

Abstract
Muller cells, the principal glia of the retina, play several key roles in normal and various retinal diseases. To date, their direct involvement in retinal innate defense against bacterial pathogens has not been investigated. Here, we show that Muller cells express Toll-like receptor-2 (TLR2), a key sensor implicated in recognizing Gram-positive bacteria. We found that intravitreal injection of TLR2 agonist Pam3Cys (Pam3) and Staphylococcus(S) aureus activated Muller glia in C57BL/6 mouse retina. Similarly, Pam3 or S. aureus elicited the expression of TLR2 and activated the NF-κB and p38 MAPK signaling cascade. Concomitant with the activation of signaling pathways, transcriptional expression and secretion of various pro-inflammatory cytokines (IL-6, TNF-α, & IL-1β), chemokines (IL-8), and antimicrobial peptide (LL-37) were also induced in Muller glia. Importantly, the culture media derived from TLR2-activated Muller glia exhibited robust bactericidal activity against S. aureus. Furthermore, utilization of neutralizing antibody, siRNA, and pharmacological inhibitors revealed that Muller glial innate response to S. aureus is mediated via theTLR2-›NF-κB axis. Collectively, this study for the first time establishes that the retinal Muller glia senses pathogens via TLR2 and contributes directly to retinal innate defense via production of inflammatory mediators and antimicrobial peptides.
INTRODUCTION
Staphylococci are a major constituent of extra-ocular flora and they often gain access to the intraocular compartments due to trauma or ocular surgery, leading to the development of endophthalmitis (1). Among staphylococci, Staphylococcus(S) aureus causes severe endophthalmitis resulting in greatly diminished or complete loss of visual acuity, despite therapeutic intervention (2). The overall incidence of endophthalmitis has been reported to be between 0.056 and 1.3% after cataract surgery (3–5) and as high as 30% after trauma in a rural setting (6). Because staphylococci are the primary organisms associated with postoperative endophthalmitis (POE) and the potential cause for severe vision loss, animal models of experimental staphylococcal endophthalmitis have been developed to investigate the pathogenesis and treatment of this disease (7, 8).
The host-pathogen interaction in the retina has been the subject of extensive research for the last several years (7) and studies have been performed to define the role of various bacterial virulence factors (toxins, cell wall components) in the pathogenesis of endophthalmitis (9). In contrast, very few studies have investigated the host response in this disease (2, 10). This is probably because classically, the retina has been viewed as an “immune-privileged” tissue i.e. when infectious organisms or tumor cells are placed into the eye; the destructive immune response is attenuated, thus allowing for the preservation of vision (11). However, recent new approaches and models have led to the increased understanding of the mechanisms of ocular inflammation and innate immunity that are operative in the abrogation of immune privilege after infection (12). The major protective mechanism which controls the infiltration of inflammatory cells and macromolecules into the posterior segment of the eye is the blood-retina-barrier (BRB) (13). Since, the production of proinflammatory cytokines and chemokines has been shown to contribute to BRB dysfunction in uveitis (14, 15), the increased BRB permeability in endophthalmitis may also be a consequence of pathogen-induced production of proinflammatory mediators (16). Thus, when BRB function is compromised, it will allow the infiltration of immune cells into the retina, resulting in massive inflammation as seen in patients with infectious endophthalmitis (17). This inflammation, if not controlled, could lead to tissue destruction and vision loss (16). How pathogens are recognized and inflammation is initiated in the retina is still not well defined. The retina, being a part of the central nervous system (CNS), must employ its resident glial cells (microglia, Muller glia or astroglia) for initial recognition and response to invading pathogens. We hypothesized that retinal cells use TLRs for early detection and initiation of innate responses, and recently showed that TLR2 is an important component in providing retinal innate defense against S. aureus, by demonstrating that pretreatment of the mice with TLR2 ligands, prevented the development of staphylococcal endophthalmitis (18).
Since we have found that multiple cells (microglia, Muller glia and retinal pigment epithelium) in the retina express TLR2 (18), they should be capable of responding towards S. aureus. To date, the relative contribution of each cell type in retinal innate defense is not known. Muller cells are the most abundant glial cell type in the retina. They span the entire thickness of the retina and have secondary processes that closely wrap around neuronal cell bodies and dendrites. Muller cell gliosis has been proposed to be neuroprotective in the early stages after retinal injury, perhaps reflecting a cellular response to protect the tissue from further damage (19, 20). Muller cells have also been shown to act as modulators of immune and inflammatory responses, by producing proinflammatory cytokines (21). General signs of Muller cell activation are cellular hypertrophy, upregulation of glial fibrillary acidic protein (GFAP) and the intermediate filament, vimentin (22). Although some studies have shown that GFAP is increased in the retina during bacterial endophthalmitis (23) and viral retinitis (24), no study has reported hitherto the direct involvement of Muller glia in pathogen recognition. Thus, the current study was aimed to assess the innate responses of Muller glia to S. aureus or a specific TLR2 ligand. Furthermore, we investigated the effect of TLR2 activation on the innate defense against bacterial growth. Our findings demonstrate an active role of the retinal Muller glia against bacterial infection and that TLR2-activated Muller glia-derived factors possess strong bactericidal properties.
MATERIALS AND METHODS
Bacterial strain and reagents
S. aureus strains RN 6390 & NCTC 8325 were maintained in tryptic soya broth (Sigma-Aldrich, St. Louis, MO). Bacterial lipopeptide Pam3Cys-Ser-(Lys)4 hydrochloride (Pam3Cys), a synthetic lipopeptide that acts as an exclusive TLR2 agonist, was purchased from Invivogen (San Diego, CA). The NF-κB inhibitor Isohelenin and p38 MAP kinase inhibitor SB203580 were purchased from Calbiochem (Gibbstown, NJ). A mouse TLR-2-neutralizing monoclonal antibody (clone 2392) was a gift from Genentech (San Francisco, CA). Anti-phospho-p38 MAPK mAb, anti-p38 antibody, anti-phospho-IκB-α, anti-IκB-α, anti-TLR2 and HSP-90 antibodies were purchased from Cell Signaling Technology (Beverly, MA). A mouse monoclonal anti-β-actin antibody was purchased from Sigma-Aldrich (St.Louis, MO). Polyclonal anti-LL-37 antibody was obtained from PANATecs GmbH (Germany). Secondary horseradish peroxidase (HRP)-conjugated anti-mouse or anti-rabbit IgG antibodies were purchased from Bio-Rad (Hercules, CA).
Cell culture, treatment and transfection
Spontaneously immortalized Human Muller Cell Line (MIO-M1), kindly provided by Dr. G. Astrid Limb were maintained in DMEM (Hyclone, South Logan, UT ) supplemented with L-glutamax (10%), FBS (10%, BioAbChem Inc, Ladson, SC) and antibiotic cocktail in a humidified 5% CO2 incubator at 37°C (25). At the time of treatment, the culture medium was replaced with antibiotic free and growth factor free fresh DMEM without phenol red (Hyclone, South Logan). At the indicated times (as shown in figures), cells were processed for RNA extraction, protein preparation, and the conditioned media were collected for cytokine assays and antimicrobial assays. In blocking experiments, cells were incubated with 10 μg/ml TLR2-neutralizing or Isotype-matched negative control antibodies for 1 h at 37°C, prior to stimulation with ligand or bacteria. In some experiments, cells were pretreated with NF- B and p38 MAP kinase pathway inhibitors for 30 min before challenge.
