

Abstract
We recently identified germline methylation of KILLIN, a novel p53-regulated tumor suppressor proximal to PTEN, in >1/3 Cowden or Cowden syndrome-like (CS/CSL) individuals who are PTEN mutation negative. Individuals with germline KILLIN methylation had increased risks of renal cell carcinoma (RCC) over those with PTEN mutations. Therefore, we tested the hypothesis that KILLIN may be a RCC susceptibility gene, silenced by germline methylation. We found germline hypermethylation by combined bisulfite restriction analysis (COBRA) in at least one of the four CpG-rich regions in 23/41 (56%) RCC patients compared to 0/50 controls (P<0.0001). Of the 23, 11 (48%) demonstrated methylation in the -598bp to -890bp region in respect to the KILLIN transcription start site. Furthermore, 19 of 20 advanced RCC showed somatic hypermethylation upstream of KILLIN, with the majority hypermethylated at more than one CpG island (13/19 vs 3/23 with germline methylation, p<0.0001). qRT-PCR revealed that methylation significantly downregulates KILLIN expression (P=0.05), and demethylation treatment by 5-aza-2’deoxycytidine significantly increased KILLIN expression in all RCC cell lines while only increasing PTEN expression in one line. Furthermore, targeted in vitro methylation revealed a significant decrease in KILLIN promoter activity only. These data reveal differential epigenetic regulation by DNA promoter methylation of this bidirectional promoter. In summary, we have identified KILLIN as a potential novel cancer predisposition gene for nonsyndromic ccRCC, and the epigenetic mechanism of KILLIN inactivation in both the germline and somatic setting suggests the potential for treatment with demethylating agents.
Keywords: Heritable kidney neoplasia, PTEN, KILLIN, DNA methylation
Introduction
Renal cell carcinoma (RCC) is the most common adult-onset kidney cancer and accounts for ~3% of all adult cancers (Banumathy and Cairns 2010). This malignancy claims 102,000 individuals each year, with 210,000 new cases annually diagnosed worldwide and the incidence has been rising over the past 30 years. There are 4 major histological subtypes of RCC including clear cell (~80%), papillary (~15%), chromophobe (5%), and collecting duct (1%) (Banumathy and Cairns 2010). In general, most cases of RCC are sporadic, with smoking, obesity, hypertension, and chemical exposure as the leading environmental causes (Pascual and Borque 2008; Rini and Atkins 2009). As with most solid tumors, hereditary RCCs comprise 3-5% of all RCCs but are still of great biological importance (Pfaffenroth and Linehan 2008). Despite heritable RCC being associated with 10 different autosomal dominant cancer syndromes, there are many RCC's that bear the “red flags” of heredity (young age of onset, bilateral disease, multifocal disease, family history) that are not accounted for by these syndromic predisposition genes.
Amongst syndromes with RCC, von Hippel-Lindau (VHL) disease is the most prominent and well-studied (Coleman 2008), with approximately 50% of VHL patients exhibiting RCCs, in a genotype-specific fashion (truncating mutations) (Lonser et al. 2003; Zbar et al. 1996). This disease affects multiple organ systems and is 90% penetrant by age 5 years (Woodward et al. 2008). Clear cell RCC (ccRCC) is the type of RCC that is unique to VHL, and metastatic ccRCC is the most prevalent cause of death in VHL patients (Kaelin 2007). Germline mutations in the VHL tumor suppressor gene predispose to VHL disease. In VHL-related tumors, such as ccRCC, the remaining wildtype allele is also often affected by somatic loss-of-heterozygosity (LOH) or hypermethylation (Pavlovich and Schmidt 2004). Furthermore, additional somatic mutations or epigenetic modifications of other loci have been identified as well, reflecting the multistep mechanism of RCC pathogenesis {Morris, 2010 #3130}. RCC appears to be a minor component neoplasia of Cowden syndrome (CS) characterized by germline PTEN mutations (Marsh et al. 1998).
PTEN negatively regulates PI3K/AKT, making it and/or its signaling pathway a very likely candidate involved in RCC. Indeed, somatic alterations of PTEN have been identified in a subset of RCCs (Cairns et al. 1998). PTEN is a well-characterized tumor suppressor dual specificity phosphatase that is involved in cellular regulation (Stambolic et al. 1998) via G1 cell cycle arrest and/or apoptosis (Weng et al. 2001). Recently, a newly identified gene, KILLIN, also residing in the 10q23.31 chromosomal region, was shown to be involved in cell cycle arrest and is regulated by p53, similar to PTEN (Cho and Liang 2008). PTEN and KILLIN share the same transcription start site, and presumably are regulated by the same bidirectional promoter, but are transcribed in opposite directions. KILLIN has been shown to be necessary and sufficient for p53-induced apoptosis (Cho and Liang 2008). This high-affinity DNA-binding protein inhibits eukaryotic DNA synthesis in vitro and causes S phase arrest before apoptosis in vivo (Cho and Liang 2008).
