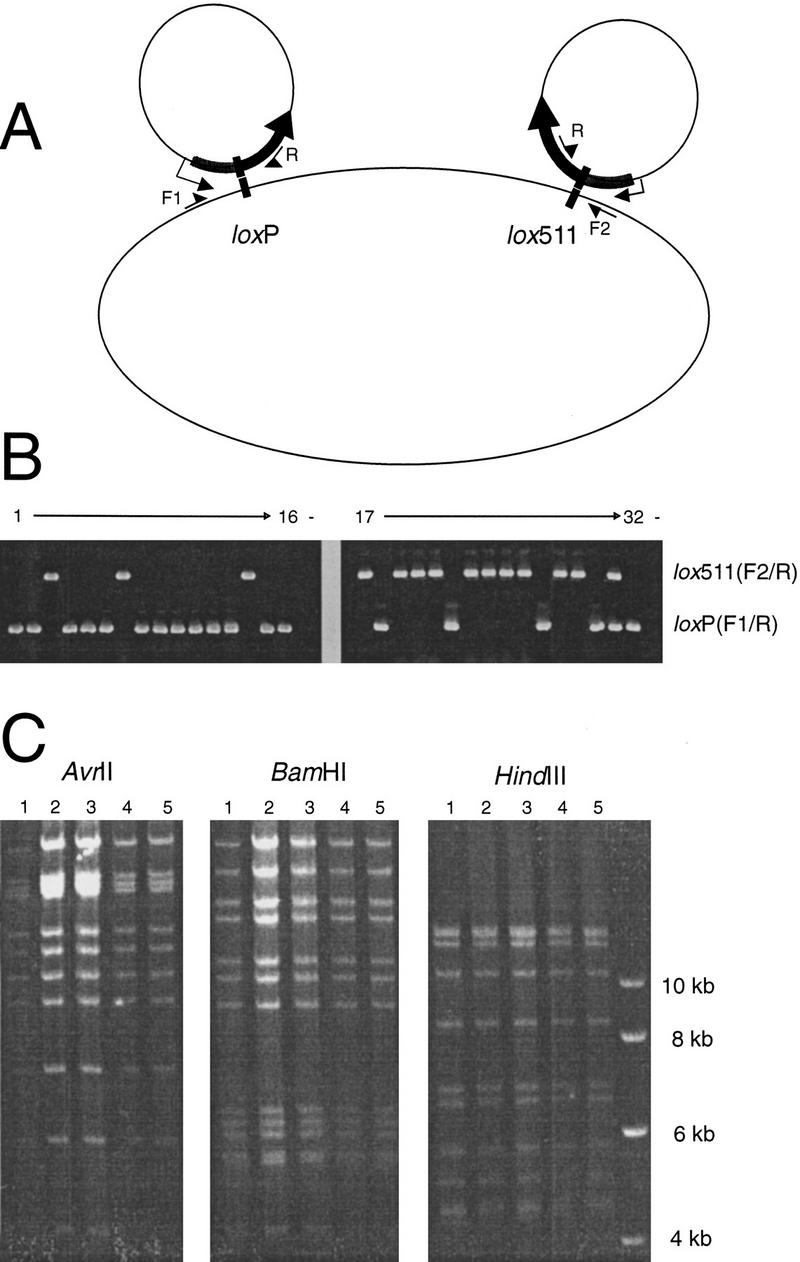
Representative results of retrofitted clones. (A) Schematic representation of possible recombinants from retrofitting pBACe3.6-generated BACs with pRetroES, showing primers used in colony PCR: F1 and F2 on the BAC vector and Retro-R (shown as R) on the retrofitting construct. (B) Colony PCR results showing the efficiency of the retrofitting and the frequency of integration at loxP and lox511 sites. Thirty-two chloramphenicol/ampicillin double-resistant colonies were PCR amplified with primers that identified integrations either at loxP site (F1 and R) or at lox511 site (F2 and R). As shown, 31 of the 32 clones have pRetroES integrated either at loxP (13/32) or at lox511 (19/32) site, and one of them, Clone 31, has a double integration. (C) Restriction digestion of five different retrofitted clones of the same BAC showing the fidelity of the retrofitting process. Five randomly picked cam/amp double-resistant colonies (numbered 1 through 5) were grown in liquid media and DNA prepared and digested with restriction enzymes AvrII, BamHI, and EcoRI. Electrophoresis was performed on 0.6% agarose at low voltage for about 24 h and the gel was stained with ethidium bromide.