For siRNA transfection, MIO-M1 cells plated on six-well dishes were transfected with non-specific targeted or TLR2-specific siRNA (Dharmacon, Lafayette, CO) with Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. After 48 h of transfection, cells were kept in DMEM containing L-glutamax (10%) and FBS (10%) overnight, followed by challenge with bacteria.
RNA extraction and RT-PCR analysis
Total RNA was isolated from MIO-M1 cells using the TRIzol solution according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA) and 2 μg of total RNA was reverse-transcribed with a first-strand synthesis system for RT-PCR (SuperScript; Invitrogen). cDNA was amplified using human specific primers (Table 1). The PCR products and internal control GAPDH were subjected to electrophoresis on 1.2 % agarose gels containing ethidium bromide. Stained gels were captured using a digital camera (EDAS 290 system; Eastman Kodak, Rochester, NY) and band intensity was quantified using Image J analysis software (NIH).
Table 1.
Sequences and product sizes of PCR primers
| Gene | Primer Sequence | Product Size (bp) | Temp (°C) |
|---|---|---|---|
| TLR1 | F: TTACTCCCGGAGGCAATGCTGCT R: TAAGTGTGTGGTGCTCAACCCCA |
662 | 55 |
| TLR2 | F: GTGGCCAGCAGGTTCAGGATG R: AGGACTTTATCGCAGCTCTCAG |
641 | 55 |
| TLR6 | F: CCTCAACCACATAGAAACGAC R: CACCACTATACTCTCAACCCAA |
531 | 55 |
| TNF-α | F: GAAAGCATGATCCGGGACGTG R: GATGGCAGAGAGGAGGTTGAC |
510 | 55 |
| IL-1β | F: AAACAGATGAAGTGCTCCTTCCAGG R: TGGAGAACACCACTTGTTGCTCCA |
390 | 55 |
| IL-6 | F: CTCCTTCTCCACAAGCGCCTTC R: GCGCAGAATGAGATGAGTTGTC |
583 | 55 |
| IL-8 | F: GCAGTTTTGCCAAGGAGTGCTA R: GCATCTGGCAACCCTACAACAAG |
347 | 55 |
| LL-37 | F: ATCATTGCCCAGGTCCTCAG R: GTCCCCATACACCGCTTCAC |
251 | 62 |
| GAPDH | F: CACCACCAACTGCTTAGCAC R: CCCTGTTGCTGTAGCCAAAT |
515 | 55 |
Flow cytometry analysis
MIO-M1 cells treated with or without ligands were dispensed in eppendorf tubes and were centrifuged at 100 g at 4°C for 5 min. Cells were then resuspended in PBS containing 1% bovine serum albumin (BSA). The cells were then blocked in 10% serum for 30 minutes at room temperature. The cells were then washed, resuspended in PBS containing anti-TLR2 antibody or Isotype matched IgG (SantaCruz Biotech, Santa Cruz, CA) (1:20 dilution) and were incubated overnight at 4°C. Cells were washed and incubated with the corresponding secondary FITC-conjugated antibody (Invitrogen) (1:200 dilution) for 30 min. A BD LSR II flow cytometer (Immunocytometry Systems, Becton and Dickinson, San Jose, CA) was used for cytometric analysis.
Western and Dot Blot analysis
MIO-M1 cell lysates were prepared with radioimmunoprecipitation assay (RIPA) buffer [150 mM NaCl, 100 mM Tris-HCl, pH 7.5, 1% deoxycholate, 0.1% sodium dodecyl sulphate (SDS), 1% Triton X-100, 50 mM NaF, 100 mM sodium pyrophosphate, 3.5 mM sodium orthovanadate, proteinase inhibitor cocktails (Sigma-Aldrich, St.Louis, MO), and 0.1 mM phenylmethylsulphonyl fluoride (PMSF)], and protein concentration was determined using the BCA assay (micro BCA; Pierce, Rockford, IL). Proteins (30–40 μg/well) were separated by sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS-PAGE) in Tris/glycine/SDS buffer (25 mM Tris, 250 mM glycine and 0.1% SDS) and electro-blotted onto nitrocellulose transfer membranes (Bio-Rad Laboratories, Hercules, CA). After blocking for 1–2 h in phosphate buffered saline with Tween-20 (PBST) (20 mM Tris-HCl, 150 mM NaCl and 0.5% Tween) containing 5% non-fat milk, the blots were probed overnight at 4°C with desired antibodies as described by the manufacturer (Cell Signaling Technology; Sigma Aldrich). NF- B activation was determined in terms of inhibitory I B-α phosphorylation and degradation using anti-IκB-α and antiphospho-IκB-α antibodies. After washing three times in PBST, membranes were incubated with secondary horseradish peroxidase (HRP)-conjugated anti-mouse or anti-rabbit IgG antibodies (Bio-Rad) for 1 h. After washing with PBST four times, for 10 min each, proteins were visualized with Super signal reagents from Pierce (Rockford, IL).
Accumulation of LL-37 in the culture media of TLR2 ligand and bacteria treated MIO-M1 cells was detected by immunoblotting as described previously (26). Briefly, culture media were collected post treatment, centrifuged, and 100 μl was applied to a nitrocellulose membrane (0.2 μm; Bio-Rad) by vacuum using a dot-blot apparatus (Bio-Rad). The membrane was fixed by incubating with 10% formalin for 2 h at room temperature followed by blocking in Tris buffered saline (TBS) containing 5% nonfat powdered milk for 1 h at room temperature. The membrane was then incubated overnight at 4 °C with rabbit anti-LL-37 diluted 1:4000 in TBS containing 5% nonfat powdered milk, 5% goat serum, 0.05% Tween-20, and 0.02% sodium azide. After washing, the membrane was incubated for 1 h at room temperature with goat anti-rabbit IgG conjugated to horseradish peroxidase diluted 1:2000 with 5% nonfat powdered milk. Immunoreactivity was visualized with Super signal reagents from Pierce.
Enzyme linked immunosorbent assay (ELISA) for cytokine analysis
MIO-M1 cells were plated (5 × 106 cells per well) in six-well plates. After growth factor starvation, cells were challenged with Pam3Cys or live/heat killed S. aureus for various times, and culture media were collected for measurement of human TNF-α, IL-1β, IL-6, and IL-8 by ELISA. The cells were lysed and protein concentration was determined. In some experiments prior to stimulation, MIO-M1 cells were pretreated with TLR2 neutralizing antibody or NF-κB/P-38 MAPK pathway inhibitors as described above. ELISAs were performed according to the manufacturer’s instructions (R&D Systems, Minneapolis, MN). The amounts of cytokines in culture media were expressed as picograms per mg of cell lysate. All values were expressed as mean ± standard deviation (SD). Statistical analysis was performed using Student’s t test.
Immunoflourescence staining
Retinal cryosections were rinsed in PBS and blocked for 1 h in blocking buffer [10% (vol./vol.) normal goat serum, 0.3% (vol./vol.) triton X-100 in PBS] at room temperature. The slides were then incubated overnight with anti-GFAP (Dako, Denmark) or anti vimentin (Sigma Aldrich) antibody at 4°C. Following removal of the primary antibodies, slides were extensively washed and incubated for 1 h in fluorescent-conjugated secondary antibodies (FITC) at room temperature. After several washing steps in PBS the slides were mounted in Vectashield anti-fade mounting medium (Vector Laboratories, Burlingame, CA, USA) and visualized using a confocal system (Leica Microsystems, Wetzlar, Germany).
