

Abstract
Alternative splicing of premessenger RNA is an important layer of regulation in eukaryotic gene expression. Splice variation of a large number of genes has been implicated in various cell growth and differentiation processes. To measure tissue-specific splicing of genes on a large scale, we collected gene expression data from 11 rat tissues using a high-density oligonucleotide array representing 1600 rat genes. Expression of each gene on the chip is measured by 20 pairs of independent oligonucleotide probes. Two algorithms have been developed to normalize and compare the chip hybridization signals among different tissues at individual oligonucleotide probe level. Oligonucleotide probes (the perfect match [PM] probe of each probe pair), detecting potential tissue-specific splice variants, were identified by the algorithms. The identified candidate splice variants have been compared to the alternatively spliced transcripts predicted by an EST clustering program. In addition, 50% of the top candidates predicted by the algorithms were confirmed by RT-PCR experiment. The study indicates that oligonucleotide probe-based DNA chip assays provide a powerful approach to detect splice variants at genome scale.
Alternative splicing is an essential biological process that generates multiple different transcripts from the same precursor mRNA. It is an important regulatory mechanism for high eukaryotic gene expression ( Smith et al. 1989; Lopez 1998; Elliott 2000). It is estimated that at least 35% of human genes undergo alternative splicing during development, cellular differentiation, and other cellular processes (Wolfsberg and Landsman 1997; Mironov et al. 1999; Brett et al. 2000; International Human Genome Sequencing Consortium 2001). Alternative splicing is tightly regulated with temporal and tissue-specific pattern. Some aberrant splicing of precursor transcripts has been associated with various human diseases (Mottes and Iverson 1995; Wilson et al. 1997; Crook et al. 1998; Weissensteiner 1998; Jiang and Wu 1999). Analysis of tissue- and disease-specific splice variations will provide important insights into the molecular mechanism of normal cellular physiology as well as these disease processes.
It has been a daunting task to elucidate the tissue-specific pattern of alternative splicing of tens of thousands of genes using traditional molecular biology approaches. The current knowledge of splice variants in the public database is fragmented. Recent efforts have been made to collect this information from annotated databases (such as SWISSPROT) and expressed sequence tag (EST) databases (Wolfsberg and Landsman 1997; Gelfand et al. 1999). It has been shown that by using a clustering procedure, a rich source of splice variants can be identified from EST sequences (Mironov et al. 1999).
Recent technological advances such as the high-density oligonucleotide arrays allow biologists to study gene expression at genome scale (Chee et al. 1996; Lipshutz et al. 1999). The Affymetrix DNA chip technology is based on hybridization of labeled RNA probes with gene-specific oligonucleotide arrays on the surface of a glass chip. By detecting the intensity of hybridizing probes on the chip, one can analyze the expression level of thousands of genes simultaneously. Because each gene is measured by a number of pairs of oligonucleotide probes spanning the 3′ region of each mRNA, DNA chips offer a unique opportunity to assess 3′ splice variants.
Here we present an exploratory study of predicting alternatively spliced transcripts using primary DNA chip expression data generated from a custom oligonucleotide array of 1600 rat genes in which expression of each gene on the chip is measured by 20 pairs of perfect match and mismatch probes (Chee et al. 1996; Lipshutz et al. 1999). Chip hybridization data were collected from 10 normal rat tissues, including bladder, eye, heart, kidney, large intestine, small intestine, liver, pancreas, placenta, testis, and skeletal muscle. To predict potential tissue-specific splice variants, we have developed algorithms to normalize and then compare the chip hybridization signals at the oligonucleotide probe level. The first algorithm, termed SPLICE, is used to transform raw hybridization signals to normalized values across all the tissues. The algorithm examines tissue-specific expression signals for each probe pair and selects candidate probes (the perfect match [PM] probe of each probe pair). These selected probes represent the initial prediction of probes hitting potential alternative splicing regions. To improve the accuracy of the initial call, we developed a second algorithm called NEIGHBORHOOD to evaluate probes whose sequences are adjacent. The process of the analysis can then be visualized using Spotfire Pro 4.0 software. For validation purposes, we compared the candidate splice variants to the alternatively spliced transcripts predicted by the Compugen LEADS EST clustering program. Some of the top candidates have also been confirmed by RT-PCR experiment.
RESULTS
Workflow of Splice Variant Prediction from DNA Chip Expression Data
As described previously, each probe set on a high-density oligonucleotide array consists of a number of oligonucleotide probes complementary to the 3′ sequences within a target mRNA (Lockhart et al. 1996). A schematic representation of 20 probe pairs aligned to the 3′ sequence of gene X is shown in Figure 1. The average hybridization signal of a probe set reflects the overall abundance of the target mRNA. In addition, the hybridization signal from an individual probe pair correlates with the expression level of the transcript region complementary to that particular probe. This establishes the basis for using an array of multiple oligonucleotide probes to differentiate alternatively spliced transcripts.
Figure 1.

