

Abstract
Many missense substitutions are identified in single nucleotide polymorphism (SNP) data and large-scale random mutagenesis projects. Each amino acid substitution potentially affects protein function. We have constructed a tool that uses sequence homology to predict whether a substitution affects protein function. SIFT, which sorts intolerant from tolerant substitutions, classifies substitutions as tolerated or deleterious. A higher proportion of substitutions predicted to be deleterious by SIFT gives an affected phenotype than substitutions predicted to be deleterious by substitution scoring matrices in three test cases. Using SIFT before mutagenesis studies could reduce the number of functional assays required and yield a higher proportion of affected phenotypes. SIFT may be used to identify plausible disease candidates among the SNPs that cause missense substitutions.
Identifying substitutions that affect protein function is of major interest for those studying proteins and their implications in disease. Disease-causing mutations tend to occur in structurally and functionally important sites, and a significant fraction of polymorphism sites are located in these regions (Sunyaev et al. 2000). It is estimated that each person is heterozygous for 24,000–40,000 amino acid-altering substitutions (Cargill et al. 1999). Predicting substitutions at these sites as deleterious or neutral may help identify disease-associated alleles. A recent single nucleotide polymorphism (SNP) study used an amino acid substitution scoring matrix, BLOSUM62, to classify each amino acid substitution caused by a SNP in a coding region as conservative or nonconservative (Cargill et al. 1999). However, use of a substitution scoring matrix may be inappropriate for predicting whether an amino acid substitution will affect a protein's function or structure because it generalizes and does not incorporate information specific to the protein of interest.
Substitution scoring matrices, such as BLOSUM62, have not been tested against experimental data for their ability to predict protein-altering substitutions. The BLOSUM62 matrix, like most matrices, is intended for database searching and pairwise alignment (Henikoff and Henikoff 1992), which is a different task than predicting deleterious substitutions. Substitution matrix scores are typically calculated from a log odds ratio of target frequencies, obtained by counting pairs of aligned amino acids, with the background frequencies of the amino acids. Substitutions to a more abundant amino acid have a lower score relative to a less abundant amino acid because the background frequency is lower for the less abundant amino acid. However, the overall abundance of an amino acid is irrelevant when considering whether an amino acid change is tolerated. On average, 14 out of the 19 possible substitutions for a given amino acid have negative scores from the BLOSUM62 matrix and are deemed nonconservative by Cargill et al. (1999). If nonconservative substitutions are predicted to be deleterious, then many substitutions will be predicted to affect phenotype. However, proteins actually contain many positions that have a high degree of plasticity in accommodating amino acid substitutions, as shown in previous mutagenesis studies (Bowie and Sauer 1989; Climie et al. 1990; Huang et al. 1992; Markiewicz et al. 1994). Therefore, experimentally testing all changes deemed nonconservative by a substitution matrix would be time-consuming and wasteful because of this overprediction, especially for large-scale studies such as examination of nonsynonymous SNPs (Lander 1996; Irizarry et al. 2000) or in genome-wide random mutagenesis projects (Bentley et al. 2000; Chen et al. 2000; McCallum et al. 2000).
Given a protein query, aligned sequences from the protein's family give position-specific information, which a substitution scoring matrix lacks. Residues that are conserved completely in the protein family are expected to be important for function, and even a conservative substitution at one of these residues may affect protein function. A substitution matrix may underestimate the severity of deleterious substitutions at these crucial positions. At some positions, any amino acid change can be tolerated in the protein if these positions are not involved in protein function or structure. Because these are expected to be neutral substitutions, one might expect amino acids in these positions of a protein alignment to be diverse. Therefore, the accuracy for predicting the phenotype that results from an amino acid substitution based on sequence alignment of protein family members should be better than using a generalized substitution scoring matrix.
SIFT is a sequence homology-based tool that sorts intolerant from tolerant amino acid substitutions and predicts whether an amino acid substitution at a particular position in a protein will have a phenotypic effect. SIFT predicts the phenotype resulting from a substitution more accurately than substitution scoring matrices for three data sets. In some exceptional cases, a substitution is predicted by SIFT to be neutral but experimentally does have a deleterious effect; these can be accounted for by query-specific interactions that are not conserved among the protein family members.
RESULTS
Rationale
SIFT takes a query sequence and uses multiple alignment information to predict tolerated and deleterious substitutions for every position of the query sequence. SIFT is a multistep procedure that, given a protein sequence, (1) searches for similar sequences, (2) chooses closely related sequences that may share similar function, (3) obtains the multiple alignment of these chosen sequences, and (4) calculates normalized probabilities for all possible substitutions at each position from the alignment. Substitutions at each position with normalized probabilities less than a chosen cutoff are predicted to be deleterious; those greater than or equal to the cutoff are predicted to be tolerated.
To test the procedure against experimental data, we chose unbiased data sets in which mutagenesis was performed throughout the entire protein, and both wild-type and negative phenotypes were assayed. There were only three data sets that we could find in the literature that fit the above criteria: LacI (Markiewicz et al. 1994; Suckow et al. 1996), HIV-1 protease (Loeb et al. 1989), and bacteriophage T4 lysozyme (Rennell et al. 1991). The scarcity of unbiased data sets indicates how difficult characterization of mutant proteins on a large scale can be.
The goal of the prediction program is to identify less severe but nonetheless affected phenotypes as well as null phenotypes from wild-type. Therefore, phenotypes that exhibited weakened activity in the functional assays were grouped with loss-of-function phenotypes. SIFT and substitution scoring matrices, BLOSUM55, BLOSUM62, and BLOSUM80, were tested for the ability to predict these substitutions as deleterious. SIFT parameters used on the HIV-1 protease and bacteriophage T4 lysozyme data sets were the same as those determined to work well for the LacI mutation data, so SIFT analysis can be generalized to any protein for which homologous sequences are available.