For in vitro staining MIO-M1 cells cultured on glass chamber slides (Fisher Scientific, Rochester, NY) were stimulated with Pam3Cys or live/heat killed bacteria for indicated time periods. Cells were washed three times with phosphate-buffered saline (PBS) and then fixed for 15 min in PBS with 4% paraformaldehyde. After washing with gentle shaking, cells were permeabilized for 5 min with ice-cold methanol and washed. The fixed cells were then blocked in 5% (wt/vol) serum for 1h at room temperature followed by incubation with TLR2 (Santa Cruz) (1:200 dilution), LL-37 (PANATecs GmbH) (1:100 dilution) and vimentin (Sigma-Aldrich) (1:200 dilution) antibodies overnight at 4°C. In control experiments, cells were incubated with non-immune IgG as isotype controls (Santa Cruz, CA; Imgenex, San Diego, CA). Following removal of the primary antibodies, the cells were then extensively washed with PBS and incubated for 1h with specific fluorescein isothiocyanate (FITC)-conjugated secondary antibodies (1: 200 dilution) at room temperature. Finally, the cells were washed extensively with PBS, and the slides were mounted in Vectashield anti-fade mounting medium (Vector Laboratories) and visualized using a confocal system (Leica Microsystems).
Zone of Inhibition Assay
The antibacterial activity of conditioned media derived from control or TLR2 activated MIO-M1 cells was tested against S. aureus strains (RN 6390 and NCTC 8325) by measuring the zone of inhibition (IZD) using disc diffusion method (27). Briefly, bacterial inoculum was prepared from broth cultures and approximately 1 × 106 CFU colony-forming units per milliliter (CFU/ml) were applied as a lawn using plate spreader. The sterile discs soaked with 10μl culture media derived from one million MIO-M1 cells grown in 1 ml media were placed on the surface of bacterial lawn culture. Gentamycin (10 μg/disc) was used as standard antibiotic positive control. The plates were incubated overnight at 37°C and diameters of inhibition zones were measured. P values were calculated using Student’s t test.
RESULTS
S. aureus and TLR2 ligand activate Muller glia in C57BL/6 mouse retina
Glial fibrillary acidic protein (GFAP)-positive staining in Müller cells has been used as a reliable indicator of acute and chronic retinal pathology (19). Under normal conditions Muller glia express GFAP only in their endfeet. But when pathologic events occur, they alter their characteristics to become reactive glial cells and express GFAP in cell bodies and processes. To assess whether TLR2 activation influences GFAP levels in vivo, mice were given intravitreal injection of a TLR2 ligand, Pam3Cys, or S. aureus (SA). The cell bodies and radially oriented processes (arrows in Fig. 1) of the Muller cells were not stained with GFAP antibody in the control, PBS injected mice. In contrast, Muller glial processes became GFAP positive in mice challenged with Pam3Cys or S. aureus. Moreover, the response was concentration-dependent as increasing Pam3Cys dosage increased GFAP expression accordingly in Muller glia processes.
Figure 1. S. aureus and TLR2 agonist enhance GFAP levels in mouse retina.

C57BL/6 mice were given intravitreal injections of PBS (vehicle), TLR2 agonist (Pam3Cys) or S. aureus (SA). After 16 h eyes were enucleated, embedded in OCT and cryosections were stained with rabbit polyclonal anti-glial fibrillary acidic protein (GFAP) antibody. Immunoreactivities are increased in Pam3 or SA challenged retina compared with the PBS controls. Moreover, Muller cell bodies and radial running processes (arrowheads) in the INL and ONL were immunopositive. H & E staining was used as a guide to the retinal layers. GCL, ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer; ELM, external limiting membrane.
S. aureus and Pam3Cys induce TLR2 expression in Muller glia
TLR2 along with TLR1 and TLR6 plays a key role in innate defense against Gram-positive bacteria. To determine whether Muller glia expresses these receptors, RT-PCR analysis was performed using the human retinal Muller glia cell line, MIO-M1. As shown in the figure 2A, compared to the control (untreated) cells, the expression of TLR2 mRNA was significantly increased in both Pam3Cys and S. aureus stimulated cells at 4 h and remained elevated up to 24 h (data not shown). In contrast, the expression levels of TLR1 and TLR6 did not change during the course of the study. We also performed the semi-quantitative densitometric analysis by normalizing induced TLR2 mRNA to internal control GAPDH mRNA. Clearly, stimulation of MIO-M1 cells by both S. aureus and Pam3Cys for 4 h up-regulated TLR2 expression by > 2.5 fold (Fig. 2B). The induced expression of TLR2 at the protein level was detected by Western blotting using an anti-TLR2 antibody (Fig. 2C); the exposure of MIO-M1 cells to S. aureus or Pam3Cys for 4 and 8 h resulted in an increase in the levels of TLR2. To provide further evidence of induced TLR2 expression, we also performed immunohistochemistry (Fig. 2C) and flow cytometry (Fig. 2D) on MIO-M1 cells, which showed that both Pam3Cys and S. aureus stimulation augmented TLR2 expression.
Figure 2. S. aureus and Pam3Cys up-regulate TLR2 expression in cultured retinal Muller glia.

Human retinal Muller glia cell line (MIO-M1) was challenged with Pam3Cys (10 μg/ml), live S.aureus (SA) or heat-killed S.aureus (HKSA) for indicated time points. (A) Total RNA was extracted; reverse-transcribed and subjected to PCR analysis with primers specific for various TLRs and GAPDH as the control. PCR products were separated by electrophoresis and stained with ethidium bromide. (B) The band intensity of PCR products were quantitated by densitometric analysis and presented as relative band intensity of TLR2 vs GAPDH. (C) MIO-M1 cells challenged with Pam3Cys or SA were lysed for Western blotting analysis using anti-TLR2 and β-actin (control) antibodies. (D) Immunostaining and (E) flowcytometry evidence of induced TLR-2 expression in stimulated MIO-M1 cells (see methods for detail). Results are representative of two independent experiments. MFI, mean fluorescent intensity.
S. aureus and Pam3Cys induce NF-κB and MAPK signaling in Muller glia
The expression of TLR2 in Muller glia cells suggests that these cells would be responsive to TLR2 agonist mediated signaling. To test this hypothesis, we measured the ability of Pam3Cys and S. aureus to activate NF-κB and MAPK (Fig. 3). First we challenged the MIO-M1 cells with increasing concentrations of Pam3Cys and showed that these cells were responsive to Pam3Cys at levels as low as 0.01 μg/mL as measured by IκB-α phosphorylation, an indicator of NF-κB activation. Treatment of MIO-M1 cells with 1 or 10 μg/mL Pam3Cys resulted in the maximum phosphorylation of IκB-α (Fig. 3A). A similar concentration-dependent trend was observed on the phosphorylation of p38, a member of the MAPK family.
Figure 3. NF-κB and p38 MAPK signaling is activated in Pam3Cys or S. aureus challenged Muller glia.