Fidelity of In Vivo Retrofitting
One important question is the fidelity of the retrofitting process, that is, if deletion could occur to the genomic insert. Because we rely on transfecting overlapping BAC clones into ES cells for both functional screening and fine mapping the location of the gene in the large inserts, it is very important to maintain the integrity of the transferred genomic clones. By using pRetroES-t for in vitro retrofitting, we observed that many clones have deletions after digestion with restriction enzymes and electrophoresis, most frequently with BACs of large inserts. Therefore, after in vivo retrofitting, we extensively analyzed the retrofitted clones with restriction digestions to see if any deletions occurred to the genomic inserts. Of dozens of clones from over 10 BACs retrofitted with insert sizes ranging from 150 kb to 246 kb, none was shown to have deletions. An example of such analysis with five retrofitted clones of a BAC is shown in Figure 2C. We postulate that the high fidelity observed in in vivo retrofitting resulted from the lack of any contaminating nuclease and the lack of selective transformation of deleted molecules.
Our purpose for BAC retrofitting is to screen for BACs bearing Ssm1, and eventually to clone Ssm1, through ES cell transfection and differentiation. Ssm1 has been finely mapped to distal chromosome 4 between two microsatellite markers (Engler and Storb 2000). Candidate BACs were obtained from C57BL/6 BAC libraries through hybridization with flanking markers and retrofitted to insert PGKneo for transfecting ES cells derived from HRD transgenic mice of DBA/2 background. Retrofitted BACs were transfected into ES cells and stable clones were obtained through G418 selection. Multiple clones from each BAC were analyzed, showing that all had integrated the corresponding BAC (data not shown).
In summary, in vivo BAC retrofitting with pRetroES has been proven to be simple, fast, and of high fidelity. Although it is intended for use in mouse ES cells, we anticipate that PGKneo would also work for certain other cell types. It would also be easy to replace PGKneo with a selection marker more suitable for other cell types with unique restriction sites flanking PGKneo (Fig. 1A). With proper modification of the protocol to fit the 96-well block format currently used for BAC strain culturing and DNA preparation (Kelley et al. 1999), we believe it would be fairly easy to retrofit large numbers of clones or even an entire BAC library, which would be a tremendous asset to functional genomics studies. Lastly, the inserted PGKneo cassette can also be deleted in stably transfected cell lines through transient expression of the cre gene to prevent possible interference to the specific gene expression.
METHODS
The in vitro retrofitting construct, pRetroES-t, was built by inserting a loxP fragment (courtesy of Dr. Kevin Fuller) into the MCS II region of pGT-N28 (New England Biolabs), followed by replacing the M13 and colE1 replication origins with the conditional replication origin oriR6Kγ from plasmid pWM91 (Metcalf et al. 1996). The resulting plasmid was cloned and amplified in JM109λpir (Penfold and Pemberton 1992). In constructing the in vivo retrofitting vector, pRetroES, a self-catalytic, suicidal cre fusion gene, was constructed with the universal plasmid fusion system (UPS, Liu et al. 1998) in the following way: the NcoI/EcoRI fragment from pGST-cre bearing the cre recombinase gene was ligated onto NcoI/BbsI digested pUNI10, Klenow-filled, religated, and transformed into competent JM109λpir. The resulting plasmid was fused with pHB2-GST by using cre recombinase purified from pGST-cre containing E. coli cells as described (Liu et al. 1998) and transformed into competent DH5α cells. To construct the in vivo retrofitting vector, we excised the tac-GST-loxP-cre fusion gene from the fused plasmid and inserted it into pRetroES-t by replacing its loxP. The detailed map of the resulting plasmid pRetroES is shown in Figure 1.