Comparison of SIFT with BLOSUM62 Predictions on LacI Mutation Data
LacI is a DNA-binding protein that normally represses transcription of the lac operon. Upon binding of a β-galactoside sugar inducer, LacI no longer binds to DNA, thus allowing the organism to use lactose as an energy source. Positions in the Escherichia coli lac repressor gene were mutated individually to amber nonsense codons (Markiewicz et al. 1994; Suckow et al. 1996). In each mutant, nonsense suppressor tRNAs that would insert 1 of 13 different amino acids at the engineered amber codon had been introduced so that >4000 amino acid substitutions were analyzed. Using a β-galactosidase colorimetric assay, each protein with a single amino acid substitution had been tested for its ability to (1) repress transcription at the lac operator and (2) cease repression upon binding of IPTG, the inducer sugar. More than 50% of the sites were generally tolerant to substitutions, and the regions that were sensitive to amino acid replacements were primarily at the DNA and inducer binding sites and at the dimer interface (Pace et al. 1997). We compared predictions from SIFT and the substitution scoring matrices with the resulting phenotypes from the substitutions examined in the mutagenesis studies.
For SIFT to predict on LacI substitutions, it must first select sequences related to the repressor. Combining the results of sequences found in the SWISS-PROT/TrEMBL 38 protein database (Bairoch and Apweiler 2000) and in the translated microbial genomes, SIFT found 55 sequences similar to LacI. Those chosen from SWISS-PROT/TrEMBL were annotated as belonging to the LacI family of transcriptional regulators. Although the chosen sequences are generally involved in transcriptional repression relieved by an inducer, the operators and inducers that interact with these proteins are different from that of LacI. For example, RBSR_ECOLI represses the ribose operon and relieves repression by addition of ribose. Another selected sequence, PURR_HAEIN, binds to the PUR operator in the presence of guanine and loses affinity for the operator without the corepressor. With this collection of proteins, overall structure is expected to be conserved, but not necessarily residues involved in binding DNA or inducer.
The collection of LacI-related sequences was used to measure the correlation between sequence conservation and tolerance to substitutions. To predict whether an amino acid substitution is deleterious based on sequence homology, the degree of conservation at a position should be correlated positively with the number of deleterious substitutions at this position. From information theory (Schneider et al. 1986), conservation can be measured at each position and ranges from zero bits at a position equally represented by all 20 amino acids to 4.3 bits at an invariant position. Strongly conserved positions are expected to be unable to tolerate most substitutions, whereas weakly conserved positions are expected to tolerate more substitutions (see Fig. 1 for an example). Conservation was calculated for each position using an alignment of the 55 chosen sequences. The Pearson correlation coefficient between conservation and the number of deleterious substitutions determined experimentally at each position is 0.550. This is a conservative estimate because proteins in the alignment bind to different inducers and operators, so that positions important for inducer and DNA binding may not necessarily be conserved throughout the protein sequences. Also, the experimental data contain only 12 or 13 substitutions at each position, whereas up to 20 amino acids are represented in the alignment. The high correlation between experimental mutation data and conservation supports the idea that we can predict from sequence data whether a given substitution affects protein function or structure.
Figure 1.

Sequence conservation corresponds to intolerant positions. (Top) Sequence logo representation (Schneider and Stephens 1990) of the LacI multiple alignment for positions 5–38, a region involved in binding DNA. At each position, the stack of letters indicates which amino acids appear in the alignment, and the total height of the stack is a measure of conservation. (Bottom) Number of substitutions deleterious to LacI function at the corresponding positions (Markiewicz et al. 1994; Suckow et al. 1996). Positions with high conservation, such as 19–23, do not tolerate substitutions. Positions with low conservation, such as 26–28, can tolerate most substitutions. Positions 17 and 18 appear diverse in the alignment but cannot tolerate most substitutions. The side chains of these residues are involved in DNA-specific recognition (Chuprina et al. 1993) that is not conserved among the paralogous sequences.
SIFT made predictions from the LacI sequence alignment (Fig. 2A) and showed higher total and experimental prediction accuracy over BLOSUM62 (Fig. 2B), as summarized in Table 1. SIFT predicted 1747 out of the 2254 (78%) experimentally tolerated substitutions. For substitutions with an affected phenotype, SIFT correctly predicted 989 of 1750 (57%) of these accurately. Amino acid substitutions with BLOSUM62 scores ≥0 are classified as conservative substitutions (Cargill et al. 2000) and occur more or as frequently than expected by chance in a database of alignments; these substitutions are predicted as tolerated. Substitutions with negative scores are classified as nonconservative changes (Cargill et al. 2000), and these changes are observed less frequently than expected by chance; these substitutions are predicted as deleterious. BLOSUM62 predicted 84% (1475/1750) of the deleterious changes because many of its amino acid substitution scores are negative (Fig. 2B, positions 1–50). BLOSUM62 predicted only 31% of the tolerated substitutions accurately and performed poorly in regions that can tolerate many substitutions (Fig. 2B, positions 101–150). This substitution scoring matrix alone did not distinguish between conserved and variable positions, mispredicting substitutions as deleterious at tolerant positions. BLOSUM80 and BLOSUM45 were also tested for prediction and performed poorly compared to SIFT in a similar manner to BLOSUM62 (data not shown). Because SIFT uses sequence-specific information, it can distinguish between the conserved and variable positions to get better prediction performance.
Figure 2.



(A) SIFT predictions for substitutions in LacI. The effects of 12–13 substitutions at each position were assayed (Markiewicz et al. 1994; Suckow et al. 1996). The number of substitutions above the X-axis are those that gave a wild-type phenotype; the number of substitutions below the X-axis gave an affected phenotype. SIFT makes a prediction for every possible substitution, but only substitutions predicted correctly by SIFT are depicted here and are colored in black. Gray bars above the x-axis indicate false positive error; these substitutions were predicted to be deleterious by SIFT, when experimentally they gave wild-type phenotype. Gray bars below the x-axis indicate true negative error; these substitutions were predicted to be neutral, but in fact gave an affected phenotype. Amino acid side chains that have been identified as involved in interactions (Chuprina et al. 1993; Bell and Lewis 2000) are labeled as follows: (double helix) those that interact with DNA, (double cylinders) those participating in the dimer interface. (Hexagons) Positions having six or more substitutions that are unable to respond to the inducer (Markiewicz et al. 1994; Pace et al. 1997). Many of the intolerant positions that were predicted to tolerate substitutions correspond to these query-specific positions. (Asterisks) Positions that can tolerate at least six substitutions, but SIFT predicted more than half of these substitutions as deleterious. The consensus sequence and the original query sequence, LACI_ECOLI, are shown. (B) BLOSUM62 prediction for substitutions in LacI for positions 1–50 and 101–150. BLOSUM62 performs well in the DNA-binding region (residues 1–50) because this region cannot tolerate many substitutions. However, in a region that tolerates substitutions, such as positions 101–150, BLOSUM62 performs poorly, predicting many experimental false positives (large gray bars above the X-axis).