We recently identified germline hypermethylation of the KILLIN promoter of the -600 to -900bp region (in respect to the KILLIN translational start site) in CS and CS-like (CSL) patients who are PTEN mutation negative (Bennett et al. 2010). Furthermore, patients with germline methylation and downregulation of KILLIN expression had a higher frequency of renal cell carcinoma than in those with germline PTEN mutations (Bennett et al. 2010). Therefore, we sought to determine whether germline KILLIN promoter hypermethylation and downregulation were also present in apparently non-syndromic RCC patients.
Material and Methods
Patients
Germline DNA samples were obtained from a total of 41 patients with ccRCC, diagnosed between the ages of 20 and 74. Power calculations revealed that a minimum sample size of 20 would achieve P>0.8 if germline KILLIN methylation occurred only in 5% assuming 0% methylation occurred in the germline of population controls, as we have previously shown (Bennett et al. 2010). In addition to the germline samples, we also screened a set of somatic DNA from RCC tissues: 20 ccRCC tumor samples from 19 different individuals. Two of these 20 cases represent primary ccRCC (0302C-146C) and metastatic tumor (0302C150) from the same individual (whose germline DNA SU-55 derives from peripheral leukocytes). Additionally, 6 of these 20 tumors also had paired normal renal tissue. Germline DNA derived from peripheral blood leukocytes from 50 unaffected individuals accrued at the Cleveland Clinic Genomic Medicine Institute served as population controls.
Among the 41 patients in the germline study, 8 had bilateral disease and 33 had unilateral disease. Of the 19 patients in the somatic study, 13 were known to have unilateral disease (the status was unknown for the 6 patients with matched normal tissue). Of all 59 patients, only 1 patient, BA12, had a known history of another cancer (cutaneous melanoma).
All samples were collected in accordance with respective IRB protocols. We were unable to obtain family histories on every research participant due to the constraints of IRB/informed consent rules deriving from one protocol (RG, principal investigator).
Analysis of Germline Hypermethylation of the Bidrectional Promoter at 10q23.13
Combined Bisulfite Restriction Analysis (COBRA) (Bennett et al. 2007) and bisulfite sequencing were performed as previously described (Tada et al. 2006).
Cell Lines and Plasmids
The patient and control cell lines used in this study were generated from renal cell carcinoma patients: RC-6, RC-9, RC-13, and RC-45 were all primary cell lines (Ebert et al. 1990) whereas 786-0, ACHN, CAKI-1, and CAKI-2 were obtained from ATCC (Manassas, VA). Renal cell cancer cell line 786-0 was used for luciferase assay. CAKI-1 and CAKI-2 were maintained in McCoy's media supplemented with 2 mM L-glutamine, nonessential amino acids (1X), 1 mM sodium pyruvate, 10% fetal bovine serum (FBS), and 2% antibiotics. All other cell lines were maintained in RPMI supplemented with 1% L-glutamine, nonessential amino acids (1X), 1 mM sodium pyruvate, and 10% FBS and 2% antibiotics.
The in vitro methylated constructs used for the luciferase assay were generated by first digesting 90μg of the original PTEN and KILLIN promoter constructs (containing 1 to 1344bp upstream of the PTEN translational start site cloned in either direction) with BglII (NEB, Ipswich, MA) and BbvCI (NEB). The linearized, digested inserts and vectors were gel extracted. The insert DNAs, which contain the sequence that is methylated in vivo in CS/CSL patients, were then methylated with CpG SssI methylase (NEB) for 4 hours. Following in vitro methylation, the insert was religated with its corresponding vector using a 3:1 insert to vector ratio with 2μg total DNA. For comparison, the unmethylated counterpart was digested and re-ligated in parallel.
Luciferase Assays
Luciferase assays were performed as previously described using the 786-0 cells (Yu et al. 2004).
Reverse-Transcription Polymerase Chain Reaction (RT-PCR)
The quantitative RT-PCRs were performed as previously described (Bennett et al. 2007).
Demethylation Treatment
Demethylation treatment was performed with a cytosine analog, 5-aza-2’deoxycytidine (5-aza; Sigma), for 96 hours at 0.5 μM concentration with ~40% confluent cells. The drug was changed daily, and the cells collected for RNA isolation.
