

Abstract
We have discovered a photocatalytic intramolecular hetero-Diels–Alder reaction of tethered bis(enones). This transformation involves the intermediacy of an enone radical anion and constitutes the formal coupling of an electron-deficient heterodiene with an electronically mismatched enone dienophile. The diastereoselectivity and regioselectivity of the process are high, and the dihydropyran products are amenable to a variety of synthetically useful transformations.
1. Introduction
Tetrahydropyrans and related six-membered heterocycles are ubiquitous substructures found in a variety of carbohydrates, polyketide natural products, and other bioactive compounds. Among the most powerful methods for the rapid construction of densely functionalized six-membered oxaheterocycles are formal hetero-Diels–Alder cycloadditions. These methods have been the subject of numerous reviewsi and have been extensively utilized as key steps in the synthesis of many complex organic structures.ii Nevertheless, hetero-Diels–Alder cycloadditions proceed efficiently under mild conditions only when the components are electronically well matched, which involves the reaction of either electron-rich dienes with electron-deficient carbonyl compounds or electron-poor heterodienes with electron-rich olefins. Electronically mismatched hetero-Diels–Alder cycloadditions between two electron-deficient components typically require forcing conditions that limit their utility in synthesis.iii
We,iv along with several other research groups,v have recently begun to explore the ability of metal polypyridyl photocatalysts to promote a variety of synthetically useful transformations upon irradiation with visible light.vi In particular, our lab has become interested in exploiting the ability of Ru(bpy)32+ and related photoredox catalysts to initiate one-electron transfer processes without the need for strong stoichiometric reductants or oxidants. The facility with which radical cations and radical anions can be generated under photocatalytic conditions has enabled us to explore the chemistry of these reactive intermediates, whose utility in synthesis has been underdeveloped in comparison to that of neutral radicals. In this paper, we report that high-yielding and highly diastereoselective radical anion hetero-Diels–Alder cycloadditions between electronically mismatched enones can be conducted using our group’s strategy for visible light photocatalysis.
2. Results and Discussions
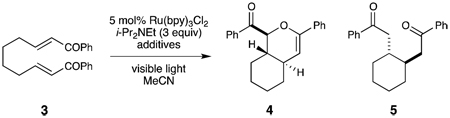
Our interest in the hetero-Diels–Alder cycloaddition began during an exploration of the scope of the photocatalytic intramolecular [2+2] enone cycloaddition developed in our labs. We observed that the length of the aliphatic tethering group had a dramatic influence on the intrinsic reactivity of the system (Scheme 1). Bis(enone) 1 bearing a three-carbon tether undergoes efficient [2+2] cycloaddition upon irradiation with visible light in the presence of Ru(bpy)32+ with LiBF4 and i-Pr2NEt as additives.iva However, when bis(enone) 3, in which the tether length was increased by one methylene unit, was subjected to the same conditions, the expected [2+2] cycloadduct was not formed. Instead, the major products are the hetero-Diels–Alder cycloadduct 4 and the product of reductive monocyclization (5). Both products are formed with high diastereoselectivity. While analogous compounds were reported to be side products in the electrochemically induced [2+2] cycloadditions of 1 reported by Bauld and Krische,vii we did not observe their formation in our studies of the photocatalytic cycloaddition of 1. Intrigued by this unexpected reactivity, we elected to initiate an examination of this hetero-Diels–Alder process by developing conditions that allow selective access to [4+2] cycloadduct 4.
Scheme 1.

Photocatalytic [2+2] and [4+2] cycloadditions.
We noted that under our initial conditions the selectivity for formation of 4 over the undesired reductive cyclization product 5 was high at relatively low conversions but steadily decreased over the course of the reaction. We hypothesized, therefore, that 5 might be a decomposition product arising from over-reduction and reductive cleavage of 4. Indeed, when 4 was isolated and resubjected to the reaction conditions for 24 h, we observed formation of 5 in 33% yield, which suggested that the long reaction times were in part responsible for the formation of 5. We therefore sought conditions that would accelerate the overall rate of conversion and limit the formation of this undesired side product.
In an initial screen of solvents, we found that the presence of water had a profound influence on the rate of the reaction. Upon addition of 10 equiv of water, the reaction time decreased dramatically from 9.5 h to 1 h (Table 1, entries 1 and 2). Importantly, very little of the undesired over-reduction product 5 was formed, and the hetero-Diels–Alder cycloadduct could be isolated in 86% yield. Water proved to be a uniquely effective protic additive;viii while both methanol and trifluoroethanol also afforded an increase in the rate of the reaction, neither provided good yields of the desired [4+2] cycloadduct. Control studies indicated that LiBF4 was an essential additive. No consumption of the starting bis(enone) occurred when LiBF4 was either omitted from the reaction or was replaced by Bu4N+BF4−. This indicates that the Lewis acidity of the lithium cation is crucial for successful cycloaddition,ix as it is in the analogous [2+2] cycloaddition reactions reported by our group.iva
Table 1.