Expression probe layout and alignment with gene sequence. Twenty probe pairs (a probe set) were designed against the 3′ region of each gene. Each probe pair contains a perfect match (PM) and a mismatch (MM) probe. The mutation in the mismatch probe is shown as ×. Representative full-length and alternatively spliced forms of a transcript are indicated.
To predict tissue-specific splice variants from DNA chip expression data, we developed algorithms to normalize chip hybridization data at the single oligonucleotide probe level. Raw intensities of each perfect match (PM) or mismatch (MM) probe were first extracted from the .cel files generated by the Affymetrix system. After subtracting background intensity, a global scaling method was used to normalize the values to each chip experiment. Normalized difference values (PM − MM) and ratio values (PM/MM) can be generated and stored in a combined signal strength (CSS) table. To compare tissue-specific expression of transcripts at the individual probe level, relative signal strength (RSS) of each probe pair was calculated for each tissue by normalizing the PM − MM difference value to the probe itself across all the tissues. RSS value was then converted to a final log ratio (FR) to facilitate comparison of RSS values across different tissues. Based on the FR value, candidate probes hitting potential splice variants in a particular tissue can be predicted using the SPLICE algorithm. To further improve the accuracy of the call and minimize artifacts caused by any single probe pair, we used the NEIGHBORHOOD algorithm to enrich the neighboring probes corresponding to an extended alternative-splicing region. Figure 2 shows a schematic representation of the workflow of data normalization and splice-variant prediction process.
Figure 2.

Schematic representation of the work flow chart of splice variant prediction algorithms. Raw chip hybridization intensities are extracted from Affymetrix. Cel files followed by chip background subtraction and chip intensity normalization. Normalized difference and ratio tables are generated by subtracting mismatch (MM) probe signals from perfect match (PM) probe signals and dividing PM by MM, respectively. Combined signal strength (CSS) table is created by assigning default difference value of zero to probe pairs with corresponding ratio values ≤ 1.2. To normalize expression level across tissues, relative signal strength (RSS) table is generated and followed by converting to final log ratio (FR) to further amplify the difference of relative probe signals across tissues. Candidate probes recognizing potential tissue-specific splice variants are predicted by the SPLICE algorithm. To improve the accuracy of the initial prediction, the NEIGHBORHOOD algorithm is used to assess the relative position of probes on the transcript and to generate a final prediction.
Splice Variant Detection from DNA Chip Data of Three Rat Tissues
To test our algorithm and improve the heuristics used in the prediction, we first collected expression data from RNA of three rat tissues using a custom-designed Affymetrix rat chip, on which each gene is monitored by 20 pairs of 25-mer oligonucleotide probes (Fig. 1). The probes were selected from the 3′ sequence of each gene. RNA samples were extracted from three normal rat heart, liver, and skeletal muscle. From each tissue, three independent probe labeling and chip hybridization experiments were performed. To optimize the prediction algorithms, SPLICE and NEIGHBORHOOD methods were applied to the data set at different selection strengths. Table 1a shows the results of the prediction on the repeated data set of the same tissue. Table 1b shows the results of prediction on the data set of the three different tissues. The triplicate data set (Table 1a) on a single tissue was used as a negative control to tune the parameters in the SPLICE algorithm. By increasing the selection ratio value (R) from 5- to 10-fold, the number of total genes selected from all three tissues using both algorithms (SP + NB) decreases from 20 to nine (Table 1a). However, further increasing of the R-value does not effectively decrease the number of prediction, suggesting that a 10-fold threshold may represent the residual background noise in the data set (see below). In contrast to the predictions from the triplicate samples, the algorithms generated a much greater number of candidates from the data set of different tissues (Table 1b). For example, 69 candidate genes were predicted as compared to nine genes at an R-value of 10 cutoff. The observed difference may represent tissue-specific expression of alternative transcripts. To eliminate background noise and retain prediction sensitivity, R = 10 was used as the default selection strength value for the following predictions in the paper. Other heuristics in the algorithms may also affect the prediction result but in a minor way as compared to the selection ratio (data not shown). The default values we described in Methods have generated consistent prediction results.
Table 1.