Statistical Analysis
The statistical significance of the results from qRT-PCR and luciferase assays was calculated by unpaired Student's t test, with P<0.05 being considered statistically significant. Differences in the frequencies of promoter methylation and differences of demographic and clinical characteristics were calculated by Fisher's 2-tailed exact test with alpha<0.05.
Results
Germline Methylation in Renal Cell Carcinoma Patients
We used COBRA to analyze peripheral blood leukocyte-derived germline genomic DNA from 41 ccRCC patients and 50 unaffected individuals for hypermethylation within the bidirectional promoter for PTEN and KILLIN. In order to provide a comprehensive analysis of the methylation status across the CpG islands, we screened four different regions (+102bp to -220bp; -200bp to -400bp; -598bp to -890bp; -1200bp to -1400bp, all with respect to the KILLIN translation start site) [Fig. 1a]. Overall, 23/41 (56%) of the specimens exhibited germline methylation in at least one site analyzed compared to no methylation at any site in all 50 controls (P<0.0001). Of the 23 with germline methylation, 3 (13%) harbored promoter methylation in two or more regions (Fig. 1b, Supplemental Table 1). The most frequent regions harboring germline methylation among the 23 with methylation were at +102bp to -220bp in 10/23 (43%) specimens and -598bp to -890bp in 11/23 (48%), followed by -200bp to -400bp in 3/23 (13%), and -1200bp to -1400bp in 2/23 (9%) [Fig. 1b; Suppl. Fig. 1]. There were no differences in frequencies of germline methylation in those diagnosed under the age of 60 years compared to those over 60 (P=1.0), nor in those with bilateral compared to unilateral disease (P=0.26; 6/8 with bilateral disease have methylation vs 16/33 with unilateral disease with methylation).
Figure 1. Germline and somatic DNA methylation of PTEN/KILLIN regulatory region in RCC patients and tumors.
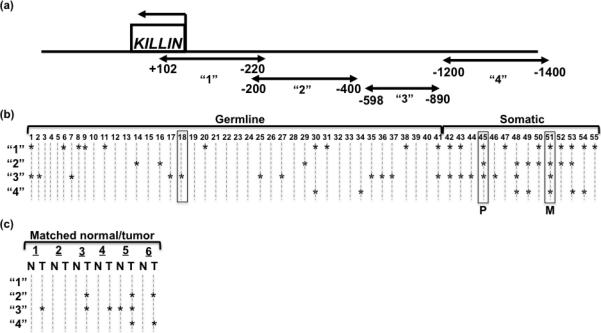
(a) The schematic shows the KILLIN gene in relationship to the methylated regions analyzed depicted by the arrow-ended lines. (b) Combined Bisulfite Restriction Analysis (COBRA) was used on germline DNA from 41 ccRCC patients, 13 primary tumor tissue specimens, one metastatic tumor specimen, and 50 unaffected individuals. From all of the patient specimens, three were matched from one individual: one germline sample (#18), one primary tumor (#45), and one metastatic tumor (#51). (COBRA gels shown in Supplemental Figure 1). The results of the COBRA analysis are displayed in this plot, where “*” indicates methylation in that region for that sample. “1” is the region spanning +102bp to -220bp, “2” is the region spanning -200bp to -400bp, “3” is the region spanning -598bp to -890bp, and “4” is the region spanning -1200bp to -1400bp. Samples enclosed in boxes indicate specimens from the same individual. “P” indicates primary tumor, “M” indicates metastatic tumor. “Germline” specimens are obtained from the leukocytes of ccRCC patients, while “Somatic” DNAs are acquired from the ccRCC tumors. (c) COBRA analysis on 6 paired normal/tumor specimens from ccRCC individuals are displayed in this plot, where “*” indicates methylation in that region for that sample. Regions “1” through “4” are denoted above under (a). “N” represents normal and “T” represents tumor.
Somatic Methylation in Renal Cell Carcinomas
Somatic hypermethylation of the bidirectional promoter was examined in genomic DNA samples from 19 primary ccRCC tumor specimens and 1 metastatic RCC. Of the 20 total tumor samples analyzed, 19 (95%) showed methylation in at least one of the 4 regions (Fig. 1b and c) in contrast to 23/41 (56%) of the leukocyte-derived germline DNA from ccRCC patients (P=0.0025). To determine if methylation frequencies and patterns of normal kidney tissue differed from peripheral leukocyte-derived germline genomic DNA, we analyzed 6 paired ccRCC-normal kidney samples (Fig. 1c). While the sample size is small, it was important to note that normal kidney methylation was no higher than that of peripheral leukocyte-derived DNA.