Table 1.
Summary of Prediction Results for SIFT and BLOSUM62
| Test set | Method | Tolerant prediction accuracy | Deleterious prediction accuracy | Total prediction accuracy | Experimental prediction accuracy |
|---|---|---|---|---|---|
| LacI* n = 4004 | SIFT | 78% (1747/2254) | 57% (989/1750) | 68% (2736/4004) | 66% (989/1496) |
| BLOSUM62 | 31% (696/2254) | 84% (1475/1750) | 54% (2171/4004) | 49% (1475/3033) | |
| HIV-1 Protease n = 336 | Automated SIFT | 70% (78/111) | 82% (184/225) | 78% (262/336) | 85% (184/217) |
| SIFT without RSV, avian sequences | 68% (75/111) | 88% (197/225) | 81% (272/336) | 85% (197/233) | |
| BLOSUM62 | 63% (70/111) | 73% (165/225) | 70% (235/336) | 80% (165/206) | |
| Bacteriophage T4 | SIFT | 59% (817/1377) | 72% (460/638) | 63% (1277/2015) | 45% (460/1020) |
| Lysozyme n = 2015 | BLOSUM62 | 30% (406/1377) | 85% (542/638) | 47% (948/2015) | 36% (542/1513) |
The effect of 4004 substitutions was assayed for LacI (Markiewicz et al. 1994; Pace et al. 1997), 336 substitutions for HIV-1 protease (Loeb et al. 1989), and 2015 substitutions for bacteriophage T4 lysozyme (Rennell et al. 1991). These three data sets are used to test prediction performance. Tolerant prediction accuracy is the number of substitutions correctly predicted to have no effect divided by the total number of substitutions that gave a wild-type phenotype under experimental test conditions. Subtracting the numerator from the denominator gives the number of substitutions that have been predicted to be deleterious but gave a wild-type phenotype under experimental conditions. Deleterious prediction accuracy is the number of substitutions correctly predicted to have an effect on the protein divided by the number of substitutions that affected protein. Subtracting the numerator from the denominator gives the number of substitutions that were predicted to have wild-type phenotype but gave a deleterious phenotype under experimental conditions. Total prediction accuracy is the total number of substitutions correctly predicted divided by the total number of substitutions. Experimental prediction accuracy is the number of substitutions that were experimentally shown to affect protein function divided by the number of substitutions predicted to affect function. For the biologist investigating substitutions predicted to have a deleterious effect, the experimental prediction accuracy reflects the proportion of predictions that will yield affected phenotypes experimentally.
SIFT offers prediction for positions 5–329 of the LacI repressor because fewer than half of the sequences are represented at positions 1–4 and 330–360.
The total number of correctly predicted substitutions by SIFT exceeds that of BLOSUM62 by 14% (Table 1, difference in total prediction accuracies). Of substitutions predicted to be deleterious by SIFT, 66% will yield a deleterious phenotype experimentally by the β-galactosidase assay (Table 1, experimental prediction accuracy). In comparison, only 49% of the substitutions predicted to be deleterious by BLOSUM62 will yield a deleterious phenotype experimentally. A higher proportion of substitutions predicted to be deleterious will give deleterious phenotypes experimentally if SIFT, rather than BLOSUM62, is used for prediction. The number of substitutions predicted to be deleterious is smaller for SIFT (1496) than for BLOSUM62 (3033). Not only does SIFT predict more accurately, but also the number of predicted deleterious substitutions is smaller. These numbers indicate that if SIFT-predicted deleterious substitutions rather than BLOSUM62-predicted deleterious substitutions are used as a guide for conducting experiments on mutant proteins, then (1) fewer experiments would have to be performed, and (2) a higher proportion of the experiments will yield affected phenotypes.
Although SIFT does well at most positions, it misses predicting substitutions involved in LacI-specific recognition. There are 158 positions that cannot tolerate six or more substitutions, yet SIFT predicted 56 of them to tolerate more than half of the deleterious substitutions. The side chains at four of these positions are involved in DNA-binding contacts (Fig. 1, positions 17–18; Fig. 2A, double helices); the side chains at nine other positions participate at the dimer interface (Fig. 2A, double cylinders; Chuprina et al. 1993; Bell and Lewis 2000). Other specific contacts might involve IPTG binding, but these are unknown because the structure solved for this complex had low resolution so that side-chain interactions could not be identified (Lewis et al. 1996). Nevertheless, of the 158 positions that do not tolerate six or more substitutions, there are 31 positions (20%) where at least six of the substitutions cannot respond to the inducer IPTG. If the 56 positions that were mispredicted as tolerant to substitutions were distributed randomly, then one would expect approximately 11 (0.20 × 56) positions to coincide with the positions sensitive to inducer. Instead, 20 positions (36%) were observed to coincide with inducer-sensitive positions, indicating that many SIFT mispredictions of intolerant positions to be tolerant are due to lack of conservation in the alignment. SIFT mispredicts when the alignment does not reflect the constraints on the individual protein.
SIFT's prediction is based on paralogous sequences in the LacI family. Although these sequences share a similar function to LacI, they do not have the same DNA operators or sugar inducers. Residues involved directly in LacI repressor's function may not be conserved throughout the alignment. Such positions will appear variable in an alignment of paralogous sequences and cannot be identified as important from sequence alone. The lack of conservation at these positions leads SIFT to miss these intolerant positions.
There are well conserved positions in the alignment that can tolerate substitutions according to the β-galactosidase assay. A substitution occurring at one of these positions will be predicted to affect protein function, although experimentally it will have no effect; this would be a false positive in a functional assay. Interestingly, a majority of the positions with high false positive error cluster at one face on the C-terminal subdomain (red residues in Fig. 3). The structure of the core tetramer does not implicate this face to be involved in tetramerization (Friedman et al. 1995), and other repressors in the alignment function as dimers. Perhaps this C-terminal face is involved in as yet undiscovered interaction.