Optimization of [4+2] cycloaddition of 3.a
 | |||
|---|---|---|---|
| Entry | Additives | Time | % Yield (4/5)b,c |
| 1 | LiBF4 (2 equiv) | 9.5 h | 28/41 |
| 2 | LiBF4 (2 equiv), H2O (10 equiv) | 1 h | 86d/<5 |
| 3 | LiBF4 (2 equiv), MeOH (10 equiv) | 1 h | 25/19e |
| 4 | LiBF4 (2 equiv), CF3CH2OH (10 equiv) | 1 h | 61/25 |
| 5 | H2O (10 equiv) | 1 h | 0/0 |
| 6 | Bu4N+BF4− (2 equiv), H2O (10 equiv) | 1 h | 0/0 |
Reactions conducted in degassed MeCN (0.1 M) under irradiation with a 200 W tungsten filament light bulb at a distance of 30 cm.
Yields determined by 1H NMR spectroscopy against an internal standard unless otherwise noted.
The products were formed in >10:1 d.r. unless otherwise noted.
Isolated yield.
5 was formed as a 2:1 mixture of diastereomers in this experiment.
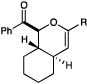
We next conducted a survey of the scope of the hetero-Diels–Alder cycloaddition, and the results are summarized in Table 2. A number of symmetrical aryl bis(enones) were found to be excellent substrates for this reaction. Both electron-deficient (entries 2–3) and electron-rich (entry 4) aryl enones react in high yield and diastereoselectivity, as do polyaromatic (entry 5) and heteroaryl enones (entry 6). Fluoride substitution at the ortho position is well tolerated (entry 7), although larger groups at this position significantly hinder reactivity (entry 8). In all cases, the desired [4+2] cycloadduct was formed with excellent diastereoselectivity.
Table 2.
Scope of the photocatalytic [4+2] cycloaddition.a
| Entry | Methodb | Substrate | Product | Time | Yieldc |
|---|---|---|---|---|---|
 |
|||||
| 1 | A | Ar = Ph | 1 h | 86% | |
| 2 | A | Ar = 4-Cl-C6H4 | 30 min | 70% | |
| 3 | A | Ar = 4-CF3-C6H4 | 30 min | 83%d | |
| 4 | A | Ar = 4-AcO-C6H4 | 1 h | 76% | |
| 5 | A | Ar = 2-naphthyl | 1.5 h | 77% | |
| 6 | A | Ar = 2-furyl | 30 min | 77% | |
| 7 | A | Ar = 2-F-C6H4 | 30 min | 84% | |
| 8 | A | Ar = 2-Me-C6H4 | 6 h | 5% e | |
 |
|||||
| 9 | A | R = CH2OBn | 20 min | 76% | |
| 10 | A | R = Me | 2 h | 39% | |
| 11 | B | R = Me | 10 min | 73% | |
| 12 | B | R = i-Pr | 1.5 h | 58% | |
| 13 | B | R = t-Bu | 6 h | 12%e | |
| 14 | B | 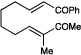 |
 |
5 h | 17% e |
All reactions were irradiated with a 200 W tungsten filament light bulb at a distance of 30 cm.
Method A: Bis(enone) substrate (1 equiv), Ru(bpy)3Cl2(0.05 equiv), LiBF4 (2 equiv), i-Pr2NEt (3 equiv), and H2O (10 equiv) in degassed MeCN (0.1 M). Method B: Bis(enone) substrate (1 equiv), Ru(bpy)3Cl2 (0.05 equiv), Mg(ClO4)2 (2 equiv), and i-Pr2NEt (5 equiv) in degassed MeCN (0.025 M).
Data represent the average isolated yields from two reproducible experiments, unless otherwise noted.
Isolated yield of a single experiment.
Yield of a single experiment, determined by 1H NMR spectroscopy against an internal standard.
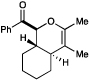
We also became interested in exploring the cycloadditions of unsymmetrical bis(enones) in which two possible constitutional isomers could reasonably be formed. We examined the reactions of a number of substrates bearing one aryl enone and one alphatic enone under our optimized reaction conditions. An α-benzyloxy enone underwent efficient cycloaddition to afford a single regioisomer of the hetero-Diels–Alder product (entry 9). Other substrates were less successful. A methyl enone required significantly longer reaction times and consequently afforded lower yields of the desired cycloadduct, although the regioselectivity of this process was also excellent (entry 10). Upon careful optimization, we were able to increase the efficiency of this reaction to 73% by removing water and replacing the LiBF4 additive with Mg(ClO4)2 (entry 11). Other aliphatic enones also underwent cycloaddition under these conditions, although the yields of these reactions decreased with increasing steric demand (entries 12–14). Reactions involving enoates and α,β-unsaturated thioesters were unsuccessful under both sets of conditions.