It is notable that while the germline DNAs rarely exhibited methylation in multiple regions for any given sample (3 of the 23 [13%] with methylation), the somatic DNAs revealed methylation of multiple regions in nearly every sample (13 of the 19 with methylation or 68%, P<0.0001) [Fig. 1b-c]. The 3 specimens that were all from the same individual showed different methylation patterns, such that the germline DNA was methylated for only one region (Fig. 1b, #18), the primary tumor was methylated for three regions (Fig. 1b, #45), and the metastatic tumor was methylated at all four regions (Fig. 1b, #51).
Methylation in RCC Cell Lines
In addition to the patient specimens, a panel of 8 renal cell carcinoma cell lines and one normal kidney cell line was screened for methylation in these 4 regions as well as -470bp to -638bp. The 8 RCC cell lines have varying VHL mutation and/or methylation status (Fig. 2a). Methylation was only detected in the cell lines in the -470bp/-638bp and -598bp/-890bp regions. In the region between -470bp and -638bp, only 3/8 (38%) of the RCC cell lines were methylated (786-0, RC-6, and CAKI-1), and the HEK293T control was completely unmethylated across all tested regions (Figure 2b). Notably, 100% of the RCC cell lines showed methylation at the region spanning -598bp to -890bp, whereas the control cell line HEK293T again revealed no methylation (Fig. 2b).
Figure 2. DNA methylation analysis by COBRA in RCC cell lines.
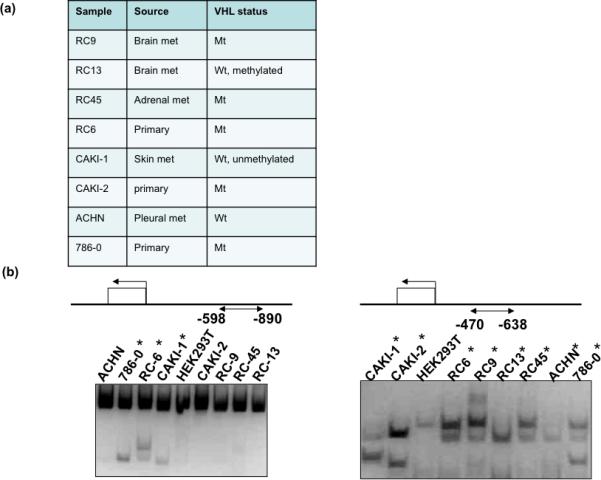
(a) Source and VHL gene status of the 8 RCC cell lines. (b) Schematic demonstrates the region analyzed for methylation by COBRA. COBRA was performed on DNA from 8 RCC cell lines and one kidney cell line used as a control (HEK293T). An increase in the intensity of smaller, digested bands indicates methylation in that tumor DNA. “*” indicates methylation in that RCC tumor specimen.
Methylation and Effect on PTEN and KILLIN Expression
Promoter methylation should result in decreased expression of the relevant gene. In order to validate the pathogenic relevance of this methylation, the expression of PTEN and KILLIN was analyzed in the 8 RCC cell lines and one kidney cell line used as a control for normalization and proof-of-principle. All 8 RCC cell lines showed downregulation of KILLIN, and 7 also showed a decrease in PTEN expression compared to the control line (Fig. 3). Furthermore, the overall extent of downregulation was 100-fold greater for KILLIN compared to that for PTEN (P=0.008, Fig. 3).
Figure 3. Quantitative mRNA analysis of PTEN and KILLIN expression in 8 RCC cell lines.
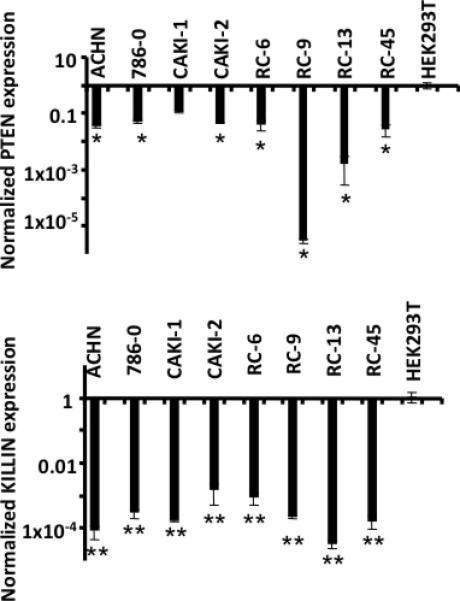
qRT-PCR analysis of 1 control (HEK293T) and 8 RCC cell lines. All samples were first normalized to their own internal control (GAPDH). The control, set to 1, was used for normalization for all samples’ values. The top panel displays the expression for PTEN. The bottom panel reveals significantly decreased KILLIN expression in all patient samples analyzed. * P < 0.05, **P < 8.4 × 10-7.