Splice Variant Prediction from Three Rat Tissues
| A. | |||||||||||
| Selection ratio (R) | 5-fold | 7-fold | 10-fold | 15-fold | 20-fold | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Tissue type | Algorithm | gene | probe | gene | probe | gene | probe | gene | probe | gene | probe |
| Heart | SP | 183 | 216 | 138 | 165 | 100 | 117 | 60 | 73 | 46 | 58 |
| Heart | SP + NB | 10 | 29 | 8 | 21 | 5 | 13 | 5 | 12 | 5 | 12 |
| Liver | SP | 55 | 67 | 43 | 54 | 22 | 32 | 13 | 21 | 9 | 17 |
| Liver | SP + NB | 3 | 9 | 3 | 9 | 3 | 9 | 3 | 9 | 3 | 9 |
| Skeletal muscle | SP | 126 | 144 | 85 | 92 | 48 | 52 | 26 | 28 | 19 | 21 |
| Skeletal muscle | SP + NB | 8 | 18 | 2 | 4 | 2 | 4 | 1 | 2 | 1 | 2 |
| Total | SP | 328 | 414 | 243 | 303 | 158 | 197 | 93 | 120 | 68 | 94 |
| Total | SP + NB | 20 | 54 | 12 | 32 | 9 | 24 | 8 | 21 | 8 | 21 |
| B. | |||||||||||
| Selection ratio (R) | 5-fold | 7-fold | 10-fold | 15-fold | 20-fold | ||||||
| Tissue type | Algorithm | gene | probe | gene | probe | gene | probe | gene | probe | gene | probe |
| 3 tissues (HLS) | SP | 864 | 2216 | 680 | 1411 | 469 | 819 | 283 | 419 | 208 | 269 |
| 3 tissues (HLS) | SP + NB | 227 | 1192 | 133 | 624 | 69 | 281 | 35 | 114 | 17 | 58 |
(A) Splice variant prediction from triplicate control experiment. Total RNA was extracted from rat heart, liver, and skeletal muscle tissues. Independent RNA labeling and chip hybridization experiments were performed as triplicate for each tissue sample. Potential splice variants were predicted from each set of triplicate data using SPLICE (SP) algorithm alone or in combination with NEIGHBORHOOD (NB) algorithm. Total number of predictions from each tissue set was calculated. (B) Splice variant prediction from three different rat tissues. To generate the data set of three different tissues, the mean CSS value of each tissue triplicate was calculated and appended into the same table. Splice variant predictions were performed using the combined data set from the three tissues.
Example of Predicted Splice Variant—Visualization and Validation
For the candidate transcripts, we searched the rat EST database using the Compugen LEADS program. Figure 3b shows an example of a rat EST cluster. The expression data set of the above three different rat tissues was pivoted and imported into Spotfire Pro 4.0, a visualization software tool that greatly facilitates the analysis and validation of the predictions. To confirm the predicted splice variants, we searched public rat EST database (NCBI release rat113–rat115) using Compugen LEADS 2.0. By mapping the oligonucleotide probe sequences onto the corresponding clusters, some of the splice variant predictions were confirmed. However, it is noted that only a small percentage (7%) of the predictions matches the information in the EST database. This result could be partly attributed to the limited number of rat ESTs in the public databases, and especially the limited tissue-specific EST information. To show the process of data visualization and validation, Figure 3 shows an example of a predicted splice variant and its validation in Compugen LEADS program. Figure 3a shows the visualization of CSS, RSS, and FR values for the transcript of rat phospholipid hydroperoxide glutathione peroxidase (PHGP, L24896). In all three panels, similar patterns of expression across all probes are shown between heart and skeletal muscle, suggesting the same transcript is present in these two tissues. However, the expression pattern in liver is quite different for several probes from that of heart and skeletal muscle. The gap shown in the FR graph indicates a potential alternatively spliced transcript present in liver. To validate the prediction, we examined the EST cluster corresponding to rat phospholipid hydroperoxide glutathione peroxidase (PHGP) gene (Figure 3b). The gap was found in one of the transcripts, suggesting the presence of an alternative spliced form of the gene. Interestingly, the probes found by the algorithms (L24896_55_251 and L24896_55_252) are located in the middle of the alternatively spliced region of the transcript.
Figure 3.

Example of data visualization and validation. (A) Data visualization in Spotfire. Data tables (CSS, RSS, FR) generated from the analysis was pivoted, imported into Spotfire Pro and visualized by 2-D line graphs. The X-axis represent the sequential order of probe pairs on a transcript and the Y-axis represent CSS, RSS, and FR values in each separate panel, respectively. As an example, L24896 is the probe set shown in the figure. Line connections in the graphs are based on tissue types and separately colored. Candidate probes detecting potential tissue-specific splice variants are represented by the gap region in FR panel. (B) Data validation of Compugen LEADS search result. Candidate probe sequences from probe set L24896 were mapped to the rat EST cluster (NCBI release 115). EST clusters were generated by LEADS. Two major alternative transcripts are shown at the top of the cluster RATPHGP. The alternative splicing region is indicated in the second transcript by the gap.
Splice Variant Prediction from 10 Different Normal Rat Tissues
The splice variant prediction method described above is based on the relative gene expression levels among different tissues at the single oligonucleotide probe level. It is reasonable to assume that the more tissue types included in the data set, the more potential splice variants can be detected. To confirm this hypothesis and further test our prediction algorithms, we have collected chip (Rat1600 chip) expression data from 10 different rat tissues, including bladder, eye, heart, kidney, large intestine, small intestine, liver, pancreas, placenta, and testis. By using a selection ratio (R) of 10, the SPLICE plus NEIGHBORHOOD algorithms predicted that a total of 268 out of 1600 genes might have alternatively spliced transcripts with alternative splicing affecting 1218 probes (Table 2). As expected, the numbers are significantly higher than those obtained from three tissues. It shows that potential splice variants can be detected across all tissues analyzed. Moreover, there is a higher chance of detecting potential splice variants in pancreas, testis, placenta, and liver tissues.