To retrofit BACs, we made competent cells from a miniculture of a BAC in the following way: 2 mL LB containing 15 μg/mL chloramphenicol was inoculated with a single BAC colony and the culture was incubated at 37°C overnight with shaking; 50 μL of the overnight culture was added to 5 mL room temperature LB with chloramphenicol in a 14-mL Falcon tube, and the tube was incubated with shaking at 37°C for exactly 3 h; the culture was then cooled on ice for 10 min and spun in a refrigerated desktop centrifuge at 3500 rpm for 10 min. The following steps were performed in a cold room: the supernatant was removed and the pellet was washed three times with 1 mL ice-cold distilled water and once with 1 mL 10% glycerol in a 1.7-mL Eppendorf tube and then drained carefully. The competent cells were resuspended in 30 μL 10% glycerol and left on ice. After moving back to a room temperature bench, 200 ng of pRetroES was immediately added to the competent cells and electroporated in a 0.1-cm cuvette at 2000 volts and 200 ohms by using a BioRad Gene Pulser. The contents were transferred to a 14-mL Falcon tube after adding 2 mL LB with 15 μg/mL chloramphenicol, and incubated with shaking at 37°C for 90 min. One-tenth of the transformed cells were spread onto an agar plate containing 50 μg/mL ampicillin and 15 μg/mL chloramphenicol and the rest onto a second plate, and the plates were incubated at 37°C for 16 h.
Colony PCR was used to confirm if the double resistant colonies were correct recombinants. We designed one primer on the retrofitting vector and the others on the BAC vector, both close to the loxP site. The primer on pRetroES, Retro-R (R in Fig. 2) has the sequence 5′-ATCGACCGGTAATGCAGGCA. The primer on pBeloBAC11 has a sequence 5′-AGGAAACGACAGCTCAA, which, together with pRetro-R, will amplify a 243-bp fragment from retrofitted BACs constructed with pBeloBAC11. The BAC vector pBAC36e.1 has a wild-type loxP and a mutant form lox511, differing by one nucleotide (Frengen et al. 1999). When retrofitting BACs cloned in pBACe3.6, pRetroES could be integrated into either of these two lox sites. To distinguish the two types of recombinants, primer Retro-R was used in combination with either primer F1 for integrations at the loxP site or primer F2 for those at the lox511 site (Fig. 2A). The sequences for F1 and F2 primers are 5′-TCAGCGTGAGACTACGATTC and 5′-GTTGCTACGCCTGAATAAGTG, and the amplified products are 376 bp and 328 bp, respectively.
DNA from retrofitted BAC clones was prepared with Qiagen Plasmid Maxi Kit by following the manufacturer's suggested protocol for BAC DNA preparation. Transfection of the retrofitted BACs into ES cells was performed with lipofection by using DOTAP lipofection reagent (Roche) following a protocol as described (Bauchwitz and Costantini 1998).
Acknowledgments
We acknowledge the following individuals: Dr. S.J. Elledge for the UPS constructs (through Dr. F.L. Liu); Dr. S.Y. Kim and C.A. Westbrook for pRetroBAC; Dr. B.L. Warner for pWM91; Dr. J.M. Pemberton for JM109 λpir; Dr. K. Fuller for plox11; and Dr. N. Michael for critical reading of the manuscript. The study was supported by NIH grant AI24780. A.L. is supported by a postdoctoral fellowship from the Cancer Research Institute.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL stor@midway.uchicago.edu; FAX (773) 702–3172.
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.159001.
REFERENCES
- Bauchwitz R, Costantini F. YAC transgenesis: A study of conditions to protect YAC DNA from breakage and a protocol for transfection. Biochim Biophys Acta. 1998;1401:21–37. doi: 10.1016/s0167-4889(97)00107-9. [DOI] [PubMed] [Google Scholar]
- Chatterjee PK, Sternberg NL. Retrofitting high molecular weight DNA cloned in P1: Introduction of reporter genes, markers selectable in mammalian cells and generation of nested deletions. Genet Anal. 1996;13:33–42. doi: 10.1016/1050-3862(95)00147-6. [DOI] [PubMed] [Google Scholar]
- Engler P, Storb U. A linkage map of distal mouse chromosome 4 in the vicinity of Ssm1, a strain-specific modifier of methylation. Mamm Genome. 2000;11:694–695. doi: 10.1007/s003350010124. [DOI] [PubMed] [Google Scholar]