If, in fact, DNA promoter methylation downregulates KILLIN expression, then demethylation should restore KILLIN expression. Therefore, we investigated whether reversal of DNA methylation would restore KILLIN expression in the cell lines. The RCC cell lines were treated with the demethylating drug 5-aza-2’-deoxycytidine with the exception of the primary culture “RC-6”, which did not grow well enough to perform the demethylation treatment with this cell line. Demethylation increased KILLIN expression in 6/7 of the RCC cell lines, but only increased PTEN expression for 1 of the 7 RCC cell lines (Fig. 4a). The one cell line (RC-45) that did not show a significant increase in KILLIN expression may require additional treatment to reverse other histone modifications to allow for re-expression, or the treatment may have activated potential suppressors of KILLIN. In summary, we have shown that DNA methylation of the bidirectional promoter region appears to downregulate the expression of KILLIN only, which can be reversed by demethylation.
Figure 4. Quantitative mRNA analysis of PTEN and KILLIN expression in 8 RCC cell lines in relationship to methylation.
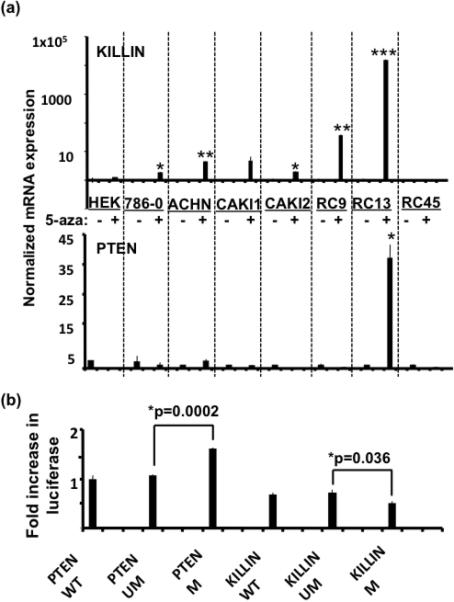
(a) Eight RCC cell lines were subjected to 5-aza-2’-deoxycytidine for 96 hours at 0.5mM concentration. Quantitative RT-PCR analysis was performed on the cDNA from cells with and without drug treatment to detect changes in expression from the demethylation and histone deacetylase inhibition treatment. All values were first normalized to their internal control (GAPDH). The fold-increase or decrease in expression in the drug treated samples is derived by normalizing to its untreated counterpart, which was set as 1. The top panel shows KILLIN expression following demethylation, which shows a significant increase in expression in 7/8 of the cell lines. *P < 0.050, **P<0.002, ***P=2.7×10-11. PTEN expression is shown on the bottom panel and reveals a significant increase in PTEN expression following demethylation in only one cell line. * P=0.020. (b) In vitro methylation with SssI methylase was performed for both the PTEN and KILLIN luciferase promoter constructs. The constructs contained the same promoter sequence (either in orientation for KILLIN or in the opposite orientation for PTEN), which includes -1 to -1344 of sequence upstream of the translation start site of PTEN (-745 to +600 in respect to the KILLIN translation start site). Luciferase promoter analysis of PTEN and KILLIN promoter activity was done using the RCC cell line 786-0. All values were first normalized to their internal control, Renilla luciferase. The fold increase is attained by comparing the methylated constructs to their unmethylated counterparts. The PTEN construct showed a significant increase in transcription following in vitro methylation (* P=0.0002). However, the KILLIN methylated construct showed significantly less activation compared to the unmethylated KILLIN luciferase construct (* P=0.036).
DNA Promoter Methylation Affects KILLIN Promoter Activation
To address whether the methylation seen in the patients directly affects KILLIN and not PTEN transcription, we artificially and purposefully methylated approximately the same CpG region in a PTEN or KILLIN promoter construct. Interestingly, we observed a significant increase in PTEN promoter activity following methylation (1.6-fold, 95% CI 1.58-1.65, P=0.0002), whereas we observed a slight but significant decrease in the level of activation with the methylated KILLIN construct (~0.3-fold, 95% CI 0.22-0.39, P=0.036) [Fig. 4b]. These results again support that the methylation seen in RCC patients and tumors specifically affects the transcriptional activation of KILLIN but not of PTEN.