Table 2.
Splice Variant Prediction from 10 Normal Rat Tissues
| Bladder | Eye | Heart | Kidney | Large intestine | Small intestine | Liver | Pancreas | Placenta | Testis | Total | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | 113 | 85 | 96 | 123 | 102 | 69 | 143 | 186 | 141 | 168 | 268 |
| Probe | 185 | 129 | 190 | 189 | 134 | 101 | 254 | 433 | 214 | 314 | 1218 |
Total RNA of 10 different rat tissues (bladder, eye, heart, kidney, large intestine, small intestine, liver, pancreas, placenta, and testis) were extracted, labeled, and hybridized to the Rat1600 chip using standard and identical procedures. Individual feature hybridization data were collected and normalized as described in Methods. Potential splice variants were predicted by SPLICE and NEIGHBORHOOD algorithm. The number of predictions for each tissue type was calculated separately. The selection ratio (R) was set at 10 and other default cutoff values were applied.
Table 3 lists the top candidate splice variants predicted from the 10 normal rat tissues. They were selected by both algorithms and ranked by a scoring matrix used in the NEIGHBORHOOD method. The final log ratios of probes of each listed transcript are graphed and visualized in Spotfire Pro 4.0 as shown in Figure 4.
Table 3.
Candidate Probes in Splice Variants Predicted from 10 Normal Rat Tissues
| Probe set | Tissue | FR | Y | X | Probes/cluster | Probes/gene |
|---|---|---|---|---|---|---|
| M26052 | Bladder | −3.754 | 77 | 52–61 | 10 | 10 |
| D13906 | Kidney | 2.398 | 15 | 112–114 | 3 | 3 |
| J03588 | Kidney | −3.106 | 37 | 131–133 | 3 | 3 |
| M32801 | Kidney | −2.941 | 85 | 24–26 | 3 | 3 |
| M32801 | Liver | 2.543 | 85 | 24–26 | 3 | 3 |
| M34007 | Testis | −2.642 | 209 | 213–215 | 3 | 3 |
| X07467 | Liver | −2.9 | 165 | 152–154 | 3 | 3 |
| X62908 | Heart | −2.6 | 179 | 133–135 | 3 | 3 |
| D16478 | Placenta | −3.634 | 17 | 194–195 | 2 | 2 |
| D30035 | Heart | −3.717 | 21 | 225–226 | 2 | 2 |
| K03245 | Kidney | −3.808 | 241 | 96–97 | 2 | 2 |
| K03245 | Liver | 3.842 | 241 | 96–97 | 2 | 2 |
| V01218 | Bladder | −4.33 | 257 | 103–104 | 2 | 2 |
| Z32519 | Testis | −3.334 | 199 | 36–37 | 2 | 2 |
The list contains the top 12 candidates selected from the pool of predicted splice variants in Table 2 based on a decision matrix: i, probes/cluster (from high to low); ii, probes/gene (set equal to probes/cluster); iii, log final ratio FR (>ln10). The identity of each probe set is represented in the first column as Gen Bank accession no.; (Tissue) the tissue type from where the splice variant was predicted; (FR) log final ratio; + and − values represent presence and absence of expression, respectively. X and Y represent coordinates of individual probes on the chip detecting a splice region. Probes/cluster and Probes/gene indicate the number of adjacent probes and the total number of predicted probes for the gene, respectively.
Figure 4.

Visualization of predicted splice variants from 10 normal rat tissues. FR values for predicted probe sets in Table 3 were visualized as 2-D line graphs. In each graph, The X-axis represents the order of the probe set and the Y-axis represents FR value. The tissue types are represented by different shapes as indicated. The type of tissue containing the predicted splice variant is highlighted and the probes are indicated by filled dark rectangles.
Confirmation of Splice Variant Prediction Using RT-PCR Experiment
To further confirm the above predicted splice variants, we performed RT-PCR experiments. Primers were designed based on the sequence of the top candidates containing predicted splice variants (Table 4A). Two pairs of primers (three primers total with one primer shared between the two) were designed for each candidate gene, one pair of primers spanning the potential alternative splicing region, and the other spanning a neighboring nonspliced region. RNA samples from two different rat tissue types were prepared and used for the RT-PCR experiment. A total of four RT-PCR reactions were performed for each candidate. Table 4B shows the comparison of the size of the predicted PCR fragments versus the actual PCR products. Three genes, M32801, M34007, and X07467, showed very good correlation between the predicted and the actual PCR products. The other three genes tested (D13906, J03588, and D30035) showed no correlation (Table 4B). Overall, 50% of the tested candidates confirmed the prediction by the algorithms.
Table 4.