MIO-M1cells were stimulated with Pam3Cys (A & B) or live S. aureus (C) for the indicated concentration or time points. The cells were lysed for Western blot analysis using antibodies against phospho-IκB-α (P-IκB) and phospho-p38 (P-p38). Antibodies against non-phosphorylated IκB and p38 were used to detect their total levels and Hsp-90 was used as an equal protein loading control. MIO-M1 cells challenged with Pam3Cys or SA showed significant activation of NF-κB, and p-38, signaling pathways, both in concentration and time-dependent manner. The data shown are representative of duplicate experiments.
Time course studies of the Muller glia response to Pam3Cys (Fig. 3B) and S. aureus (Fig. 3C) were also performed. MIO-M1 cells stimulated with 10 μg/ml Pam3Cys resulted in IκB-α phosphorylation detectable at 15 min, and its levels remained elevated at 90 min post stimulation. Accompanying the increase in IκB-α phosphorylation, IκB-degradation was observed 30 min post stimulation and was maximal at 90 min (Fig. 3B). Time-dependent IκB-α phosphorylation and degradation was also observed in MIO-M1 cells challenged with live S. aureus (Fig. 3C). The time course of p38 phosphorylation was similar to that of IκB-α, with maximal activation detected 90 min after stimulation. Together, these findings demonstrate that TLR2 stimulation of Muller glia induces phosphorylation of IκBα and MAPKs within a similar time-frame.
Muller glia produces inflammatory mediators in response to S. aureus and Pam3Cys challenge
To assess the biological relevance of induced NF-κB and p38 MAPK signaling, we determined the effects of S. aureus and Pam3Cys on the expression and secretion of proinflammatory cytokines using RT-PCR. Compared to the controls (lane marked C), both Pam3Cys and S. aureus induced the expression of IL-6, IL-8, TNF-α, and IL-1β in a time-dependent manner (Fig. 4). The mRNA levels of GAPDH (internal control) remained largely unchanged in both control and stimulated cells (Fig. 4). The protein levels of induced cytokines were assessed by ELISA. Significantly increased amounts of IL-6, IL-8, TNF-α, and IL-1β accumulated in the culture media (Fig. 5) of MIO-M1 cells challenged with Pam3Cys and live or heat-killed S. aureus (HKSA). The response is time-dependent and compared to Pam3Cys, S. aureus (live and heat -killed) appeared to be a stronger inducer of cytokine secretion in MIO-M1 cells. Taken together, these results indicate that the Muller glia is responsive to TLR2 agonists via the expression and production of proinflammatory cytokines and chemokines.
Figure 4. TLR2 activated Muller glia expresses proinflammatory cytokine genes.
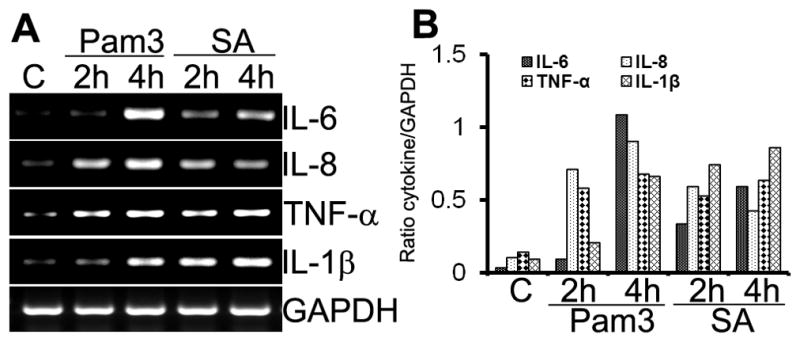
MIO-M1 cells were stimulated with Pam3Cys (10 μg/mL) or live S. aureus (SA, 50 MOI) for the indicated times. (A) Total RNA was extracted; reverse transcribed, and amplified using specific primers (Table 1), with GAPDH as the control. PCR products were separated by electrophoresis and stained with ethidium bromide. (B) The band intensity of PCR products were quantitated by densitometric analysis and presented as relative band intensity of cytokines vs GAPDH. MIO-M1cells expressed the proinflammatory cytokines/chemokines IL-6, IL-8, TNF-α, and IL-β in response to S. aureus and TLR2 agonist. Results shown are representative of three independent experiments.
Figure 5. S. aureus and Pam3Cys induced cytokine secretion in Muller glia.

MIO-M1 cells were stimulated with Pam3Cys (10 μg/ml), live S. aureus (SA) or heat-killed S. aureus (HKSA) for indicated time points. Culture supernatants were used to quantitate indicated cytokines/chemokines accumulation by ELISA and cells were lysed to determine protein concentration. The levels of cytokines are expressed in picogram/milligram (pg/mg) of cell lysate and the results represent data (mean ± SD) from three independent experiments performed in triplicate. Statistical analysis was performed using Student’s t-test and indicated statistical differences are comparisons of stimulated vs unstimulated control (C): *p < 0.05; **p < 0.001.
S. aureus-induced inflammatory response in Muller glia is mediated by TLR2 and NF-κB
Having shown that both Pam3Cys and S. aureus activated downstream signaling pathways and induced the production of inflammatory mediators, we next determined whether TLR2 is required for this response. In the presence of functional blocking antibody (Ab) against TLR2, the production of IL-8 was reduced 62% and 82% in cells challenged with live S. aureus (Fig 6. A) or HKSA (Fig 6. B), respectively. In addition to antibody-mediated inhibition of TLR2 signaling, we also used the siRNA approach to knockdown TLR2 expression. MIO-M1 cells were transfected with TLR2 siRNA or non-targeted (scrambled) siRNA control, and then challenged with S. aureus or HKSA for 8 h. As shown in figure 6C, the siRNA specifically knocked down the TLR2 expression with no effect on the mRNA levels of the housekeeping gene, GAPDH. Consistent with reduced TLR2 expression, siRNA transfected cells secreted significantly reduced levels of IL-8 compared to non-transfected or scrambled siRNA control (Fig 6. A & B). A similar trend was observed for the levels of IL-6 (Fig 6. C & D). These results indicate that functional blocking of TLR2 inhibits S. aureus-induced production of inflammatory mediators by Muller glia.
Figure 6. S. aureus induced inflammatory response is attenuated by inhibition of TLR2 and NF-kB signaling.

MIO-M1 cells were pretreated for 1h with isotype (10 μg/ml), anti-TLR2 (10 μg/ml), p38 MAPK (SB, 5 μM) and NF-κB (IS, 25 μM) pathway inhibitors followed by challenge with (A & C) live S.aureus (SA) or (B & D) heat-killed S.aureus (HKSA). After 8h of stimulation culture supernatants were collected and IL-8 & IL-6 levels were quantitated by ELISA. For siRNA transfection, MIO-M1 cells were transfected with non-targeted siRNA (NT-siRNA) or TLR2-siRNA for 48 h, followed by challenge with SA or HKSA for 8h. TLR2 knockdown was assessed by RT-PCR (E). Data are means ± SD of triplicate cultures and are representative of three independent experiments. Statistical analysis was performed using Student’s t-test and indicated statistical differences are comparisons of isotype vs TLR2 antibody, NT-siRNA vs TLR2-siRNA, and untreated vs inhibitor treated cells: *p < 0.05; **p < 0.001.