Discussion
Renal cell carcinoma affects over 100,000 individuals each year with an annual burden of approximately $4.8 billion in the US alone. RCC patients have low survival rates in advanced stages and currently have limited treatment options, making it critical to develop better tools, such as genetic and epigenetic biomarkers, for early diagnosis, risk assessment, and management. Finding mutations in RCC-predisposition genes has been an effective way of cancer risk assessment and management as illustrated by the VHL paradigm. Approximately 3-10% of RCC are heritable, representing 10,000 individuals a year, yet the existing predisposition genes only account for a fraction. In this current study, we have found germline KILLIN promoter hypermethylation in 50% of individuals diagnosed with ccRCC compared to no germline methylation in population controls. Additionally, somatic KILLIN promoter methylation occurred in 95% of the ccRCC analyzed, a frequency which is different from that of the germline. Our current observations suggest that KILLIN may be another RCC-predisposition gene with a different mechanism of inactivation, namely, epigenetic regulation. Although the sample size is relatively small in the somatic series, it is important to note that all tumors harbored methylation in the bidirectional promoter, and at more than one region.
KILLIN is a recently identified gene that had previously only been associated with colon cancer through somatic gene knockout strategies in two colon cancer cell lines (H1299 and DLD-1) (Cho and Liang 2008). Recently, we identified germline KILLIN promoter hypermethylation in 30% of CS and 40% of CSL individuals who did not carry germline PTEN mutations (Bennett et al. 2010). Of relevance, CS/CSL individuals had a two-fold increased prevalence of RCC compared to those with germline PTEN mutations. Taken together with this observation, our current study here supports our hypothesis that KILLIN methylation could be associated with syndromic and non-syndromic RCC. The region of maximal hypermethylation occurs at -600 to -900 region in all 45 individuals with CS/CSL and germline methylation. Interestingly, in the 23 ccRCC patients with germline methylation, 11 have methylation in this same region (-598bp to -890bp, p<0.0001) and 10 in the +102bp to -220bp region. Because CS/CSL patients with germline KILLIN methylation (all at -600 to -900) also have a three-fold increased prevalence of female breast cancer over those with germline PTEN mutation (Bennett et al. 2010), an important but unanswered clinical question is whether women with ccRCC with germline methylation in the -600 to -900 region should also be offered heightened breast surveillance.
In CS/CSL, germline methylation of the region appears to provide effective downregulation by blocking p53 binding and transcriptional activation of KILLIN (Bennett et al. 2010). In those ccRCC patients with similar regional methylation, the mechanism must be similar. Thus, the common denominator of epigenetic regulation in both CS/CSL and ccRCC patients is that the germline methylation effectively downregulates KILLIN rather than PTEN expression. While there was no difference in frequency of germline methylation in those diagnosed before 60 years versus those after, there may be a hint of a trend of different frequencies of germline methylation in individuals with bilateral versus unilateral disease. We also do not have information on a subset of the unilateral cases in regard to their focality, with unilateral multifocal disease being a genetic red flag. Thus, our comparison is likely a conservative one. Overall, it seems worthwhile to formally examine germline KILLIN methyation against clinical red flags in a much larger independent series to identify and validate clinico-pathologic correlations.
We found somatic methylation universally in 18 of 19 primary ccRCC and one metastatic deposit. The striking difference in methylation between the germline and somatic DNAs suggests that the hypermethylation of KILLIN in these patients may indeed impact the pathogenesis of renal cell cancer, and in a subset, germline methylation may confer low penetrance predisposition. Although the sample size is small, it is interesting to note that the patient with the primary RCC and metastatic disease carried germline methylation of one region, the primary tumor three regions and the metastatic lesion all four. This observation is worth pursuing in a larger series which should include leukocyte-derived germline DNA, normal kidney, primary RCC and metastatic deposit.
Targeting angiogenesis alone has proven insufficient for complete suppression of RCC, and genetic and epigenetic markers are now being recognized for their promise in RCC diagnosis as well as predicting therapy response (Banumathy and Cairns 2010). The mechanism of KILLIN inactivation suggests treatment or preventive measures, which include demethylating agents, for those found to be at-risk. Epigenetic therapies in combination with cytokine-based treatment or tyrosine kinase inhibitors may provide a more complete eradication in those who already have developed RCCs. Future clinical trials should interrogate the efficacy of demethylating drugs such as 5-aza-2’deoxycytidine in patients with varied histological RCC subtypes where methylation is prominent, including that of KILLIN. Ultimately, this could yield more understanding to allow for personalized treatment regimens and optimal response in ccRCC patients that meet these criteria.
Supplementary Material
Acknowledgements
We are grateful to members of the Eng lab for thoughtful discussions. This study was funded, in part, by the Breast Cancer Research Foundation, the William Randolph Hearst Foundations, the Healthnetwork Foundation (per generosity of Sharon and Roger Vail), and P01CA124570 from the National Cancer Institute (all to CE). SG is the recipient of an American Cancer Society Joseph F. Silber Undergraduate Student Summer Research Fellowship. CE is the Sondra J. and Stephen R. Hardis Chair of Cancer Genomic Medicine at the Cleveland Clinic, was a Doris Duke Distinguished Clinical Scientist, and is an American Cancer Society Clinical Research Professor, generously funded, in part, by the F.M. Kirby Foundation.