Figure 3.

(A) Structure of LacI as a homodimer (light and dark blue strands) with DNA (yellow strand). The N-terminal subdomain whose interface is important for DNA binding and the allosteric mechanism is at the upper part of the figure; the C-terminal domain is at the bottom. The 186 positions tolerant for six or more substitutions are colored in white on one monomer (Markiewicz et al. 1994; Suckow et al. 1996). For 31 of these positions, >50% of the substitutions were predicted to affect phenotype according to SIFT when experimentally they did not (see also Fig. 2, asterisks). These positions are shown as space-fill atoms in red. Noticeably, many of these occurred at the bottom face of the C-terminal domain. This structure is 1EFA from PDB (Bell and Lewis 2000). (B) Same figure rotated 90° about the Z-axis.
Comparison of SIFT with BLOSUM62 Predictions on HIV-1 Protease Mutation Data
HIV-1 protease cleaves the gag and gag-pol polyproteins into mature products and is therefore necessary for AIDS virus maturation. HIV protease must recognize nine nonhomologous sites within the HIV polyproteins. Loeb and his colleagues (1989) tested the effect of 336 single missense mutations in HIV-1 protease. Mutations were generated by random mutagenesis, sequenced, and then scored for their ability to process the Pol precursor. Missense mutants were placed in one of three categories: (1) wild-type, (2) intermediate, for which both processed and unprocessed products were observed, and (3) negative, for which no mature processed products were produced by the protease. SIFT and three substitution matrices from the BLOSUM series were tested for their ability to predict substitutions with intermediate and negative phenotypes as deleterious and substitutions with wild-type phenotype as tolerated.
The predictions returned by SIFT under default parameters are more accurate than those of BLOSUM62 for HIV-1 protease (Table 1). Because the TrEMBL database may contain mutant HIV-1 protease sequences, which are not necessarily functional, sequences were chosen from the SWISS-PROT database. Thirty-eight proteases were chosen, with the most distantly related sequence being 30% identical to the query sequence. SIFT performed better than BLOSUM62 for predicting both neutral and deleterious substitutions (Table 1). Out of 215 substitutions predicted by SIFT to be deleterious, 85% give an affected phenotype (Table 1, experimental prediction accuracy) using the protease assay by Loeb et al. (1989).
Although the total prediction accuracy of SIFT exceeds that of BLOSUM62 by 8% (Table 1), performance can be improved further by basing predictions on an alignment of sequences with similar substrate specificity. The SIFT alignment contained protease sequences from the Rous sarcoma virus (RSV) and avian myeloblastosis virus (AMV), which differ from each other in only one residue. Although their structures are very similar to HIV-protease (Wlodawer et al. 1989), AMV has been shown to have substrate specificity distinct from human HIV protease (Tomasselli et al. 1990). Also, the SIFT alignment of RSV and AMV with HIV-1 protease did not match the structural alignment (Wlodawer et al. 1989) at some positions. These specificity differences and misalignments may have reduced SIFT performance. Therefore, RSV and AMV protease sequences were removed, so that the remaining 36 sequences in the alignment are proteases from humans and simians. SIV protease has substrates homologous to HIV protease and has been shown to cleave HIV-1 polyprotein substrate in a manner similar to HIV-1 (Grant et al. 1991). Thus, prediction based on this alignment should not be confounded by substrate-specific residues as much as prediction based on the alignment containing RSV and AMV protease sequences. Indeed, SIFT performance based on the alignment without RSV and AMV protease sequences was 3% better than SIFT performance on the alignment with these sequences (Table 1). BLOSUM80 and BLOSUM45 were also tested for prediction and performed poorly compared with SIFT (data not shown). The prediction accuracy for deleterious substitutions increased when AMV and RSV proteases were excluded because residues important for substrate specificity may be conserved in the alignment of human and simian viral proteases. Prediction for neutral substitutions decreases only slightly, which indicates that the remaining protease sequences are diverse enough for prediction.
We examined the literature to account for mispredictions at some positions. Several intolerant positions that SIFT predicted to tolerate substitutions cluster together in region 35–40. Residues 36–46 show large structural deviations and are implicated in HIV protease adaptation to binding of the substrates (Prabu-Jeyabalan et al. 2000), so that errors at these residues could be accounted for by substrate specificity. In general, SIFT predicts better than substitution matrices on HIV-1 protease mutation data; with careful selection of sequences and comparison of structures, performance can be improved further.
Comparison of SIFT with BLOSUM62 Predictions on Bacteriophage T4 Lysozyme Mutation Data
The final test case, which uses mutation data from bacteriophage T4 lysozyme, shows that SIFT can improve prediction remarkably when only one homologous sequence is available. Bacteriophage T4 produces a soluble lysozyme that breaks up bacterial cell walls late in the infection of E. coli. Bacteriophage T4 lysozyme was subjected to a mutagenesis study using amber suppressor tRNAs (Rennell et al. 1991). Similar to the LacI results, approximately half of the positions could tolerate all tested substitutions. Lysozyme function was assayed by plaque formation, and mutants were scored by plaque size. Mutants with plaques the same size as wild type were scored as wild-type. Intermediate phenotypes were scored for mutants with smaller plaque size. Mutants that produced no plaques were scored as null. We tested whether SIFT could predict a mutant with a wild-type phenotype as tolerated, and mutants with either intermediate or null phenotypes as deleterious.
When using the automated procedure for choosing similar proteins, the lysozyme amino acid sequence was unable to meet SIFT's criteria for choosing similar sequences. An error was returned to the user, indicating that there were not enough sequences and the user should examine the results manually. The SIFT alignment had gaps occurring in regions corresponding to secondary structure and in a core region that is conserved among distant proteins (Monzingo et al. 1996). Only VG05_BPT4, a tail-associated lysozyme in bacteriophage T4, aligned well with bacteriophage T4 soluble lysozyme (43% identity, 3% gaps). This protein is similar in function to the soluble lysozyme because a tail-associated lysozyme mutant can substitute for it (Kao and McClain 1980). The biological evidence and the global pairwise alignment supported VG05_BPT4 as a good candidate for SIFT prediction on bacteriophage T4 lysozyme.