TLR2 engagement leads to the activation of both NF-κB and p38 MAPK signaling pathways (Fig. 3). To evaluate the relative contribution of NF-κB and MAPKs signaling, MIO-M1 cells were pretreated with the NF-κB (isohelenin) or p38 MAP kinase (SB203580) inhibitors followed by S. aureus challenge. NF-κB inhibitor effectively blocked both live S. aureus and HKSA-induced production of IL-8 by 85% and 92%, respectively. In contrast, p38 MAPK inhibitor partially attenuated IL-8 secretion by 60% and 70% in Muller glia challenged with live S. aureus (Fig. 6A) or HKSA respectively (Fig. 6B). These results confirm the roles of NF-κB and p38 in S. aureus-induced inflammatory response in Muller glia.
TLR2 activation induces the expression of the antimicrobial peptide LL-37 in Muller glia
Besides inducing proinflammatory mediator release, another consequence of TLR activation is the production of antimicrobial peptides (18). Thus, we next sought to determine the expression and production of LL-37 in TLR2 activated Muller glia. As shown in figure 7A, both Pam3Cys and S. aureus challenge resulted in a time-dependent expression of LL-37 mRNA detected by RT-PCR. Consistent with up-regulation of LL-37 mRNA, the dot-blot assay revealed that the secreted (active) form of LL-37 accumulated in the culture media of stimulated MIO-M1 cells in a time-dependent manner (Fig. 7B). S.aureus induced a rapid (within 1h) increase in LL-37 peptide whereas Pam3 stimulation increased levels of LL-37 after 4 h. In order to further ascertain the expression of LL-37, we performed immunohistochemistry of cultured Muller glia using an anti LL-37 antibody. Consistently, our immunofluorescence data also show the expression and up regulation of LL-37 in Muller glia (Fig. 7C).
Figure 7. S. aureus and Pam3Cys stimulated Muller glia expresses antimicrobial peptide LL-37.

MIO-M1cells were challenged with Pam3Cys (10 μg/ml), live S.aureus (SA) or heat-killed S.aureus (HKSA) for indicated time points. (A)Total RNA was extracted; reverse-transcribed and subjected to PCR analysis with primers specific for LL-37 and GAPDH. PCR products were separated by electrophoresis and stained with ethidium bromide. (B) The secretion of LL-37 in culture supernatant was detected by dot-blot and (C) the intensity of the dots were quantitated by densitometric analysis and presented as fold increase by using a value of one for the control (0h) sample. (D) Immunostaining evidence of induced LL-37 expression in Pam3Cys or HKSA stimulated MIO-M1 cells (see methods for detail). Shown are representative of two independent experiments.
Conditioned medium of TLR2-activated Muller glia possesses bactericidal activity
Since Pam3Cys and S. aureus-challenged Muller glia produce antimicrobial peptides, we speculated that TLR2-activated Muller glia would exhibit more inherent antibacterial activity than unstimulated cells. To test the bactericidal activity of Muller glia conditioned media, we used a qualitative zone of inhibition assay (28). The conditioned media derived from Pam3Cys, HKSA or S. aureus-treated MIO-M1 cells significantly inhibited the growth of two S. aureus strains NCTC 8325 and RN 6390 (Fig. 8). Importantly, stimulation with Pam3Cys exhibited the strongest anti-staphylococcal activity, followed by HKSA and live S. aureus.
Figure 8. Conditioned medium from TLR2 activated Muller glia possess bactericidal activity against S. aureus.
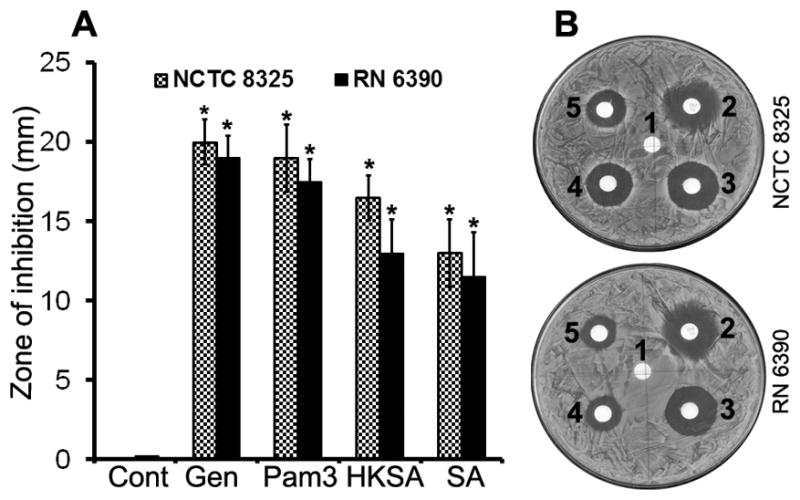
Filter discs, soaked with 10 μl of conditioned media from unstimulated (Cont, disc no.1), Pam3Cys (disc no.3), HKSA (disc no.4) and SA (disc no.5) challenged MIO-M1 cells were placed on the agar plates containing lawn culture of S. aureus strains RN6390 or NCTC 8325. Disc containing 10 μg gentamycin (Gen, disc no.2) was used as a positive control. (A) After overnight incubation, the diameters of inhibition zones around the media-soaked filters were measured and presented as means ± SD (n=5). (B) Representative plates showing inhibition zone. Student’s t-test was used and indicated statistical differences are comparisons of unstimulated (cont) vs TLR2 agonist stimulated cells: *p < 0.001.
DISCUSSION
Muller cells constitute a functional link between neurons and blood vessels, and are responsible for maintaining the homeostasis of the retinal extracellular milieu (29, 30). Despite the important role of Muller glia in various retinal diseases (31), their role in bacterial endophthalmitis is largely unknown. In this study, for the first time we report that Muller glia actively participate in retinal innate defense against bacterial pathogens via the action of Toll-like receptors (TLRs). We demonstrated that S.aureus and TLR2 ligand challenge up-regulate TLR2 expression in Muller glia. Moreover, exposure of Muller glia to S. aureus or Pam3Cys resulted in the activation of NF-κB and p38 MAPK signaling pathways and an increased expression and secretion of various proinflammatory cytokines and chemokines (IL-6, IL-8, TNF-α, and IL-1β). To better understand the involvement of TLRs in the Muller glial innate immune response, we used TLR2-neutralizing antibody and siRNA approaches and showed that the Muller glial innate response to S. aureus is TLR2-dependent. The novel finding in this study is that TLR2-activated Muller glia produced the antimicrobial peptide LL-37 and their conditioned media exhibited profound bactericidal activity against S. aureus. To the best of our knowledge this is the first study to demonstrate this phenomenon.
Three types of glial cells (Muller, astrocytes and microglia) are found in the retina. Compared to other cell types, Muller cells are specialized radial glial cells that span the entire thickness of the retina and contact/ensheath all retinal neurons and capillaries (32). Since the Muller cell endfeet (Fig. 1) lie in the inner limiting membrane (33) next to the vitreous, and in close proximity to invading pathogens, we reasoned that Muller glia should possess the ability to sense and quickly respond to infectious stimuli. Our data demonstrated that intravitreal injection of Pam3Cys or S. aureus enhanced GFAP expression in the mouse retina, an indicative of the responsiveness of Muller glia in vivo. However, it should be noted that retinal astrocytes also express GFAP. Thus, the abundant GFAP immunoreactivity, as seen in the ganglion cell layer (GCL), may also be contributed by astrocytes, due to the intertwining of astrocyte processes with the end-feet of the Muller cells (Fig. 1). Although, the radial pattern of GFAP immunoreactivity is suggestive of retinal Muller cells, to confirm their identity, we also performed additional immunohistochemical studies using an anti-vimentin antibody and found that GFAP co-localizes with vimentin (data not shown). Furthermore, using cultured Muller glial cells, the major finding of our study is that these cells recognize and respond to S. aureus by expressing and secreting proinflammatory cytokines/chemokines and the cathelicidin (LL-37) antimicrobial peptide. LL-37 may eliminate the invading bacteria through their direct antimicrobial action; the cytokines released by the Muller glia may recruit immune cells to the retina. Hence, our findings suggest an active role of Muller glia in innate defense against bacterial infection in the retina.