The high level of regioselectivity observed in the cycloaddition of unsymmetrical bis(enones) (e.g., 6) can be rationalized by the mechanism outlined in Scheme 2, which is based upon Krische and Bauld’s proposal that radical anion cyclodditions proceed in a step-wise fashion.vii Photoexcitation of Ru(bpy)32+ with visible light affords an excited state that can undergo efficient reductive quenching by i-Pr2NEt. The Lewis acid-activated enone complex ([LA]-6) can then accept an electron from the resulting Ru(bpy)3+ reductant to afford an activated radical anion intermediate (7) that should undergo β,β coupling to afford a monocyclized distonic radical anion intermediate (8). Formation of the carbon-oxygen bond could proceed to form two possible isomeric ketyl radicals (9 and 10). We speculate that the greater stabilization of the aryl ketyl radical may then serve as a driving force for selective formation of 9. Finally, the neutral hetero-Diels–Alder cycloadduct is produced upon loss of one electron, either to another equivalent of enone in a chain propagation step or to the photogenerated amine radical cation in a chain termination step.
Scheme 2.

Proposed mechanism for regioselective hetero-Diels–Alder cycloaddition of unsymmetrical bis(enone) 6.
The dihydropyrans formed in this study can be synthetically elaborated in a number of ways (Scheme 3). The enol ether functionality of 4 can be converted to an acetal (12) upon treatment with methanol and catalytic PTSA in excellent yield and diastereoselectivity. Dihydroxylation of the olefin under Upjohn conditionsx similarly provides the corresponding diol (13) with high stereochemical purity. Finally, catalytic hydrogenation of 4 reduces both the enol ether and the aryl ketone, introducing two new stereocenters with very high diastereoselectivity in the doubly reduced product (14).
Scheme 3.

Diastereoselective functionalization of 4.
The origins of the chemoselectivity for [2+2] vs [4+2] pathways in these radical anion processes are not clear at this time, but the length of the aliphatic tether appears to be critical. Subjecting the three-carbon tethered bis(enone) 1 to the optimized conditions for 2 in Table 2 produced only [2+2] cycloadduct and none of the hetero-Diels–Alder product. Five- and six-carbon tethers that would afford medium-sized rings produced neither cycloadduct. We speculate that the initial bond-forming event in the stepwise cycloaddition of 1 has a kinetic preference for formation of the cis cyclopentane isomer. Subsequent coupling of the α,α carbons would afford the [3.2.0] bicycloheptane ring structure observed in the intramolecular cyclobutanation.iva On the other hand, we speculate that the initial carbon-carbon bond formation in the cycloaddition of 3 produces a trans-substituted cyclohexane intermediate. Coupling of the α positions would afford a trans [4.2.0] ring system that we would expect to be prohibitively strained, while formation of a new C–O bond would produce a less energetically unfavorable trans oxadecalin bicycle.
In summary, our group’s investigations of visible light photocatalysis have led to the discovery of an interesting intramolecular hetero-Diels–Alder cycloaddition. This reactivity is notable for a number of reasons. First, the intermediacy of an enone radical anion facilitates the efficient coupling of a dienophile and heterodiene that are both electron-deficient, which enables the construction of a cycloadduct that is difficult to access upon thermal activation. Second, the diastereoselectivity of the process is high, and the products are amenable to a variety of further synthetic manipulations. Finally, one of the most intriguing unanswered questions is how the effect of the tether length controls the chemoselectivity for [4+2] vs. [2+2] cycloaddition. Studies to elucidate the origins of this divergent reactivity are underway in our laboratory, and these investigations provide a promising framework for further studies of the chemistry of photogenerated radical anions.
3. Experimental section
3.1 General methods
Acetonitrile, dichloromethane and tetrahydrofuran were purified by elution through alumina as described by Grubbs.xi Diisopropylethylamine was purified by distillation from CaH2 immediately prior to use. Ru(bpy)3Cl2·6H2O was purchased from Strem and used without purification. LiBF4 and Mg(ClO4)2 were purchased from Sigma-Aldrich and Strem, respectively, and stored in a glove box under an atmosphere of nitrogen. Millipore water was used in all photochemical reactions depicted in Table 2 and prepared as a stock solution in acetonitrile. A 200 W (3980 lumens) GE Crystal Clear tungsten filament light bulb was used for all photochemical reactions and the solvent required for these reactions was degassed by three freeze-pump-thaw cycles. Diastereomer ratios for all products were determined by 1H NMR analysis of the isolated products after flash column chromatography. Flash column chromatography was performed with Silicycle 40–63Å silica (230–400 mesh). All glassware was oven-dried prior to use. 1H and 13C NMR data for all previously uncharacterized compounds were obtained using a Varian Unity-500 spectrometer and are referenced to TMS (0.0 ppm) and CDCl3 (77.0 ppm), respectively. Mass spectrometry was performed with a Waters Micromass LCT (electrospray ionization, time-of-flight analyzer).