Confirmation of Predicted Splice Variants Using RT-PCR
| A | ||
| Accession | Primer name | Primer sequence |
|---|---|---|
| D13906 | D13906_1S | TCGGTGGAGCTCCACTCTCCT |
| D13906 | D13906_2A | GAAGGAGTCTATGCACTTCTCC |
| D13906 | D13906_3A* | GCTCAGGATCATGCAAACTCTG |
| J03588 | J03588_1S | TCTGAAGAGACCTGGCACACTC |
| J03588 | J03588_2A* | GTGTGATCATCTGAGGGAAGGC |
| J03588 | J03588_3A | AGCCACACTCCAGCTACAAAGG |
| M32801 | M32801_1S* | CATCGGACCTGCCTATGCCATC |
| M32801 | M32801_2S | GTGTGGAGAAGCTGGGAATTCC |
| M32801 | M32801_3A | TCCCACGAAGATGGCTCCTGTA |
| M34007 | M34007_1S | TGATCAGATGGACAACGCCAAG |
| M34007 | M34007_2A* | CACTTCTCAGTGGGTCTTGGAT |
| M34007 | M34007_3A | TTCTGGGTGTATAGTGTATGGTG |
| X07467 | X07467_1S | CTTTGGGACCATAGGCCTTAGC |
| X07467 | X07467_2A | CTCAGGGAAGTGTGGTTTGGTC |
| X07467 | X07467_3A* | TAAGGCTAGTGTGGCTATGGGC |
| D30035 | D30035_1S | GGATTCTCACTTCTGTCATCTGGC |
| D30035 | D30035_2S* | CAAGCGCACCATTGCTCAGGAT |
| D30035 | D30035_3A | TTTCTTCTGGCTGCTCAAAGCTG |
| B | |||||||
| Accession | Primer pair | Tissue 1 | Tissue 2 | ||||
|---|---|---|---|---|---|---|---|
| name | predicted (bp) | PCR (bp) | name | predicted (bp) | PCR (bp) | ||
| D13906 | D13906_1S, D13906_2A | Kidney | 350 | ∼350 | Placenta | 350 | ∼350 |
| D13906 | D13906_1S, D13906_3A | Kidney | 450 | ∼450 | Placenta | no signal | ∼450 |
| J03588 | J03588_1S, J03588_2A | Liver | 274 | ∼270 | Kidney | no signal | ∼270 |
| J03588 | J03588_1S, J03588_3A | Liver | 360 | ∼380 | Kidney | <380 | ∼380 |
| M32801 | M32801_1S, M32801_3A | Liver | 401 | ∼400 | Kidney | no signal | no signal |
| M32801 | M32801_2S, M32801_3A | Liver | 291 | ∼290 | Kidney | 291 | ∼290 |
| M34007 | M34007_1S, M34007_2A | Testis | no signal | no signal | Bladder | 424 | ∼425 |
| M34007 | M34007_1S, M34007_3A | Testis | <542 | ∼420 | Bladder | 542 | ∼550 |
| X07467 | X07467_1S, X07467_2A | Sm Intestine | 330 | ∼330 | Liver | 330 | ∼330 |
| X07467 | X07467_1S, X07467_3A | Sm Intestine | 410 | ∼400 | Liver | no signal | no signal |
| D30035 | D30035_1S, D30335_3A | Kidney | 410 | ∼400 | Heart | <410 | ∼400 |
| D30035 | D30035_2S, D30035_3A | Kidney | 324 | no signal | Heart | no signal | weak signal |
(A) The list of PCR primers and their corresponding gene (Accession). An asterisk indicates that the primer is located within the predicted alternative splicing region. All primer sequences are from 5′ to 3′. (B) Comparison of predicted and actual RT-PCR results. Two pairs of primers were used for each gene. For each gene, a pair of different tissue samples was chosen to perform the RT-PCR experiment. The Predicted column indicates the predicted size of PCR product (or no signal) based on the Splice Variant Prediction Algorithms. The PCR column shows the size of the actual RT-PCR product (or no signal) revealed by the experiment. The genes with correlated result between predicted and actual PCR are highlighted in bold.
DISCUSSION
Alternative mRNA splicing plays an important role in regulating eukaryotic qualitative gene expression, although few approaches are available to analyze alternative splicing of genes on a genome-wide scale. In this paper, we described a novel method to predict tissue-specific splice variants using large data sets generated by Affymetrix Genechips.