S. aureus produces a variety of virulence factors which are either cell wall associated molecules or secreted bacterial proteins (toxins), and their coordinated action leads to the manifestation of staphylococcal infections (34). Previous studies by Booth et al showed the importance of the toxins in the pathogenesis of staphylococcal endophthalmitis (35, 36). Since, the toxins are produced during the stationary phase of bacterial growth, the early innate immune response must be initiated by cell-wall associated components (37). Consistent with these studies, our data showed that both Pam3Cys (a synthetic bacterial lipopeptide) and heat-killed S. aureus (HKSA) induced an inflammatory response in Muller glia, indicating the role of cell wall components. How do retinal cells recognize these bacterial cell wall components? Based on previous studies from our lab (26, 38, 39) and others (40–42), we postulated that TLR2 may be a major receptor involved in responses to Gram-positive bacterial infections and demonstrated that Muller glia express TLR2 and its co-receptors TLR1 and 6. Considering the fact that the retina is usually not exposed to the outside environment, one might expect TLRs to be expressed at basal levels in normal retinal cells, and their activation will result in the up-regulation of the same or different TLRs. Indeed, we observed up-regulation of TLR2 expression in Muller glia following S. aureus and TLR2 agonist stimulation. This increased TLR2 expression may facilitate more efficient pathogen recognition and rapid initiation of the innate immune response. Thus, our data suggests that TLR2 expression by Muller glia may be involved in the host defense against S. aureus infection.
TLR2 is an important receptor for the recognition of staphylococcal cell wall components. However, based on the observation that astrocytes or macrophages from TLR2-deficient mice stimulated by S. aureus still produce proinflammatory mediators (42, 43), it was suggested that other receptors might also be involved in S. aureus recognition. In this study we showed that treatment of Muller glia with either TLR2 neutralizing antibody or siRNA knockdown of TLR2 almost completely (80–90%) abrogated the production of inflammatory cytokines induced by HKSA- and Pam3Cys- (data not shown) but not live S. aureus-challenged cells (Fig. 6), suggesting the involvement of other receptors for live S. aureus. One potential candidate receptor could be nucleotide-binding oligomerization domain containing (NOD)-like receptors (NLRs), specifically NOD2, which has been shown to regulate the innate immune response against bacterial pathogens in a TLR-independent manner (44). Moreover, studies have reported the expression of NOD2 in ocular tissues (45) and its role in uveitis (46, 47). Since TLRs are involved in the recognition of pathogens in the extracellular compartment, whereas NLRs detect the microbial ligands intracellularly (48), we propose that TLR2 is the major receptor for detection of S. aureus in Muller glia and the early innate response is generated by this receptor. Whether NOD2 plays a role in the Muller glial innate response towards S. aureus warrants further investigation.
The outcome of host-pathogen interactions frequently leads to the release of pro-inflammatory mediators which play a key role in the recruitment of immune cells to control infection. In addition to the inflammatory response, TLR activation also leads to the production of antimicrobial peptides (AMPs). The two best characterized families of AMPs are defensins (49) and cathelicidins (50, 51). Cathelicidins are constitutively expressed at high levels by neutrophils (52) and their expression at the mucosal surfaces is induced in response to infection or injury (53, 54). In the present study, we showed that stimulation of Muller glia by S. aureus and the TLR2 ligand significantly up-regulated the expression of LL-37 mRNA and protein. Furthermore, detection of the LL-37 (the active secreted form) in the conditioned media of TLR2 activated Muller glia suggests that the mechanism for processing cathelicidin to LL-37 peptide is also activated in Muller glia. This corroborates the results we observed in our recent study whereby intravitreal injection of Pam3Cys induced Camp (murine homolog of human LL-37) expression in mouse retina (18). Here we also showed that the culture media derived from TLR2-activated Muller glia profoundly inhibited S. aureus growth. Since, LL-37 is a multifunctional protein (55), it is not clear at this stage whether the observed bactericidal effect is primarily due to LL-37 or in synergism with other antimicrobial agent (56). To the best of our knowledge this is the first demonstration that retinal Muller glia secretes antimicrobial peptides.
In summary, our findings suggest that in response to Gram-positive bacterial infection, Muller glia cells up-regulate TLR2 expression. The increased expression of TLR2 may result in an increased Muller glial reactivity towards proliferating bacteria in the vitreous, leading to the secretion of antimicrobial peptides that may directly kill the invading pathogen, and the expression of inflammatory mediators may induce infiltration of neutrophil and other immune cell into the retina. Thus, with respect to infectious endophthalmitis, potential prophylactic and/or therapeutic strategies may include enhancement of retinal innate immunity by activating TLR signaling pathways.
Acknowledgments
Grant Support: This work was supported by the National Institutes of Health Grant RO1 EY19888 (to A.K.), Alliance for vision research (to A.K.), Research to prevent blindness (to Department of Ophthalmology) and core vision research grant from National Eye Institute (P30 EY04068).
We thank Dr. G. Astrid Limb (Institute of Ophthalmology and Moorfields Eye Hospital, London, UK) for providing MIO-M1 cells, Drs. Fu-shin Yu, Jia Yin, and Gi Sang Yoon (Kresge Eye Institute) for reading of the manuscript. Authors are also thankful to Jacob Blair and Travis Kochan for providing technical support in this study.