3.2 Photocatalytic hetero-Diels-Alder cycloadditions
3.2.1 General Procedure: Method A (Table 2)
To an oven-dried 25 mL Schlenk tube equipped with a magnetic stir bar was added bis(enone) (1 equiv), Ru(bpy)3Cl2·6H2O (0.05 equiv), LiBF4 (2 equiv), MeCN (0.1 M), H2O (10 equiv) as a stock solution in MeCN, and i-Pr2NEt (3 equiv). The tube was sealed and degassed by three freeze-pump-thaw cycles under nitrogen in the absence of light. The reaction mixture was then stirred in a water bath at room temperature and irradiated with a 200 W tungsten filament light bulb at a distance of 30 cm. Upon consumption of the bis(enone), the reaction mixture was passed across a short plug of silica with a mixture of either hexanes:EtOAc or hexanes:Et2O, concentrated in vacuo to approximately 4 mL and then purified immediately by flash column chromatography on silica gel.
3.2.2 General Procedure: Method B (Table 2)
To an oven-dried 50 mL Schlenk tube equipped with a magnetic stir bar was added Ru(bpy)3Cl2·6H2O (0.05 equiv), Mg(ClO4)2 (2 equiv), and MeCN (0.025 M). The mixture was stirred until homogenous and then charged with the bis(enone) (1 equiv) and i-Pr2NEt (3 equiv). The tube was sealed and degassed by 3 freeze-pump-thaw cycles under nitrogen in the absence of light. The reaction mixture was then stirred in a water bath at room temperature and irradiated with a 200 W tungsten filament light bulb at a distance of 30 cm. Upon consumption of the bis(enone), the reaction mixture was passed across a 6 inch plug of silica with a mixture of hexanes:Et2O. The reaction mixture was then concentrated in vacuo to approximately 4 mL and purified immediately by silica gel flash column chromatography.
3.2.3 (Table 2, entry 1)
Experiment 1: Conducted using Method A with 100. mg (0.314 mmol) bis(enone), 12 mg (0.016 mmol) Ru(bpy)3Cl2·6H2O, 60 mg (0.64 mmol) LiBF4, 57 µL (3.1 mmol) H2O, 164 µL (0.942 mmol) i-Pr2NEt, and 3.1 mL MeCN and an irradiation time of 60 min. Purification by flash column chromatography (20:1 hexanes:EtOAc) afforded 85 mg cycloadduct (0.27 mmol, 85%). Experiment 2: 100 mg (0.314 mmol) bis(enone), 12 mg (0.016 mmol) Ru(bpy)3Cl2·6H2O, 59 mg (0.63 mmol) LiBF4, 57 µL (3.1 mmol) H2O, 164 µL (0.942 mmol) i-Pr2NEt, and 3.1 mL MeCN. Isolated 86 mg cycloadduct (0.27 mmol, 86%) as a white solid (m.p. 60–64 °C). IR (thin film): 2928, 1684, 1447, 1270 cm−1. 1H NMR: (500 MHz, CDCl3) δ8.13 (ddd, J = 8.1, 1.3, 1.3 Hz, 2H), 7.59 (tt, J = 7.4, 1.3 Hz, 1H), 7.54 (ddd, J = 7.8, 1.3, 1.3 Hz, 2H), 7.31-7.23 (m, 3H), 5.31 (d, J = 1.7 Hz, 1H), 4.93 (d, J = 10.4 Hz, 1H), 2.17 (dddd, J = 10.5, 10.5, 2.2, 2.2 Hz, 1H), 1.95-1.74 (m, 4H), 1.56 (dddd, J = 11.9, 2.2, 2.2, 2.2 Hz, 1H), 1.39 (qt, J = 12.8, 3.3 Hz, 1H), 1.29 (qt, J = 12.8, 3.3 Hz, 1H), 1.21 (qd, J = 12.8, 3.3 Hz, 1H), 1.07 (qd, J = 12.6, 3.5 Hz, 1H); 13C NMR: (125 MHz, CDCl3) δ196.9, 149.6, 135.7, 135.1, 133.5, 129.4, 128.6, 128.1, 128.0, 124.4, 102.5, 83.1, 41.3, 38.4, 32.7, 27.7, 26.0, 25.9. HRMS (EI) calculated for [C22H22O2]+ m/z requires 318.1615, found 318.1599.