Recent advances in DNA chip technology provide great opportunities to study global gene expression in depth. Because each gene is represented by multiple oligonucleotide probes on a chip, a probe-by-probe mapping of the expression of a transcript can be conducted so that tissue-specific differential expression of splice variants can be detected. Based on the hypothesis, we developed algorithms to predict potential splice variants from the chip data. From the expression data of three different rat tissues, we have predicted that ∼4.5% (69 out of 1600) of the genes on the chip contain potential splice variants. Because this is a prediction from expression data of only three tissues, it is likely an underestimate of the actual number of genome-wide splice variants. For example, expression data from 10 rat tissues predicted a significantly greater number of potential splice variants (17%). Some recent studies based on EST clustering data suggest that 35%–40% of mammalian genes contain alternative splicing (Wolfsberg and Landsman 1997; Mironov et al. 1999; Brett et al. 2000). However, the number of human genes containing splice variants involving 3′ exons is believed to be much lower (Mironov et al. 1999). Because of limitations on the probe labeling process, current probe selection for the DNA chips is biased toward the 3′ portion of a gene, and therefore, we can only assess the status of alternative splicing in the 3′ region (usually ∼600 bp upstream of poly-A signal). The methods described in this paper can be applied easily to the expression data generated by 5′ probes when they become available. To effectively analyze alternative splicing across the whole gene, probes need to be selected that encompass a greater length of the transcript. Similar algorithms can be applied to data obtained from oligonucleotide-based microarray technology.
Here we have shown that 50% of the top predictions can be confirmed by a RT-PCR experiment. Because RT-PCR experiment is an extremely sensitive assay, one of the explanations for the three failure cases is that the nucleic acid hybridization-based chip assay is not sensitive enough to detect low abundance, minor splice variants. Alternatively, some of the nonconfirmed cases can be attributed to complicated splicing patterns in the tissues investigated.
The accuracy of the results predicted by the algorithms depends on several factors, the most important being data consistency and reproducibility. Sample variation is a major contributor to error rate (data not shown) and is usually caused by differences in tissue handling and RNA extraction protocols. To ensure consistency in sample preparation, a highly repeatable tissue preparation and RNA extraction procedure needs to be used. RNA labeling and chip hybridization processes can also introduce variations, although the data generated from the triplicate experiments suggest that the variations from independent labeling and hybridization processes can be minimized by following strict protocols. To further reduce data inconsistency, dual color experiments may prove to be a powerful approach to assess subtle transcript differences in DNA chip experiment (Chee et al. 1996; Hacia et al. 1996). The size of the data set also contributes to the effectiveness of splice variant prediction. Theoretically, the more tissue types (or samples from different developmental stages) included in the study, the more splice variants that can be detected. This is shown by the significant increase of predicted potential splice variants in 10 rat tissues as compared to those from three tissues.
Better chip design will dramatically improve the accuracy of splice variant prediction and increase the usefulness of the technique. The background noise encountered during the current prediction can be attributed partly to the physical defects on the chip, such as scratches or debris from manufacturing. By introducing duplicate or triplicate probes on the chip and using a probe scrambling technique, the data variations from those defects can be nearly eliminated. Better probe selection based on improving EST cluster information may greatly improve the efficiency of splice variant detection. Ideally, the selected oligonucleotide probes should be derived from as many different alternative transcripts as possible and evenly distributed across the overall length of the transcript. The ability to design such probes depends heavily on a comprehensive EST cluster database with a large collection of tissue-specific transcript information. Expansion of current public and private EST projects should eventually help reach this goal. At last, a robust probe selection algorithm will help design the next generation of DNA chips, including tissue-specific splice variant detection chips.
Conclusions
Alternative splicing has proved to be a critical part of gene regulation. Different splice variants provide a fresh source of target identification in future drug discovery and clinical diagnosis. Here we described a novel approach for studying alternative splicing of genes at a global scale by using DNA chip technology. We have developed algorithms to effectively predict potential splice variants from chip expression data. Future efforts to collect highly consistent data from a large number of tissue samples will help refine the algorithms. The work will also provide guidance for future tissue and/or transcript-specific DNA chip design.
METHODS
Sample Preparation and Hybridization on Affymetrix GeneChips
Total RNA from normal rat bladder, eye, heart, kidney, large intestine, small intestine, liver, pancreas, placenta, testis, and skeletal muscle was extracted using TRIZOL reagent (Life Technologies). Transcript integrity was monitored using denaturing agarose gel electrophoresis in 1X MOPS. Double-stranded cDNA was prepared from 15 μg of total RNA using a modified oligo-dT primer with a 5′ T7 RNA polymerase promoter sequence and the Superscript Choice System for cDNA Synthesis (Life Technologies). Following phenol-chloroform extraction and ethanol precipitation, one-half of the cDNA reaction (0.5–1.0 μg) was used as template in an in vitro transcription reaction (BioArray High Yield Kit, ENZO) containing T7 RNA polymerase, a mixture of unlabeled ATP, CTP, GTP, and UTP, and biotin-11-CTP and biotin-16-UTP. The resulting cRNA was purified on an affinity resin (RNeasy, QIAGEN) and quantified using the convention that 1 O.D. 260 corresponds to 40 μg/mL of RNA. Randomly fragmented were 15 μg of biotinylated cRNA to an average size of 50 nt by incubating for 35 min at 94°C in 40 mM TRIS-acetate at pH 8.1, 100 mM potassium acetate, and 30 mM magnesium acetate. The fragmented cRNA was hybridized for 16 h at 45°C on a custom Affymetrix GeneChip containing probes for 1600 individual rat genes in a solution containing 100 mM MES, 1 M [Na+], 20 mM EDTA, 0.01% TWEEN 20, 50 pM of Control Oligonucleotide B2 (Affymetrix), 0.1 mg/mL of sonicated herring sperm DNA, and 0.5 mg/mL BSA. Each hybridization included a mixture of four bacterial biotinylated-RNA transcripts (BioB, BioC, BioD, and cre) spiked at 1.5, 5, 25, and 100 pM, respectively. The hybridization reactions were processed and scanned according to standard Affymetrix protocols.