References
- 1.Graham JE, Moore JE, Jiru X, Goodall EA, Dooley JS, Hayes VE, Dartt DA, Downes CS, Moore TC. Ocular pathogen or commensal: a PCR-based study of surface bacterial flora in normal and dry eyes. Invest Ophthalmol Vis Sci. 2007;48:5616–5623. doi: 10.1167/iovs.07-0588. [DOI] [PubMed] [Google Scholar]
- 2.Whiston EA, Sugi N, Kamradt MC, Sack C, Heimer SR, Engelbert M, Wawrousek EF, Gilmore MS, Ksander BR, Gregory MS. alphaB-crystallin protects retinal tissue during Staphylococcus aureus-induced endophthalmitis. Infect Immun. 2008;76:1781–1790. doi: 10.1128/IAI.01285-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Montan PG, Koranyi G, Setterquist HE, Stridh A, Philipson BT, Wiklund K. Endophthalmitis after cataract surgery: risk factors relating to technique and events of the operation and patient history: a retrospective case-control study. Ophthalmology. 1998;105:2171–2177. doi: 10.1016/S0161-6420(98)91211-8. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Y, Zhang MN, Jiang CH, Yao Y, Zhang K. Endophthalmitis following open globe injury. Br J Ophthalmol. 2010;94:111–114. doi: 10.1136/bjo.2009.164913. [DOI] [PubMed] [Google Scholar]
- 5.Javitt JC, Vitale S, Canner JK, Street DA, Krakauer H, McBean AM, Sommer A. National outcomes of cataract extraction. Endophthalmitis following inpatient surgery. Arch Ophthalmol. 1991;109:1085–1089. doi: 10.1001/archopht.1991.01080080045025. [DOI] [PubMed] [Google Scholar]
- 6.Boldt HC, Pulido JS, Blodi CF, Folk JC, Weingeist TA. Rural endophthalmitis. Ophthalmology. 1989;96:1722–1726. doi: 10.1016/s0161-6420(89)32658-3. [DOI] [PubMed] [Google Scholar]
- 7.Callegan MC, Gilmore MS, Gregory M, Ramadan RT, Wiskur BJ, Moyer AL, Hunt JJ, Novosad BD. Bacterial endophthalmitis: therapeutic challenges and host-pathogen interactions. Prog Retin Eye Res. 2007;26:189–203. doi: 10.1016/j.preteyeres.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giese MJ, Mondino BJ. Experimental staphylococcal endophthalmitis. Ophthalmologica. 2001;215:321–336. doi: 10.1159/000050882. [DOI] [PubMed] [Google Scholar]
- 9.Callegan MC, Cochran DC, Kane ST, Ramadan RT, Chodosh J, McLean C, Stroman DW. Virulence factor profiles and antimicrobial susceptibilities of ocular bacillus isolates. Curr Eye Res. 2006;31:693–702. doi: 10.1080/02713680600850963. [DOI] [PubMed] [Google Scholar]
- 10.Engelbert M, Gilmore MS. Fas ligand but not complement is critical for control of experimental Staphylococcus aureus Endophthalmitis. Invest Ophthalmol Vis Sci. 2005;46:2479–2486. doi: 10.1167/iovs.04-1139. [DOI] [PubMed] [Google Scholar]
- 11.Streilein JW. Ocular immune privilege: therapeutic opportunities from an experiment of nature. Nat Rev Immunol. 2003;3:879–889. doi: 10.1038/nri1224. [DOI] [PubMed] [Google Scholar]
- 12.Hazlett LD, Hendricks RL. Reviews for immune privilege in the year 2010: immune privilege and infection. Ocul Immunol Inflamm. 2010;18:237–243. doi: 10.3109/09273948.2010.501946. [DOI] [PubMed] [Google Scholar]
- 13.Gregory M, Callegan MC, Gilmore MS. Role of bacterial and host factors in infectious endophthalmitis. Chem Immunol Allergy. 2007;92:266–275. doi: 10.1159/000099277. [DOI] [PubMed] [Google Scholar]
- 14.Luna JD, Chan CC, Derevjanik NL, Mahlow J, Chiu C, Peng B, Tobe T, Campochiaro PA, Vinores SA. Blood-retinal barrier (BRB) breakdown in experimental autoimmune uveoretinitis: comparison with vascular endothelial growth factor, tumor necrosis factor alpha, and interleukin-1beta-mediated breakdown. J Neurosci Res. 1997;49:268–280. doi: 10.1002/(sici)1097-4547(19970801)49:3<268::aid-jnr2>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 15.Xu H, Chen M, Forrester JV. Para-inflammation in the aging retina. Prog Retin Eye Res. 2009;28:348–368. doi: 10.1016/j.preteyeres.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 16.Moyer AL, Ramadan RT, Novosad BD, Astley R, Callegan MC. Bacillus cereus-induced permeability of the blood-ocular barrier during experimental endophthalmitis. Invest Ophthalmol Vis Sci. 2009;50:3783–3793. doi: 10.1167/iovs.08-3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doshi RR, Arevalo JF, Flynn HW, Jr, Cunningham ET., Jr Evaluating exaggerated, prolonged, or delayed postoperative intraocular inflammation. Am J Ophthalmol. 2010;150:295–304. e291. doi: 10.1016/j.ajo.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 18.Kumar A, Singh CN, Glybina IV, Mahmoud TH, Yu FS. Toll-like receptor 2 ligand-induced protection against bacterial endophthalmitis. J Infect Dis. 2010;201:255–263. doi: 10.1086/649589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karl MO, Reh TA. Regenerative medicine for retinal diseases: activating endogenous repair mechanisms. Trends Mol Med. 2010;16:193–202. doi: 10.1016/j.molmed.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Melo Reis RA, Ventura AL, Schitine CS, de Mello MC, de Mello FG. Muller glia as an active compartment modulating nervous activity in the vertebrate retina: neurotransmitters and trophic factors. Neurochem Res. 2008;33:1466–1474. doi: 10.1007/s11064-008-9604-1. [DOI] [PubMed] [Google Scholar]
- 21.Zong H, Ward M, Madden A, Yong PH, Limb GA, Curtis TM, Stitt AW. Hyperglycaemia-induced pro-inflammatory responses by retinal Muller glia are regulated by the receptor for advanced glycation end-products (RAGE) Diabetologia. 2010 doi: 10.1007/s00125-010-1900-z. [DOI] [PubMed] [Google Scholar]
- 22.Lindqvist N, Liu Q, Zajadacz J, Franze K, Reichenbach A. Retinal glial (Muller ) cells: sensing and responding to tissue stretch. Invest Ophthalmol Vis Sci. 2010;51:1683–1690. doi: 10.1167/iovs.09-4159. [DOI] [PubMed] [Google Scholar]
- 23.Ramadan RT, Ramirez R, Novosad BD, Callegan MC. Acute inflammation and loss of retinal architecture and function during experimental Bacillus endophthalmitis. Curr Eye Res. 2006;31:955–965. doi: 10.1080/02713680600976925. [DOI] [PubMed] [Google Scholar]
- 24.Drescher KM, Whittum-Hudson JA. Evidence for induction of interferon-alpha and interferon-beta in retinal glial cells of Muller. Virology. 1997;234:309–316. doi: 10.1006/viro.1997.8661. [DOI] [PubMed] [Google Scholar]
- 25.Limb GA, Salt TE, Munro PM, Moss SE, Khaw PT. In vitro characterization of a spontaneously immortalized human Muller cell line (MIO-M1) Invest Ophthalmol Vis Sci. 2002;43:864–869. [PubMed] [Google Scholar]
- 26.Kumar A, Zhang J, Yu FS. Toll-like receptor 2-mediated expression of beta-defensin-2 in human corneal epithelial cells. Microbes Infect. 2006;8:380–389. doi: 10.1016/j.micinf.2005.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barry AL, Thornsberry C, Jones RN, Gavan TL. Aztreonam: antibacterial activity, beta-lactamase stability, and interpretive standards and quality control guidelines for disk-diffusion susceptibility tests. Rev Infect Dis. 1985;7(Suppl 4):S594–604. doi: 10.1093/clinids/7.supplement_4.s594. [DOI] [PubMed] [Google Scholar]