Abbreviations
- CS
Cowden syndrome
- CSL
Cowden-like syndrome
- RCC
renal cell carcinoma
- ccRCC
clear cell renal cell carcinoma
- DNA
deoxyribonucleic acid
REFERENCES
- Banumathy G, Cairns P. Signaling pathways in renal cell carcinoma. Cancer Biol Ther. 2010;10(7) doi: 10.4161/cbt.10.7.13247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett KL, Hackanson B, Smith LT, Morrison CD, Lang JC, Schuller DE, Weber F, Eng C, Plass C. Tumor suppressor activity of CCAAT/enhancer binding protein alpha is epigenetically down-regulated in head and neck squamous cell carcinoma. Cancer Res. 2007;67(10):4657–4664. doi: 10.1158/0008-5472.CAN-06-4793. [DOI] [PubMed] [Google Scholar]
- Bennett KL, Mester J, Eng C. Germline epigenetic regulation of KILLIN in Cowden and Cowden-like syndrome. JAMA. 2010;304(24):2724–2731. doi: 10.1001/jama.2010.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns P, Evron E, Okami K, Halachmi N, Esteller M, Herman JG, Bose S, Wang SI, Parsons R, Sidransky D. Point mutation and homozygous deletion of PTEN/MMAC1 in primary bladder cancers. Oncogene. 1998;16(24):3215–3218. doi: 10.1038/sj.onc.1201855. [DOI] [PubMed] [Google Scholar]
- Cho YJ, Liang P. Killin is a p53-regulated nuclear inhibitor of DNA synthesis. Proc Natl Acad Sci U S A. 2008;105(14):5396–5401. doi: 10.1073/pnas.0705410105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman JA. Familial and hereditary renal cancer syndromes. Urol Clin North Am. 2008;35(4):563–572. v. doi: 10.1016/j.ucl.2008.07.014. [DOI] [PubMed] [Google Scholar]
- Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, Butler A, Davies H, Edkins S, Hardy C, Latimer C, Teague J, Andrews J, Barthorpe S, Beare D, Buck G, Campbell PJ, Forbes S, Jia M, Jones D, Knott H, Kok CY, Lau KW, Leroy C, Lin ML, McBride DJ, Maddison M, Maguire S, McLay K, Menzies A, Mironenko T, Mulderrig L, Mudie L, O'Meara S, Pleasance E, Rajasingham A, Shepherd R, Smith R, Stebbings L, Stephens P, Tang G, Tarpey PS, Turrell K, Dykema KJ, Khoo SK, Petillo D, Wondergem B, Anema J, Kahnoski RJ, Teh BT, Stratton MR, Futreal PA. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463(7279):360–363. doi: 10.1038/nature08672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert T, Bander NH, Finstad CL, Ramsawak RD, Old LJ. Establishment and characterization of human renal cancer and normal kidney cell lines. Cancer Res. 1990;50(17):5531–5536. [PubMed] [Google Scholar]
- Esteller M, Silva JM, Dominguez G, Bonilla F, Matias-Guiu X, Lerma E, Bussaglia E, Prat J, Harkes IC, Repasky EA, Gabrielson E, Schutte M, Baylin SB, Herman JG. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst. 2000;92(7):564–569. doi: 10.1093/jnci/92.7.564. [DOI] [PubMed] [Google Scholar]
- Gemmill RM, Zhou M, Costa L, Korch C, Bukowski RM, Drabkin HA. Synergistic growth inhibition by Iressa and Rapamycin is modulated by VHL mutations in renal cell carcinoma. Br J Cancer. 2005;92(12):2266–2277. doi: 10.1038/sj.bjc.6602646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goelz SE, Vogelstein B, Hamilton SR, Feinberg AP. Hypomethylation of DNA from benign and malignant human colon neoplasms. Science. 1985;228(4696):187–190. doi: 10.1126/science.2579435. [DOI] [PubMed] [Google Scholar]
- Kaelin WG., Jr. The von Hippel-Lindau tumor suppressor protein and clear cell renal carcinoma. Clin Cancer Res. 2007;13(2 Pt 2):680s–684s. doi: 10.1158/1078-0432.CCR-06-1865. [DOI] [PubMed] [Google Scholar]
- Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM, Oldfield EH. von Hippel-Lindau disease. Lancet. 2003;361(9374):2059–2067. doi: 10.1016/S0140-6736(03)13643-4. [DOI] [PubMed] [Google Scholar]