With sequence information from just the soluble lysozyme query and VG05_BPT4, SIFT yields better prediction results than BLOSUM62. Twice as many neutral substitutions are predicted correctly when compared with BLOSUM62 (59% vs. 30%), with a 13% reduction in predicting deleterious substitutions so that total prediction accuracy was 25% higher (Table 1). SIFT also performed better than BLOSUM80 and BLOSUM45 (data not shown). There are many tolerant positions predicted intolerant presumably due to basing the prediction on only two sequences. Some intolerant positions predicted incorrectly to tolerate substitutions may be residues that specifically recognize bacterial cell wall composition, because the soluble lysozyme destroys bacterial cell walls from the inside whereas tail-associated lysozyme recognizes cell walls from the outside (Mosig et al. 1989; Nakagawa et al. 1985). The performance on this mutation data set shows that additional information from just a single homologous sequence can yield better prediction results than predictions from substitution matrices.
DISCUSSION
SIFT is a novel tool that incorporates position-specific information by using sequence alignment and is intended specifically for predicting whether an amino acid substitution affects protein function. For all three test cases, SIFT had a higher number of correctly predicted substitutions than the substitution scoring matrices. Moreover, a higher proportion of substitutions predicted to be deleterious by SIFT had affected phenotypes in the experimental assays than substitutions predicted to be deleterious by substitution matrices. For all of the data sets, SIFT made fewer mispredictions than the substitution matrices that a substitution was deleterious when it was tolerated experimentally. For two out of the three data sets, SIFT missed more deleterious substitutions than the substitution scoring matrices. Some of these errors were accounted by query-specific interactions that are not conserved in the family.
SIFT bases its predictions on sequence data alone and does not depend on knowledge of protein structure or function. Substitutions in uncharacterized proteins can be evaluated by SIFT only when homologous sequences are available. Although SIFT can choose sequences automatically, better prediction results are obtained when a list of homologs is provided, as seen with the HIV-protease mutation data. The ideal set of sequences for SIFT prediction is well-aligned orthologous sequences. Paralogs with distinct biochemical functions will confound prediction at residues conserved only among the orthologs. However, as protein databases grow with data from whole genome sequencing, a larger number of orthologs will become available and SIFT prediction should become more accurate.
Surprisingly, few sequences are needed by SIFT to observe improvement of prediction over a substitution scoring matrix. In the case of lysozyme, we observed that with only one sequence homologous to the test protein, SIFT prediction is significantly better than using a generalized substitution scoring matrix for prediction. This indicates that with only a single diverged relative, SIFT can offer better prediction than a substitution matrix.
Our results indicate that given a set of substitutions to assay, those substitutions predicted to be deleterious by SIFT will yield a greater proportion of affected phenotypes compared with substitutions judged to be nonconservative by substitution scoring matrices. Some of the substitutions predicted to be tolerated by SIFT may in fact be deleterious; the LacI test case showed that SIFT is unable to identify residues that are important for function but have not been conserved throughout the family. Positions predicted to be intolerant by SIFT but which tolerate substitutions according to the functional assay might be involved in an unknown function that the assay does not detect. In LacI, many of the conserved positions that tolerate substitutions in the DNA- and sugar-binding assays occur together at an exposed face in the C-terminal subdomain. Because these residues have been conserved among diverged but functionally related sequences, this indicates that this C-terminal face may participate in an as yet unknown interaction. Substitutions at conserved positions, which still behave as wild-type in functional assays, nevertheless may be involved in a function in vivo for which the existing assays do not test.
The majority of scores in log-odds substitution scoring matrices are negative to prevent sequence alignments from extending spuriously in database searching (Altschul 1991). For example, on average, 14 out of the 19 possible substitutions for a given amino acid have negative scores in BLOSUM62 and are classified as nonconservative changes (Cargill et al. 2000). If functional assays are performed on substitutions deemed nonconservative by substitution scoring matrices, many of the deleterious mutants will be detected simply because the matrix is dominated by negative entries. This benefit of characterizing most of the deleterious substitutions when using matrix predictions rather than SIFT's comes at the cost of assaying substitutions that do not affect phenotype. If there are few variants to characterize, or it is important to not miss any variants that alter protein function, then characterizing all substitutions or those with negative scores in a substitution matrix is a good strategy. However, in large-scale projects in which many missense mutations are generated, it is more important to minimize the number of unnecessary experiments rather than to identify all deleterious substitutions. Hence, SIFT will be more efficient than substitution scoring matrices for large-scale projects. Preliminary data show that SIFT predicts 69% of more than 3500 disease-causing substitutions to be deleterious, indicating that SIFT may be suitable for automated prediction on the genome scale (data not shown).
Linkage disequilibrium and association studies make use of polymorphism data to find genetic factors that may cause or increase risk for a disease. Among the markers identified in association or linkage disequilibrium studies, SIFT can predict which markers that result in an amino acid change may themselves be the cause of a deleterious effect on the protein. Because of the amount of polymorphism data needed to conduct linkage disequilibrium and association studies, a plethora of missense mutations are being identified, and some of the missense variants themselves are likely to be involved in disease. Approximately half of the gene lesions known to be responsible for human inherited disease are due to amino acid substitutions (Cooper et al. 1998), showing that amino acid substitutions play a large role in diseases. In a study on nonsynonymous SNPs in proteins for which structures were known (Sunyaev et al. 2000), 45% of the missense variants mapped to structurally and functionally important regions, and it was suggested that a large fraction of nonsynonymous SNPs can have strong effects on the encoded proteins. Sunyaev et al. (2000) studied only 86 nonsynonymous SNPs because they relied on structure for their analysis. Because SIFT uses sequence homology rather than protein structure, it could potentially analyze a larger number of nonsynonymous SNPs than studies based on protein structure alone. In HGBASE (Brookes et al. 2000), a public database of human sequence variants that may or may not be involved in disease, there were 20,482 gene variants, of which 3146 caused amino acid substitutions, as of January 2000. It has been predicted that there will eventually be ∼200,000 coding sequence variants (Brookes et al. 2000), which suggests there may eventually be 30,000 missense variants in this database alone. The sheer magnitude of missense variants renders it unfeasible to test all of these substitutions for their effects on the proteins for which they code. Because SIFT is an automated, relatively quick procedure, it can be used to predict which missense variants are likely to be deleterious and thus hone in on which ones are likely candidates for disease and which proteins should be subjected to further investigation.