Individually repeated RNA preparation and chip hybridization experiments were performed for three normal rat tissue samples: heart, liver, and skeletal muscle.
Preprocessing of Data
The detailed workflow is shown in Figure 2. After chip scanning, raw intensity of each PM or MM probe on the chip is extracted from the .cel file generated by the Affymetrix software. To eliminate noise from background hybridization, the average intensity of the lowest 2% of the probe signals of each experiment is used as background noise and subtracted from each probe signal on that chip. To further normalize signals across different chips, global scaling is performed for each chip. A normalized difference table is then created by subtracting each MM signal from its corresponding PM signal. Similarly, a normalized ratio table can be generated by dividing the PM and MM signals of each probe pair. To combine the two tables, a default PM − MM difference value of zero is assigned for probe pairs with a PM/MM ratio ⋜ 1.2. The resulting difference table is called combined signal strength (CSS) table.
SPLICE Algorithm
To compare tissue-specific expression of each gene at probe level, the signal of each probe pair needs to be normalized across tissues. A tissue-specific relative signal strength (RSS) table is calculated from the CSS table. The formula of the conversion is:
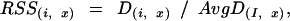 |
in which RSS(i, x) represents the relative signal strength value of probe pair i within probe set I in tissue X. D(i, x) is the PM − MM difference value of probe pair i in tissue X from the CSS table. AvgD(I, x) is the trimmed mean PM − MM difference value of all probe pairs of probe set I in tissue X.
To simplify the calculation and reduce outlier effects, several cutoff thresholds are used in the normalization. Min Diff and Max Diff are the minimum difference and maximum difference cutoff; the default is 20 and 5000, respectively. Signals that are above or below the cutoffs are replaced by the cutoff values. After applying the Min and Max cutoffs on the CSS table, the average difference of each probe set in each tissue [AvgD(I, x)] can be calculated, as well as the average difference of each probe pair across different tissues [AvgDi]. For noninformative probe threshold (NIPT) functions to take away the probe pairs with no or very low expression in all the tissues collected, the default is set at AvgDi > 30. To consider the situations in which there is no or extremely low expression of a gene in a particular tissue, a noninformative tissue type threshold (NITT) is used to eliminate those tissues from the prediction. The default value is AvgD(I, x) > 30. For cases in which a few probes give strong hybridization signals in comparison with the rest of the probe set, a single probe threshold (SPT) is used to differentiate the signals from otherwise noninformative probe set. The default value for SPT is set at 200.
After obtaining tissue-specific relative signal strength for each probe pair, the expression signal of each probe pair of the gene can be compared among different tissues. To capture and amplify the difference across tissues, we further convert the RSS value of each probe pair to a log final ratio that reflects the relative strength of the probe pair among those tissues. The formula for the conversion is:
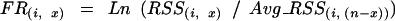 |
in which FR(i, x) is the final log ratio of probe i in tissue X. RSS(i, x) represents the relative signal strength value of probe pair i in tissue X. Avg_RSS(i, (n-x)) is the average RSS value of probe pair i in all tissues except tissue X.
The FR value is used for splice variant prediction. Probes# with absolute FR value greater than a defined ln(R) in a particular tissue are selected as candidate probes from that tissue. R is the selection ratio, the default is set at 10.
NEIGHBORHOOD Algorithm
To improve the accuracy of splice variant prediction by the SPLICE algorithm, we considered relative location of the selected probes on a gene. The assumption is that an alternatively spliced region on a gene is large enough to contain two or more adjacent probes#. The 20 oligonucleotide probe pairs for each gene were aligned so that they correlate to the physical locations of those probes matching 5′ to 3′ orientation of the gene. For the probes selected by the SPLICE algorithm, their relative locations on the gene are assessed so that singleton probes or nonadjacent probes can be filtered out. Two of the parameters used in the algorithm are probes/gene (number of identified candidate probes per gene or per probe set; 3 is default) and probes/cluster (number of identified adjacent probes, 2 is default). The adjacent probes survived the selection by the Neighborhood algorithm represent potential extended regions of alternative splicing.
RT-PCR Experiment
PCR primers were designed from the sequence of the gene fragments containing predicted splice variants. The oligonucleotide primers were synthesized by Operon Technologies. Total RNAs were extracted from normal rat tissues using TRIZOL reagent (Life Technologies). Standard RT-PCR experiments were performed using SuperScript One-Step RT-PCR System (Life Technologies) as described by the manufacturer. PCR products were separated by standard agarose gel electrophoresis and visualized under ultraviolet (UV) light after staining with ethidium bromide.