3.2.4 (Table 2, entry 2)
Experiment 1: Conducted using Method A with 99 mg (0.26 mmol) bis(enone), 10. mg (0.013 mmol) Ru(bpy)3Cl2·6H2O, 49 mg (0.53 mmol) LiBF4, 46 µL (2.6 mmol) H2O, 135 µL (0.774 mmol) i-Pr2NEt, and 2.6 mL MeCN and an irradiation time of 30 min. Purification by flash column chromatography (20:1 hexanes:EtOAc) afforded 69 mg cycloadduct (0.18 mmol, 69%). Experiment 2: 100. mg (0.258 mmol) bis(enone), 9.7 mg (0.013 mmol) Ru(bpy)3Cl2·6H2O, 48 mg (0.52 mmol) LiBF4, 46 µL (2.6 mmol) H2O, 135 µL (0.774 mmol) i-Pr2NEt, and 2.6 mL MeCN. Isolated 71 mg cycloadduct (0.18 mmol, 71%) as a white solid (m.p. 42–46 °C). IR (thin film): 2922, 1692, 1491, 1092 cm−1. 1H NMR: (500 MHz, CDCl3) δ8.05 (dt, J = 8.7, 2.0 Hz, 2H), 7.44 (dt, J = 8.7, 2.0 Hz, 4H), 7.25 (dt, J = 8.7, 2.0 Hz, 2H), 5.30 (d, J = 1.8 Hz, 1H), 4.86 (d, J = 10.5 Hz, 1H), 2.15 (dddd, J = 10.8, 10.8, 2.3, 2.3 Hz, 1H), 1.90 (dddd, J = 13.0, 2.5, 2.5, 2.5 Hz, 1H), 1.85-1.75 (m, 3H), 1.54 (dddd, J = 13.0, 2.5, 2.5, 2.5 Hz, 1H), 1.44-1.15 (m, 3H), 1.065 (dddd, J = 12.7, 12.7, 12.7, 3.8 Hz, 1H); 13C NMR: (125 MHz, CDCl3) δ195.5, 148.5, 140.1, 133.8, 133.4, 130.8, 129.0, 128.3, 125.7, 103.0, 83.3, 41.2, 38.4, 32.6, 27.6, 26.0, 25.8. HRMS (EI) calculated for m/z [C22H20Cl2O2]+ requires 386.0835, found 386.0848.
3.2.5 (Table 2, entry 3)
Conducted using Method A with 81 mg (0.18 mmol) bis(enone), 8 mg (0.01 mmol) Ru(bpy)3Cl2·6H2O, 37 mg (0.39 mmol) LiBF4, 32 µL (1.8 mmol) H2O, 92 µL (0.53 mmol) i-Pr2NEt, and 1.8 mL MeCN and an irradiation time of 30 min. Purification by flash column chromatography (30:1 hexanes:EtOAc) afforded 68 mg cycloadduct (0.15 mmol, 83%) as a white solid (m.p. 48–52 °C). IR (thin film): 2932, 1696, 1326, 1169 cm−1. 1H NMR: (500 MHz, CDCl3) δ8.22 (d, J = 8.3 Hz, 2H), 7.75 (d, J = 8.3 Hz, 2H), 7.62 (d, J = 8.3 Hz, 2H), 7.54 (d, J = 8.3 Hz, 2H), 5.44 (d, J = 1.7 Hz, 1H), 4.93 (d, J = 10.4 Hz, 1H), 2.20 (dddd, J = 10.4, 10.4, 2.3, 2.3 Hz, 1H), 1.94 (dd, J = 12.5, 2.4 Hz, 1H), 1.87-1.77 (m, 3H), 1.56 (dq, J = 10.1, 2.7 Hz, 1H), 1.47-1.18 (m, 3H), 1.10 (qd, J = 12.4, 3.7 Hz, 1H); 13C NMR: (125 MHz, CDCl3) δ198.4, 151.0, 140.9, 140.8, 137.4 (q, J = 33.0 Hz), 132.6 (q, J = 33.0), 132.3, 130.0, 129.5, 128.4 (q, J = 3.6 Hz), 127.9 (q, J = 3.6 Hz), 127.2, 125.7, 125.1, 123.5, 122.9, 107.5, 86.1, 43.7, 40.8, 35.1, 30.2, 28.6, 28.4. HRMS (EI) calculated for m/z [C24H20F6O2]+ requires 454.1362, found 454.1352.