SIFT can also be applied to large-scale, reverse-genetic projects in which mutations are introduced randomly in the genome of an experimental organism, altered genes are identified, and then the phenotype for the resulting mutants ascertained (Bentley et al. 2000; Chen et al. 2000; McCallum et al. 2000). A majority of the mutations generated in the coding regions by the chemical mutagens used in these large-scale projects cause amino acid substitutions. The rate-limiting step may be deciding which mutants to pursue for further study. The same dilemma arises when a gene is targeted for random mutagenesis. If the phenotype of a deleterious mutant is unknown or difficult to assay, SIFT can be used as a guide for which mutations are likely to be deleterious to protein function and are worth pursuing.
METHODS
Obtaining Sequences Related to a Protein of Interest
SIFT starts with a query protein sequence. Relying on the observation that proteins in the same subfamily have high conservation in conserved regions (Nevill-Manning et al. 1997), it selects sequences that are similar to the query sequence by adding the most similar sequence from a database of protein sequences iteratively to the growing collection until conservation in the conserved regions decreases. We use Rc to measure the conservation at position c where Rc = log220 − Σ20aa pcalog pca where pca is the frequency at which amino acid a appears in position c (Schneider et al. 1986).
PSI-BLAST (Altschul et al. 1997) with parameters −e 0.0001 and −h 0.002 is run for four iterations to collect a pool of sequences similar to the query from a protein sequence database such as SWISS-PROT (Bairoch and Apweiler 2000). The sequences found by PSI-BLAST are then grouped together if they are >90% identical in the regions aligned by PSI-BLAST, and a consensus sequence is made for each group by choosing the amino acid that occurs most frequently at each position. Next, the motif-finding algorithm MOTIF (Smith et al. 1990; Henikoff and Henikoff 1991) is used to find conserved regions among the query sequence and consensus sequences that were derived from at least two sequences. Consensus sequences that were derived from only one sequence are removed in the motif-finding step to increase efficiency. Once the conserved regions in the query sequence have been identified by MOTIF, these regions are extracted from the sequences aligned by PSI-BLAST. The conserved regions are grouped together if they are >90% identical, and a consensus sequence is made for each group. The conserved regions of the query sequence and those consensus sequences >90% identical are converted to a PSI-BLAST checkpoint file. This checkpoint file is the seed to which additional sequences will be added.
The checkpoint file is given to PSI-BLAST to search among the remaining conserved regions of the consensus sequences not included in the seed checkpoint file. The top hit is added to the alignment corresponding to the seed checkpoint file, and the conservation over the entire alignment of conserved regions, ΣcRc, is calculated. If Rc is greater than or equal to the Rc of the seed checkpoint file, then conservation has not decreased by adding this consensus sequence. Therefore, this consensus sequence is added to the alignment, and the checkpoint file is rebuilt. The process repeats: The checkpoint file is used as a query for PSI-BLAST to search among the conserved regions of the remaining consensus sequences, and the decision to add the highest-scoring hit depends on whether the hit does not decrease conservation. After this process terminates, the sequences found in the initial PSI-BLAST search that correspond to the consensus sequences in the final checkpoint file are used in subsequent steps of prediction. These sequences tend to align globally with the query sequence and usually belong to a small clade within the query protein's family.
Position-Specific Probability Estimation
The multiple alignment of the query sequence with sequences that were chosen as described in the previous paragraph is extracted from the initial PSI-BLAST results. PSI-BLAST alignments have been shown to be fairly accurate and long in comparison to other sequence alignment tools (Sauder and Dunbrack 2000). The alignment is converted into a position-specific scoring matrix (PSSM; Gribskov et al. 1987). A PSSM is an l × 20 matrix where l is the length of the protein sequence. Each matrix entry, pca, is the probability of amino acid a at position c of the protein where c ranges from 1 to l and a is any one of the 20 amino acids. The probability of amino acid a appearing at position c is estimated by the following general formula (Henikoff and Henikoff 1996).
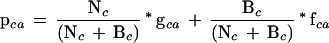 |
1 |
Nc is set to the total number of sequences in the alignment and gca is the sequence-weighted frequency that amino acid a appears at position c in the alignment (Henikoff and Henikoff 1994). If an alignment position includes gaps, they are distributed among the amino acids as follows: If gc- is the frequency of gaps observed at position c, then for all 20 amino acids a, the count gca is incremented by 1/20 of gc-.
Because the observed sequences similar to the query are only those available in the sequence database searched, pseudocounts fca are added to the observed counts for each amino acid in each column of the alignment (Henikoff and Henikoff 1996). fca is calculated from a 13-component Dirichlet mixture (Sjolander et al. 1996), and Bc is the total number of pseudocounts. Thus, pca is a weighted average of the observed amino acid frequencies in the alignment and estimated unobserved frequencies. For SIFT, we wanted to give pseudocounts more weight relative to observed counts when the amino acids present at a position are more diverse. To achieve this, we chose Bc to be an exponential function of a weighted diversity measure, Dc. Let the reference amino acid in a position be the amino acid that appears with the highest frequency and let ra be the rank that amino acid a has in an ordered list from the highest to lowest score from a substitution matrix for the reference amino acid. (BLOSUM62 is used to compute ra, but other substitution matrices should give similar results). So ra = 1 for the reference amino acid. Then Dc = Σa (ra * gca). At an invariant position, we set Bc = 0, otherwise Bc = exp(Dc).
Prediction
To automate SIFT, we wanted to apply one cutoff to all columns of the PSSM calculated in the previous section. In the most diverse alignment column possible, all 20 amino acids might appear in a position with equal probability of 0.05 = 1/20, whereas in a conserved position only two amino acids might appear, one with probability 0.05 and the other with 0.95. But if, for example, 0.05 were chosen as a cutoff for pca so that substitution to amino acid a in column c is predicted to be deleterious if pca ≤ 0.05, then substitution to any amino acid would be predicted as deleterious in the column where pca = 0.05 for all a, when this is obviously a very tolerant column. So a cutoff cannot be applied to pca alone. Instead, the pcas are normalized on the consensus amino acid in each column, which is the amino acid with the highest pca. The consensus amino acid may be different from the reference amino acid defined in the previous section because pseudocounts are now included.