The data and the algorithms in this work are available from the authors on request.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL KenGang.Hu@pfizer.com; FAX (734) 622-1468.
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.165501.
REFERENCES
- Brett D, Hanke J, Lehmann G, Haase S, Delbruck S, Krueger S, Reich J, Borka P. EST comparison indicates 38% of human mRNAs contain possible alternative splice forms. FEBS Lett. 2000;474:83–86. doi: 10.1016/s0014-5793(00)01581-7. [DOI] [PubMed] [Google Scholar]
- Chee M, Yang R, Hubbell E, Berno A, Huang XC, Stern D, Winkler J, Lockhart DJ, Morris MS, Fodor SPA. Accessing genetic information with high-density DNA arrays. Science. 1996;274:610–614. doi: 10.1126/science.274.5287.610. [DOI] [PubMed] [Google Scholar]
- Crook R, Verkkoniemi A, Perez-Tur J, Mehta N, Baker M, Houlden H, Farrer M, Hutton M, Lincoln S, Hardy J, et al. A variant of Alzheimer's disease with spastic paraparesis and unusual plaques due to deletion of exon 9 of presenilin 1. Nat Med. 1998;4:452–455. doi: 10.1038/nm0498-452. [DOI] [PubMed] [Google Scholar]
- Elliott DJ. Splicing and the single cell. Histol Histopathol. 2000;15:239–249. doi: 10.14670/HH-15.239. [DOI] [PubMed] [Google Scholar]
- Gelfand MS, Dubchak I, Dralyuk I, Zorn M. ASDB: Database of alternatively spliced genes. Nucleic Acids Res. 1999;27:301–302. doi: 10.1093/nar/27.1.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacia JG, Brody LC, Chee MS, Fodor SPA, Collins FS. Detection of heterozygous mutations in BRCA1 using high density oligonucleotide arrays and two-colour fluorescence analysis. Nat Genet. 1996;14:441–447. doi: 10.1038/ng1296-441. [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Jiang ZH, Wu JY. Alternative splicing and programmed cell death. Proc Soc Exp Biol Med. 1999;220:64–72. doi: 10.1046/j.1525-1373.1999.d01-11.x. [DOI] [PubMed] [Google Scholar]
- Lipshutz RJ, Fodor SPA, Gingeras TR, Lockhart DJ. High density synthetic oligonucleotide arrays. Nat Genet Suppl. 1999;21:20–24. doi: 10.1038/4447. [DOI] [PubMed] [Google Scholar]
- Lockhart DJ, Dong H, Byrne MC, Follettie MT, Gallo MV, Chee MS, Mittmann M, Wang C, Kobayashi M, Horton H, et al. Expression monitoring by hybridization to high-density oligonucleotide arrays. Nat Biotechnol. 1996;14:1675–1680. doi: 10.1038/nbt1296-1675. [DOI] [PubMed] [Google Scholar]
- Lopez AJ. Alternative splicing of pre-mRNA: developmental consequences and mechanisms of regulation. Annu Rev Genet. 1998;32:279–305. doi: 10.1146/annurev.genet.32.1.279. [DOI] [PubMed] [Google Scholar]
- Mironov AA, Fickett JW, Gelfand MS. Frequent alternative splicing of human genes. Genome Res. 1999;9:1288–1293. doi: 10.1101/gr.9.12.1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mottes JR, Iverson LE. Tissue-specific alternative splicing of hybrid Shaker/lacZ genes correlates with kinetic differences in Shaker K + currents in vivo. Neuron. 1995;14:613–623. doi: 10.1016/0896-6273(95)90318-6. [DOI] [PubMed] [Google Scholar]
- Smith CW, Patton JG, Nadal-Ginard B. Alternative splicing in the control of gene expression. Annu Rev Genet. 1989;23:527–577. doi: 10.1146/annurev.ge.23.120189.002523. [DOI] [PubMed] [Google Scholar]
- Weissensteiner T. Prostate cancer cells show a nearly 100-fold increase in the expression of the longer of two alternatively spliced mRNAs of the prostate-specific membrane antigen. Nucleic Acids Res. 1998;26:687. [PMC free article] [PubMed] [Google Scholar]
- Wilson CA, Payton MN, Elliott GS, Buaas FW, Cajulis EE, Grosshans D, Ramos L, Reese DM, Slamon DJ, Calzone FJ. Differential subcellular localization, expression and biological toxicity of BRCA1 and the splice variant BRCA1-delta11b. Oncogene. 1997;14:1–16. doi: 10.1038/sj.onc.1200924. [DOI] [PubMed] [Google Scholar]
- Wolfsberg TG, Landsman D. A comparison of expressed sequence tags (ESTs) to human genomic sequences. Nucleic Acids Res. 1997;25:1626–1632. doi: 10.1093/nar/25.8.1626. [DOI] [PMC free article] [PubMed] [Google Scholar]