3.2.6 (Table 2, entry 4)
Experiment 1: Conducted using Method A with 101 mg (0.232 mmol) bis(enone), 9 mg (0.01 mmol) Ru(bpy)3Cl2·6H2O, 42 mg (0.45 mmol) LiBF4, 41 µL (2.3 mmol) H2O, 120 µL (0.690 mmol) i-Pr2NEt, and 2.3 mL MeCN and an irradiation time of 30 min. Purification by flash column chromatography (5:1 hexanes:acetone) afforded 77 mg cycloadduct (0.18 mmol, 76%). Experiment 2: 92.0 mg (0.212 mmol) bis(enone), 8 mg (0.01 mmol) Ru(bpy)3Cl2·6H2O, 41 mg (0.44 mmol) LiBF4, 39 µL (2.2 mmol) H2O, 114 µL (0.651 mmol) i-Pr2NEt, and 2.2 mL MeCN. Isolated 70 mg cycloadduct (0.16 mmol, 76%) as a white solid (m.p. 42–46 °C). IR (thin film): 2929, 1758, 1685, 1599, 1506, 1370, 1198 cm−1. 1H NMR: (500 MHz, CDCl3) δ8.17 (dt, J = 8.8, 1.9 Hz, 2H), 7.54 (dt, J = 8.8, 1.9 Hz, 2H), 7.20 (dt, J = 8.8, 1.9 Hz, 2H), 7.01 (dt, J = 8.8, 1.9 Hz, 2H), 5.28 (d, J = 1.9 Hz, 1H), 4.86 (d, J = 10.4 Hz, 1H), 2.33 (s, 3H), 2.28 (s, 3H), 2.15 (dddd, J = 10.6, 10.6, 2.5, 2.5 Hz, 1H), 1.90 (dddd, J = 12.7, 2.3, 2.3, 2.3 Hz, 1H), 1.86-1.75 (m, 3H), 1.54 (dddd, J = 12.7, 2.3, 2.3, 2.3 Hz, 1H), 1.45-1.15 (m, 3H), 1.07 (qd, J = 12.7, 3.9 Hz, 1H); 13C NMR: (125 MHz, CDCl3) δ195.5, 169.4, 168.8, 154.6, 150.5, 148.8, 133.1, 132.8, 131.2, 125.6, 121.8, 121.3, 102.7, 83.6, 41.2, 38.4, 32.6, 27.6, 26.0, 25.8, 21.1, 21.1. HRMS (EI) calculated for [C26H26O6]+ requires m/z 434.1724, found 434.1714.
3.2.7 (Table 2, entry 5)
Experiment 1: Conducted using Method A with 101 mg (0.240 mmol) bis(enone), 9 mg (0.01 mmol) Ru(bpy)3Cl2·6H2O, 44 mg (0.47 mmol) LiBF4, 43 µL (2.4 mmol) H2O, 125 µL (0.717 mmol) i-Pr2NEt, and 2.4 mL MeCN and an irradiation time of 1.5 hours. Purification by flash column chromatography (20:1 hexanes:Et2O) afforded 75 mg cycloadduct (0.18 mmol, 75%). Experiment 2: 100. mg (0.238 mmol) bis(enone), 9 mg (0.01 mmol) Ru(bpy)3Cl2·6H2O, 45 mg (0.48 mmol) LiBF4, 43 µL (2.4 mmol) H2O, 125 µL (0.717 mmol) i-Pr2NEt, and 2.4 mL MeCN. Isolated 79 mg cycloadduct (0.19 mmol, 79%) as a colorless oil. IR (thin film): 2928, 1683, 1279, 1123 cm−1. 1H NMR: (500 MHz, CDCl3) δ8.75 (s, 1H), 8.19 (dd, J = 8.8, 1.7 Hz, 1H), 8.03 (s, 1H), 7.93 (t, J = 7.9 Hz, 2H), 7.89 (d, J = 8.1 Hz, 1H), 7.80-7.67 (m 4H), 7.61 (td, J = 7.9, 0.9 Hz, 1H), 7.52 (td, J = 7.9, 0.9 Hz, 1H), 7.43-7.37 (m, 2H), 5.49 (d, J = 1.7 Hz, 1H), 5.16 (d, J = 10.4 Hz, 1H), 2.27 (dddd, J = 10.4, 10.4, 1.5, 1.5 Hz, 1H), 1.96 (qd, J = 10.5, 3.1 Hz, 2H), 1.82 (dt, J = 12.4, 2.4 Hz, 1H), 1.77 (dt, J = 12.4, 2.4 Hz, 1H), 1.61 (dd, J = 12.2, 2.4 Hz, 1H), 1.49-1.22 (m, 3H), 1.12 (qd, J = 13.0, 3.7 Hz, 1H); 13C NMR: (125 MHz, CDCl3) δ196.9, 149.7, 135.8, 133.2, 133.2, 133.1, 132.5, 132.4, 131.5, 129.9, 128.8, 128.5, 128.4, 127.8, 127.7, 127.5, 126.8, 126.1, 125.9, 124.8, 123.4, 122.6, 103.4, 83.1, 41.6, 38.7, 32.7, 27.8, 26.1, 25.9. HRMS (EI) calculated for [C30H26O2]+ requires m/z 418.1928, found 418.1936.