- Engler P, Haasch D, Pinkert CA, Doglio L, Glymour M, Brinster R, Storb U. A strain-specific modifier on mouse chromosome 4 controls the methylation of independent transgene loci. Cell. 1991;65:939–947. doi: 10.1016/0092-8674(91)90546-b. [DOI] [PubMed] [Google Scholar]
- Frengen E, Weichenhan D, Zhao B, Osoegawa K, van Geel M, de Jong PJ. A modular, positive selection bacterial artificial chromosome vector with multiple cloning sites. Genomics. 1999;58:250–253. doi: 10.1006/geno.1998.5693. [DOI] [PubMed] [Google Scholar]
- Hartung M, Kisters-Woike B. Cre mutants with altered DNA binding properties. J Biol Chem. 1998;273:22884–22891. doi: 10.1074/jbc.273.36.22884. [DOI] [PubMed] [Google Scholar]
- Hejna JA, Johnstone PL, Kohler SL, Bruun DA, Reifsteck CA, Olson SB, Moses RE. Functional complementation by electroporation of human BACs into mammalian fibroblast cells. Nucleic Acids Res. 1998;26:1124–1125. doi: 10.1093/nar/26.4.1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen JR, Meng A, McFarlane RJ, Paw BH, Zon LI, Smith GR, Lin S. Modification of bacterial artificial chromosomes through χ-stimulated homologous recombination and its application in zebrafish transgenesis. Proc Natl Acad Sci. 1998;95:5121–5126. doi: 10.1073/pnas.95.9.5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley JM, Field CE, Craven MB, Bocskai D, Kim UJ, Rounsley SD, Adams MD. High throughput direct end sequencing of BAC clones. Nucleic Acids Res. 1999;27:1539–1546. doi: 10.1093/nar/27.6.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SY, Horrigan SK, Altenhofen JL, Arbieva ZH, Hoffman R, Westbrook CA. Modification of bacterial artificial chromosome clones using Cre recombinase: Introduction of selectable markers for expression in eukaryotic cells. Genome Res. 1998;8:404–412. doi: 10.1101/gr.8.4.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Li MZ, Leibham D, Cortez D, Elledge SJ. The univector plasmid-fusion system, a method for rapid construction of recombinant DNA without restriction enzymes. Curr Biol. 1998;8:1300–1309. doi: 10.1016/s0960-9822(07)00560-x. [DOI] [PubMed] [Google Scholar]
- Mejia JE, Monaco AP. Retrofitting vectors for Escherichia coli-based artificial chromosomes (PACs and BACs) with markers for transfection studies. Genome Res. 1997;7:179–186. doi: 10.1101/gr.7.2.179. [DOI] [PubMed] [Google Scholar]
- Metcalf WW, Jiang W, Daniels LL, Kim SK, Haldimann A, Wanner BL. Conditionally replicative and conjugative plasmids carrying lacZ α for cloning, mutagenesis, and allele replacement in bacteria. Plasmid. 1996;35:1–13. doi: 10.1006/plas.1996.0001. [DOI] [PubMed] [Google Scholar]
- Muyrers JP, Zhang Y, Testa G, Stewart AF. Rapid modification of bacterial artificial chromosomes by ET- recombination. Nucleic Acids Res. 1999;27:1555–1557. doi: 10.1093/nar/27.6.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan K, Williamson R, Zhang Y, Stewart AF, Ioannou PA. Efficient and precise engineering of a 200 kb β-globin human/bacterial artificial chromosome in E. coli DH10B using an inducible homologous recombination system. Gene Ther. 1999;6:442–447. doi: 10.1038/sj.gt.3300901. [DOI] [PubMed] [Google Scholar]
- O'Shea KS. Embryonic stem cell models of development. Anat Rec. 1999;257:32–41. doi: 10.1002/(SICI)1097-0185(19990215)257:1<32::AID-AR6>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Osoegawa K, Tateno M, Woon PY, Frengen E, Mammoser AG, Catanese JJ, Hayashizaki Y, de Jong PJ. Bacterial artificial chromosome libraries for mouse sequencing and functional analysis. Genome Res. 2000;10:116–128. [PMC free article] [PubMed] [Google Scholar]
- Penfold RJ, Pemberton JM. An improved suicide vector for construction of chromosomal insertion mutations in bacteria. Gene. 1992;118:145–146. doi: 10.1016/0378-1119(92)90263-o. [DOI] [PubMed] [Google Scholar]
- Sheng Y, Mancino V, Birren B. Transformation of Escherichia coli with large DNA molecules by electroporation. Nucleic Acids Res. 1995;23:1990–1996. doi: 10.1093/nar/23.11.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XW, Model P, Heintz N. Homologous recombination based modification in Escherichia coli and germline transmission in transgenic mice of a bacterial artificial chromosome. Nat Biotechnol. 1997;15:859–865. doi: 10.1038/nbt0997-859. [DOI] [PubMed] [Google Scholar]