Positions with normalized probabilities <0.05 are predicted to be deleterious; those ≥0.05 are predicted to be tolerated. This cutoff was chosen for the LacI data set and then used on the bacteriophage T4 lysozyme and HIV protease data sets. A user may decide from examining the probability distribution whether a substitution with a probability near the cutoff should be reclassified as deleterious if predicted tolerated, or vice versa.
Because protein alignment may not extend to the ends of a protein, so that N- and C-terminal positions may contain insufficient sequence information, we arbitrarily chose to predict at positions where >50% of the sequences were represented. Normalized probabilities and predictions are returned for every position; a user can judge whether enough sequences are represented at the position to rely on the prediction.
Availability
A sequence, related sequences, or a sequence alignment can be submitted for SIFT prediction at the BLOCKS Web site: http://blocks.fhcrc.org/∼pauline/SIFT.html. If a sequence is submitted, related sequences are returned along with SIFT predictions so that the user can manually refine the sequences and the alignment and resubmit for prediction. The LacI and HIV-1 protease alignments and results from BLOSUM45 and BLOSUM80 predictions can also be obtained at this site.
Acknowledgments
This work would not have been possible without the encouragement and advice offered by Jorja Henikoff, Harmit Malik, and Elizabeth Greene. Kami Ahmad and Jim Smothers offered thoughtful suggestions on the manuscript. A NSF and a DOE Computational Science Graduate Fellowship supported P.C.N. This work was supported by the NIH.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL steveh@muller.fhcrc.org; FAX (206) 667-5889.
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.176601.
REFERENCES
- Altschul SF. Amino acid matrices from an information theoretic perspective. J Mol Biol. 1991;219:555–565. doi: 10.1016/0022-2836(91)90193-A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. GappedBLASTand PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch A, Apweiler R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acid Res. 2000;28:45–48. doi: 10.1093/nar/28.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell C, Lewis M. A closer view of the conformation of the Lac repressor bound to operator. Nat Struct Biol. 2000;7:209–214. doi: 10.1038/73317. [DOI] [PubMed] [Google Scholar]
- Bentley A, MacLennan B, Calvo J, Dearolf CR. Targeted recovery of mutations in Drosophila. Genetics. 2000;156:1169–1173. doi: 10.1093/genetics/156.3.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie JU, Sauer RT. Identifying determinants of folding and activity for a protein of unknown structure. Biochemistry. 1989;86:2152–2156. doi: 10.1073/pnas.86.7.2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes AJ, Lehvaslaiho H, Siegfried M, Boehm JG, Yuan YP, Sarkar CM, Bork P, Ortigao F. HGBASE: A database of SNPs and other variations in and around human genes. Nucleic Acids Res. 2000;28:356–360. doi: 10.1093/nar/28.1.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cargill M, Altshuler D, Ireland J, Sklar P, Ardlie K, Patil N, Lane C, Lim EP, Kalyanaraman N, Nemesh J, et al. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat Genet. 1999;22:231–238. doi: 10.1038/10290. [DOI] [PubMed] [Google Scholar]
- Chen Y, Yee D, Dains K, Chatterjee A, Cavalcoli J, Schneider E, Om J, Woychick RP, Magnusohn T. Genotype-based screen for ENU-induced mutations in mouse embryonic stem cells. Nat Genet. 2000;24:314–317. doi: 10.1038/73557. [DOI] [PubMed] [Google Scholar]
- Chuprina VP, Rullmann JA, Lamerichs RM, van Boom JH, Boelens R, Kaptein R. Structure of the complex of lac repressor headpiece and an 11 base-pair half-operator determined by nuclear magnetic resonance spectroscopy and restrained molecular dynamics. J Mol Biol. 1993;234:446–462. doi: 10.1006/jmbi.1993.1598. [DOI] [PubMed] [Google Scholar]
- Climie S, Ruiz-Perez L, Gonzalez-Pacanowska D, Prapunwattana P, Cho S, Stroud R, Santi D. Saturated site-directed mutagenesis of thymidylate synthase. J Biol Chem. 1990;265:18776–18779. [PubMed] [Google Scholar]
- Cooper DN, Ball EV, Krawczak M. The human gene mutation database. Nucleic Acids Res. 1998;26:285–287. doi: 10.1093/nar/26.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman AM, Fischmann TO, Steitz TA. Crystal structure of lac repressor core tetramer and its implication for DNA looping. Science. 1995;268:1721–1727. doi: 10.1126/science.7792597. [DOI] [PubMed] [Google Scholar]
- Grant SK, Deckman IC, Minnich MD, Culp J, Franklin S, Dreyer GB, Tomaszek TA, Jr, Debouck C, Meek TD. Purification and biochemical characterization of recombinant simian immunodeficiency virus protease and comparison to human immunodeficiency virus type 1 protease. Biochemistry. 1991;30:8424–8434. doi: 10.1021/bi00098a021. [DOI] [PubMed] [Google Scholar]
- Gribskov M, MacLachlan AD, Eisenberg D. Profile analysis: Detection of distantly related proteins. Proc Natl Acad Sci. 1987;84:4355–4358. doi: 10.1073/pnas.84.13.4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S, Henikoff JG. Automated assembly of protein blocks for database searching. Nucleic Acids Res. 1991;19:6565–6572. doi: 10.1093/nar/19.23.6565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ————— Position-based sequence weights. J Mol Biol. 1994;243:574–578. doi: 10.1016/0022-2836(94)90032-9. [DOI] [PubMed] [Google Scholar]
- ————— Using substitution probabilities to improve position-specific scoring matrices. CABIOS. 1996;12:135–143. doi: 10.1093/bioinformatics/12.2.135. [DOI] [PubMed] [Google Scholar]