3.2.8 (Table 2, entry 6)
Experiment 1: Conducted using Method A with 102 mg (0.342 mmol) bis(enone), 13 mg (0.018 mmol) Ru(bpy)3Cl2·6H2O, 62 mg (0.66 mmol) LiBF4, 60 µL (3.4 mmol) H2O, 175 µL (1.01 mmol) i-Pr2NEt, and 3.4 mL MeCN and an irradiation time of 30 min. Purification by flash column chromatography (15:1 hexanes:EtOAc) afforded 79 mg cycloadduct (0.26 mmol, 77%). Experiment 2: 102 mg (0.340 mmol) bis(enone), 13 mg (0.017 mmol) Ru(bpy)3Cl2·6H2O, 62 mg (0.67 mmol) LiBF4, 60 µL (3.4 mmol) H2O, 175 µL (1.01 mmol) i-Pr2NEt, and 3.4 mL MeCN. Isolated 77 mg cycloadduct (0.26 mmol, 76%) as a colorless oil. IR (thin film): 2930, 1668, 1464 cm−1. 1H NMR: (500 MHz, CDCl3) δ7.67 (dd, J = 1.6, 0.6 Hz, 1H), 7.40 (dd, J = 3.7, 0.6 Hz, 1H), 7.36 (dd, J = 1.4, 1.4 Hz, 1H), 6.55 (dd, J = 3.7, 1.6 Hz, 1H), 6.39-6.36 (m, 2H), 5.29 (d, J = 1.8 Hz, 1H), 4.86 (d, J = 10.6 Hz, 1H), 2.14 (dddd, J = 10.6, 10.6, 2.2, 2.2 Hz, 1H), 1.90 (dddd, J = 13.0, 1.3, 1.3, 1.3 Hz, 1H), 1.79 (dddd, J = 12.4, 2.2, 2.2, 2.2 Hz, 1H), 1.69 (dddd, J = 10.5, 10.5, 10.5, 3.0 Hz, 1H), 1.60-1.53 (m, 1H), 1.44-1.08 (m, 4H); 13C NMR: (125 MHz, CDCl3) δ185.6, 150.8, 149.5, 147.4, 142.7, 142.0, 120.4, 112.3, 111.1, 105.9, 101.7, 83.3, 42.1, 38.0, 32.5, 27.2, 25.9, 25.8. HRMS (EI) calculated for m/z [C18H18O4]+ requires 298.1200, found 298.1187.
3.2.9 (Table 2, entry 7)
Experiment 1: Conducted using Method A with 99.7 mg (0.281 mmol) bis(enone), 11 mg (0.014 mmol) Ru(bpy)3Cl2·6H2O, 52 mg (0.56 mmol) LiBF4, 51 µL (2.8 mmol) H2O, 148 µL (0.847 mmol) i-Pr2NEt, and 2.8 mL MeCN and an irradiation time of 30 min. Purification by flash column chromatography (20:1 hexanes:Et2O) afforded 84 mg cycloadduct (0.24 mmol, 84%). Experiment 2: 99.8 mg (0.282 mmol) bis(enone), 11 mg (0.015 mmol) Ru(bpy)3Cl2·6H2O, 52 mg (0.55 mmol) LiBF4, 51 µL (2.8 mmol) H2O, 148 µL (0.847 mmol) i-Pr2NEt, and 2.8 mL MeCN. Isolated 84 mg cycloadduct (0.24 mmol, 84%) as a white solid (m.p. 34–36 °C). IR (thin film): 2928, 1693, 1610, 1490, 1451, 1267, 1211 cm−1. 1H NMR: (500 MHz, CDCl3) δ7.86 (td, J = 7.6, 1.7 Hz, 1H), 7.54 (m, 1H), 7.45 (td, J = 7.9, 1.7 Hz, 1H), 7.25 (td, J = 7.8, 1.1 Hz, 1H), 7.21-7.12 (m, 2H), 7.04-6.98 (m, 2H), 5.39 (bs, 1H), 5.01 (d, J = 10.4 Hz, 1H), 2.19 (dddd, J = 11.0, 11.0, 2.3, 2.3 Hz, 1H), 1.91-1.75 (m, 4H), 1.70 (dddd, J = 12.6, 2.7, 2.0, 2.0 Hz, 1H), 1.45-1.08 (m, 4H); 13C NMR: (125 MHz, CDCl3) δ196.9 (d, J = 3.2 Hz), 161.4 (d, J = 254.0 Hz), 159.8 (d, J = 251.2 Hz), 144.7 (d, J = 3.5 Hz), 134.6 (d, J = 9.8 Hz), 130.9 (d, J = 2.3 Hz), 128.9 (d, J = 8.6 Hz), 128.1 (d, J = 2.2 Hz), 125.8 (d, J = 13.1 Hz), 124.5 (d, J = 3.2 Hz), 123.8 (d, J = 3.6 Hz), 123.3 (d, J = 10.8 Hz), 116.7 (d, J = 23.2 Hz), 115.8 (d, J = 23.2 Hz), 107.9 (d, J = 12.1 Hz), 83.4 (d, J = 5.0 Hz), 40.9, 38.5, 27.4, 26.1, 25.9, 32.6. HRMS (EI) calculated for [C22H20F2O2]+ m/z requires 354.1426, found 354.1423.