- 28.Jorgensen JH, Ferraro MJ. Antimicrobial susceptibility testing: a review of general principles and contemporary practices. Clin Infect Dis. 2009;49:1749–1755. doi: 10.1086/647952. [DOI] [PubMed] [Google Scholar]
- 29.Garcia M, Vecino E. Role of Muller glia in neuroprotection and regeneration in the retina. Histol Histopathol. 2003;18:1205–1218. doi: 10.14670/HH-18.1205. [DOI] [PubMed] [Google Scholar]
- 30.Jadhav AP, Roesch K, Cepko CL. Development and neurogenic potential of Muller glial cells in the vertebrate retina. Prog Retin Eye Res. 2009;28:249–262. doi: 10.1016/j.preteyeres.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bringmann A, Pannicke T, Grosche J, Francke M, Wiedemann P, Skatchkov SN, Osborne NN, Reichenbach A. Muller cells in the healthy and diseased retina. Prog Retin Eye Res. 2006;25:397–424. doi: 10.1016/j.preteyeres.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 32.Bringmann A, Skatchkov SN, Pannicke T, Biedermann B, Wolburg H, Orkand RK, Reichenbach A. Muller glial cells in anuran retina. Microsc Res Tech. 2000;50:384–393. doi: 10.1002/1097-0029(20000901)50:5<384::AID-JEMT7>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 33.Wolburg H, Willbold E, Layer PG. Muller glia endfeet, a basal lamina and the polarity of retinal layers form properly in vitro only in the presence of marginal pigmented epithelium. Cell Tissue Res. 1991;264:437–451. doi: 10.1007/BF00319034. [DOI] [PubMed] [Google Scholar]
- 34.Fournier B, Philpott DJ. Recognition of Staphylococcus aureus by the innate immune system. Clin Microbiol Rev. 2005;18:521–540. doi: 10.1128/CMR.18.3.521-540.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Booth MC, Atkuri RV, Nanda SK, Iandolo JJ, Gilmore MS. Accessory gene regulator controls Staphylococcus aureus virulence in endophthalmitis. Invest Ophthalmol Vis Sci. 1995;36:1828–1836. [PubMed] [Google Scholar]
- 36.Booth MC, Cheung AL, Hatter KL, Jett BD, Callegan MC, Gilmore MS. Staphylococcal accessory regulator (sar) in conjunction with agr contributes to Staphylococcus aureus virulence in endophthalmitis. Infect Immun. 1997;65:1550–1556. doi: 10.1128/iai.65.4.1550-1556.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Callegan MC, Booth MC, Jett BD, Gilmore MS. Pathogenesis of gram-positive bacterial endophthalmitis. Infect Immun. 1999;67:3348–3356. doi: 10.1128/iai.67.7.3348-3356.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li Q, Kumar A, Gui JF, Yu FS. Staphylococcus aureus lipoproteins trigger human corneal epithelial innate response through toll-like receptor-2. Microb Pathog. 2008;44:426–434. doi: 10.1016/j.micpath.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumar A, Zhang J, Yu FS. Innate immune response of corneal epithelial cells to Staphylococcus aureus infection: role of peptidoglycan in stimulating proinflammatory cytokine secretion. Invest Ophthalmol Vis Sci. 2004;45:3513–3522. doi: 10.1167/iovs.04-0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun Y, Hise AG, Kalsow CM, Pearlman E. Staphylococcus aureus-induced corneal inflammation is dependent on Toll-like receptor 2 and myeloid differentiation factor 88. Infect Immun. 2006;74:5325–5332. doi: 10.1128/IAI.00645-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ribeiro CM, Hermsen T, Taverne-Thiele AJ, Savelkoul HF, Wiegertjes GF. Evolution of recognition of ligands from Gram-positive bacteria: similarities and differences in the TLR2-mediated response between mammalian vertebrates and teleost fish. J Immunol. 2010;184:2355–2368. doi: 10.4049/jimmunol.0900990. [DOI] [PubMed] [Google Scholar]
- 42.Takeuchi O, Hoshino K, Akira S. Cutting edge: TLR2-deficient and My D88-deficient mice are highly susceptible to Staphylococcus aureus infection. J Immunol. 2000;165:5392–5396. doi: 10.4049/jimmunol.165.10.5392. [DOI] [PubMed] [Google Scholar]
- 43.Esen N, Tanga FY, DeLeo JA, Kielian T. Toll-like receptor 2 (TLR2) mediates astrocyte activation in response to the Gram-positive bacterium Staphylococcus aureus. J Neurochem. 2004;88:746–758. doi: 10.1046/j.1471-4159.2003.02202.x. [DOI] [PubMed] [Google Scholar]
- 44.Brodsky IE, Monack D. NLR-mediated control of inflammasome assembly in the host response against bacterial pathogens. Semin Immunol. 2009;21:199–207. doi: 10.1016/j.smim.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 45.Rodriguez-Martinez S, Cancino-Diaz ME, Jimenez-Zamudio L, Garcia-Latorre E, Cancino-Diaz JC. TLRs and NODs mRNA expression pattern in healthy mouse eye. Br J Ophthalmol. 2005;89:904–910. doi: 10.1136/bjo.2004.056218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rosenzweig HL, Kawaguchi T, Martin TM, Planck SR, Davey MP, Rosenbaum JT. Nucleotide oligomerization domain-2 (NOD2)-induced uveitis: dependence on IFN-gamma. Invest Ophthalmol Vis Sci. 2009;50:1739–1745. doi: 10.1167/iovs.08-2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rosenzweig HL, Martin TM, Jann MM, Planck SR, Davey MP, Kobayashi K, Flavell RA, Rosenbaum JT. NOD2, the gene responsible for familial granulomatous uveitis, in a mouse model of uveitis. Invest Ophthalmol Vis Sci. 2008;49:1518–1524. doi: 10.1167/iovs.07-1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shaw PJ, Lamkanfi M, Kanneganti TD. NOD-like receptor (NLR) signaling beyond the inflammasome. Eur J Immunol. 2010;40:624–627. doi: 10.1002/eji.200940211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ganz T. Defensins and host defense. Science. 1999;286:420–421. doi: 10.1126/science.286.5439.420. [DOI] [PubMed] [Google Scholar]
- 50.Schauber J, Gallo RL. Antimicrobial peptides and the skin immune defense system. J Allergy Clin Immunol. 2009;124:R13–18. doi: 10.1016/j.jaci.2009.07.014. [DOI] [PubMed] [Google Scholar]
- 51.Zanetti M. Cathelicidins, multifunctional peptides of the innate immunity. J Leukoc Biol. 2004;75:39–48. doi: 10.1189/jlb.0403147. [DOI] [PubMed] [Google Scholar]
- 52.Cowland JB, Johnsen AH, Borregaard N. hCAP-18, a cathelin/pro-bactenecin-like protein of human neutrophil specific granules. FEBS Lett. 1995;368:173–176. doi: 10.1016/0014-5793(95)00634-l. [DOI] [PubMed] [Google Scholar]
- 53.Redfern RL, McDermott AM. Toll-like receptors in ocular surface disease. Exp Eye Res. 2010;90:679–687. doi: 10.1016/j.exer.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bals R, Weiner DJ, Moscioni AD, Meegalla RL, Wilson JM. Augmentation of innate host defense by expression of a cathelicidin antimicrobial peptide. Infect Immun. 1999;67:6084–6089. doi: 10.1128/iai.67.11.6084-6089.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gao N, Kumar A, Jyot J, Yu FS. Flagellin-Induced Corneal Antimicrobial Peptide Production and Wound Repair Involve a Novel NF-κB–Independent and EGFR-Dependent Pathway. PLoS One. 2010;5:e9351. doi: 10.1371/journal.pone.0009351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bals R, Wilson JM. Cathelicidins--a family of multifunctional antimicrobial peptides. Cell Mol Life Sci. 2003;60:711–720. doi: 10.1007/s00018-003-2186-9. [DOI] [PMC free article] [PubMed] [Google Scholar]