- Marsh DJ, Coulon V, Lunetta KL, Rocca-Serra P, Dahia PL, Zheng Z, Liaw D, Caron S, Duboue B, Lin AY, Richardson AL, Bonnetblanc JM, Bressieux JM, Cabarrot-Moreau A, Chompret A, Demange L, Eeles RA, Yahanda AM, Fearon ER, Fricker JP, Gorlin RJ, Hodgson SV, Huson S, Lacombe D, Eng C, et al. Mutation spectrum and genotypephenotype analyses in Cowden disease and Bannayan-Zonana syndrome, two hamartoma syndromes with germline PTEN mutation. Hum Mol Genet. 1998;7(3):507–515. doi: 10.1093/hmg/7.3.507. [DOI] [PubMed] [Google Scholar]
- Morris MR, Ricketts C, Gentle D, Abdulrahman M, Clarke N, Brown M, Kishida T, Yao M, Latif F, Maher ER. Identification of candidate tumour suppressor genes frequently methylated in renal cell carcinoma. Oncogene. 2010;29(14):2104–2117. doi: 10.1038/onc.2009.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual D, Borque A. Epidemiology of kidney cancer. Adv Urol. 2008:782381. doi: 10.1155/2008/782381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlovich CP, Schmidt LS. Searching for the hereditary causes of renal-cell carcinoma. Nat Rev Cancer. 2004;4(5):381–393. doi: 10.1038/nrc1364. [DOI] [PubMed] [Google Scholar]
- Pfaffenroth EC, Linehan WM. Genetic basis for kidney cancer: opportunity for disease-specific approaches to therapy. Expert Opin Biol Ther. 2008;8(6):779–790. doi: 10.1517/14712598.8.6.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rini BI, Atkins MB. Resistance to targeted therapy in renal-cell carcinoma. Lancet Oncol. 2009;10(10):992–1000. doi: 10.1016/S1470-2045(09)70240-2. [DOI] [PubMed] [Google Scholar]
- Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, Mak TW. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95(1):29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- Tada Y, Brena RM, Hackanson B, Morrison C, Otterson GA, Plass C. Epigenetic modulation of tumor suppressor CCAAT/enhancer binding protein alpha activity in lung cancer. J Natl Cancer Inst. 2006;98(6):396–406. doi: 10.1093/jnci/djj093. [DOI] [PubMed] [Google Scholar]
- Weng LP, Gimm O, Kum JB, Smith WM, Zhou XP, Wynford-Thomas D, Leone G, Eng C. Transient ectopic expression of PTEN in thyroid cancer cell lines induces cell cycle arrest and cell type-dependent cell death. Hum Mol Genet. 2001;10(3):251–258. doi: 10.1093/hmg/10.3.251. [DOI] [PubMed] [Google Scholar]
- Woodward ER, Ricketts C, Killick P, Gad S, Morris MR, Kavalier F, Hodgson SV, Giraud S, Bressac-de Paillerets B, Chapman C, Escudier B, Latif F, Richard S, Maher ER. Familial non-VHL clear cell (conventional) renal cell carcinoma: clinical features, segregation analysis, and mutation analysis of FLCN. Clin Cancer Res. 2008;14(18):5925–5930. doi: 10.1158/1078-0432.CCR-08-0608. [DOI] [PubMed] [Google Scholar]
- Yu L, Liu C, Bennett K, Wu YZ, Dai Z, Vandeusen J, Opavsky R, Raval A, Trikha P, Rodriguez B, Becknell B, Mao C, Lee S, Davuluri RV, Leone G, Van den Veyver IB, Caligiuri MA, Plass C. A NotI-EcoRV promoter library for studies of genetic and epigenetic alterations in mouse models of human malignancies. Genomics. 2004;84(4):647–660. doi: 10.1016/j.ygeno.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Zbar B, Kishida T, Chen F, Schmidt L, Maher ER, Richards FM, Crossey PA, Webster AR, Affara NA, Ferguson-Smith MA, Brauch H, Glavac D, Neumann HP, Tisherman S, Mulvihill JJ, Gross DJ, Shuin T, Whaley J, Seizinger B, Kley N, Olschwang S, Boisson C, Richard S, Lips CH, Lerman M, et al. Germline mutations in the Von Hippel-Lindau disease (VHL) gene in families from North America, Europe, and Japan. Hum Mutat. 1996;8(4):348–357. doi: 10.1002/(SICI)1098-1004(1996)8:4<348::AID-HUMU8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.