- Huang A, Lee J, King SC, Wilson TH. Amino acid substitution in the lactose carrier protein with the use of amber suppressors. J Bact. 1992;174:5436–5441. doi: 10.1128/jb.174.16.5436-5441.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry K, Kustanovich V, Li C, Brown N, Nelson S, Wong W, Lee CJ. Genome-wide analysis of single-nucleotide polymorphism in human expressed sequences. Nat Genet. 2000;26:233–236. doi: 10.1038/79981. [DOI] [PubMed] [Google Scholar]
- Kao SH, McClain WH. Baseplate protein of bacteriophage T4 with both structural and lytic functions. J Virol. 1980;34:95–103. doi: 10.1128/jvi.34.1.95-103.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES. The new genomics: Global views of biology. Science. 1996;274:536–539. doi: 10.1126/science.274.5287.536. [DOI] [PubMed] [Google Scholar]
- Lewis M, Chang G, Horton NC, Kercher MA, Pace HC, Schumacher MA, Brennan RG, Lu P. Crystal structure of the lactose operon repressor and its complexes with DNA and inducer. Science. 1996;271:1247–1254. doi: 10.1126/science.271.5253.1247. [DOI] [PubMed] [Google Scholar]
- Loeb DD, Swanstrom R, Everitt L, Manchester M, Stamper SE, Hutchison CA., III Complete mutagenesis of the HIV-1 protease. Nature. 1989;340:397–400. doi: 10.1038/340397a0. [DOI] [PubMed] [Google Scholar]
- Markiewicz P, Kleina L, Cruz C, Ehret S, Miller JH. Genetic studies of the lac repressor XIV. Analysis of 4000 altered Escherichia coli lac repressors reveals essential and non-essential residues, as well as “spacers” which do not require a specific sequence. J Mol Biol. 1994;240:421–433. doi: 10.1006/jmbi.1994.1458. [DOI] [PubMed] [Google Scholar]
- McCallum CM, Comai L, Greene EA, Henikoff S. Targeting induced local lesions IN genomes (TILLING) for plant functional genomics. Plant Physiol. 2000;123:439–442. doi: 10.1104/pp.123.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monzingo AF, Marcotte EM, Hart PJ, Robertus JD. Chitinases, chitosanases, and lysozymes can be divided into prokaryotic and eucaryotic families sharing a conserved core. Nat Struct Biol. 1996;3:133–140. doi: 10.1038/nsb0296-133. [DOI] [PubMed] [Google Scholar]
- Mosig G, Lin GW, Franklin J, Fan WH. Functional relationships and structural determinants of two bacteriophage T4 lysozymes: A soluble (gene e) and a baseplate-associated (gene 5) protein. New Biol. 1989;1:171–179. [PubMed] [Google Scholar]
- Nakagawa H, Arisaka F, Ishii S. Isolation and characterization of the bacteriophage T4 tail-associated lysozyme. J Virol. 1985;54:460–466. doi: 10.1128/jvi.54.2.460-466.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevill-Manning CG, Sethi KS, Wu TD, Brutlag DL. Enumerating and ranking discrete motifs. ISMB. 1997;5:202–209. [PubMed] [Google Scholar]
- Pace HC, Kerchr MA, Lu P, Markiewicz P, Miller JH, Chang G, Lewis M. Lac repressor genetic map in real space. TIBS. 1997;22:334–339. doi: 10.1016/s0968-0004(97)01104-3. [DOI] [PubMed] [Google Scholar]
- Prabu-Jeyabalan M, Nalivaika E, Schiffer CA. How does a symmetric dimer recognize an asymmetric substrate? A substrate complex of HIV-1 protease. J Mol Biol. 2000;301:1207–1220. doi: 10.1006/jmbi.2000.4018. [DOI] [PubMed] [Google Scholar]
- Rennell D, Bouvier SE, Hardy LW, Poteete AR. Systematic mutation of bacteriophage T4 lysozyme. J Mol Biol. 1991;222:67–87. doi: 10.1016/0022-2836(91)90738-r. [DOI] [PubMed] [Google Scholar]
- Sauder JM, Dunbrack RL., Jr Genomic fold assignment and rational modeling of proteins of biological interest. ISMB. 2000;8:296–306. [PubMed] [Google Scholar]
- Schneider TD, Stevens RM. Sequence logos: A new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider TD, Stormo GD, Gold L, Ehrenfeucht A. Information content of binding sites on nucleotide sequences. J Mol Biol. 1986;188:415–431. doi: 10.1016/0022-2836(86)90165-8. [DOI] [PubMed] [Google Scholar]
- Sjolander K, Karplus K, Brown M, Hughey R, Krogh A, Mian IS, Haussler D. Dirichlet mixtures: A method for improved detection of weak but significant protein sequence homology. CABIOS. 1996;12:327–345. doi: 10.1093/bioinformatics/12.4.327. [DOI] [PubMed] [Google Scholar]
- Smith HO, Annau TM, Chandrasegaran S. Finding sequence motifs in groups of functionally related proteins. Proc Natl Acad Sci. 1990;87:826–830. doi: 10.1073/pnas.87.2.826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suckow J, Markiewicz P, Kleina LG, Miller J, Kisters-Woike B, Muller-Hill B. Genetic studies of the Lac repressor. XV: 4000 single amino acid substitutions and analysis of the resulting phenotypes on the basis of the protein structure. J Mol Biol. 1996;261:509–523. doi: 10.1006/jmbi.1996.0479. [DOI] [PubMed] [Google Scholar]
- Sunyaev S, Ramensky V, Bork P. Towards a structural basis of human non-synonymous single nucleotide polymorphisms. Trends Genet. 2000;16:198–200. doi: 10.1016/s0168-9525(00)01988-0. [DOI] [PubMed] [Google Scholar]
- Tomasselli AG, Hui JO, Sawyer TK, Staples DJ, Bannow CA, Reardon IM, Chaudhary VK, Fryling CM, Patan I, Fitzgerald DJ, et al. Proteases from human immunodeficiency virus and avian myeloblastosis virus show distinct specificities in hydrolysis of multidomain protein substrates. J Virol. 1990;64:3157–3161. doi: 10.1128/jvi.64.7.3157-3161.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodawer A, Miller M, Jaskolski M, Sathyanarayana BK, Baldwin E, Weber IT, Selk LM, Clawson L, Schneider J, Kent SBH. Conserved folding in retroviral proteases: Crystal structure of a synthetic HIV-1 protease. Science. 1989;245:616–621. doi: 10.1126/science.2548279. [DOI] [PubMed] [Google Scholar]