3.2.10 (Table 2, entry 9)
Experiment 1: Conducted using Method A with 100. mg (0.275 mmol) bis(enone), 11 mg (0.014 mmol) Ru(bpy)3Cl2·6H2O, 52 mg (0.56 mmol) LiBF4, 50 µL (2.8 mmol) H2O, 144 µL (0.828 mmol) i-Pr2NEt, and 2.8 mL MeCN and an irradiation time of 20 min. Purification by flash column chromatography (10:1 hexanes:Et2O) afforded 75 mg cycloadduct (0.21 mmol, 75%). Experiment 2: 100. mg (0.276 mmol) bis(enone), 10 mg (0.014 mmol) Ru(bpy)3Cl2·6H2O, 52 mg (0.56 mmol) LiBF4, 50 µL (2.8 mmol) H2O, 144 µL (0.828 mmol) i-Pr2NEt, and 2.8 mL MeCN. Isolated 76 mg cycloadduct (0.21 mmol, 76%) as a colorless oil. IR (thin film): 2927, 1677, 1448, 1072 cm−1. 1H NMR: (500 MHz, CDCl3) δ8.07 (dt, J = 7.5, 1.2 Hz, 2H), 7.57 (tt, J = 7.5, 1.2 Hz, 1H), 7.43 (td, J = 7.5, 1.2 Hz, 2H), 7.36-7.25 (m, 5H), 4.84 (d, J = 10.4 Hz, 1H), 4.80 (s, 1H), 4.57 (s, 2H), 3.96 (ABq, J = 12.0 Hz, 2H), 2.03 (dddd, J = 11.0, 11.0, 1.4, 1.4 Hz, 1H), 1.84-1.69 (m, 4H), 1.49 (ddd, J = 12.8, 2.3, 2.3 Hz, 1H), 1.39-1.19 (m, 2H), 1.12 (dddd, J = 12.8, 12.8, 12.8, 3.2 Hz, 1H), 1.00 (dddd, J = 12.8, 12.8, 12.8, 3.7 Hz, 1H); 13C NMR: (125 MHz, CDCl3) δ197.0, 149.0, 138.2, 135.8, 133.4, 129.3, 128.5, 128.3, 127.8, 127.6, 104.9, 82.6, 72.3, 69.9, 41.4, 37.8, 32.4, 27.7, 26.0, 25.9. HRMS (EI) calculated for [C24H26O3]+ requires m/z 362.1877, found 362.1869.
3.2.11 (Table 2, entry 11)
Experiment 1: Conducted using Method B with 101 mg (0.394 mmol) bis(enone), 14 mg (0.019 mmol) Ru(bpy)3Cl2·6H2O, 174 mg (0.779 mmol) Mg(ClO4)2, 340 µL (1.95 mmol) i-Pr2NEt, and 15.6 mL MeCN and an irradiation time of 10 min. Purification by flash column chromatography (20:1 hexanes:EtOAc) afforded 75 mg cycloadduct (0.29 mmol, 74%). Experiment 2: 100. mg (0.386 mmol) bis(enone), 14 mg (0.019 mmol) Ru(bpy)3Cl2·6H2O, 173 mg (0.775 mmol) Mg(ClO4)2, 340 µL (1.95 mmol) i-Pr2NEt, and 15.6 mL MeCN. Isolated 72 mg cycloadduct (0.28 mmol, 72%) as a colorless oil. IR (thin film): 2923, 1680, 1448, 1280 cm−1. 1H NMR: (500 MHz, CDCl3) δ8.05 (dt, J = 7.7, 1.3 Hz, 2H), 7.58 (tt, J = 7.7, 1.3 Hz, 1H), 7.47 (tt, J = 7.7, 1.3 Hz, 2H), 4.82 (d, J = 10.3 Hz, 1H), 4.43 (s, 1H), 1.95 (dddd, J = 11.3, 11.3, 1.8, 1.8 Hz, 1H), 1.79 (dd, J = 2.0, 0.8 Hz, 3H), 1.77-1.64 (m, 4H), 1.44 (dddd, J = 12.4, 2.7, 2.7, 2.7 Hz, 1H), 1.37-1.18 (m, 2H), 1.07 (dddd, J = 12.4, 12.4, 12.4, 2.7 Hz, 1H), 0.97 (dddd, J = 12.6, 12.6, 12.6, 3.6 Hz, 1H); 13C NMR: (125 MHz, CDCl3) δ197.5, 149.2, 136.0, 133.4, 129.2, 128.5, 128.5, 101.5, 82.3, 41.7, 38.1, 32.7, 27.7, 25.9, 25.9, 19.7. HRMS (EI) calculated for [C17H20O2]+ requires m/z 256.1458, found 256.1455.
3.2.12 (Table 2, entry 12)
Experiment 1: Conducted using Method B with 100. mg (0.353 mmol) bis(enone), 14 mg (0.018 mmol) Ru(bpy)3Cl2·6H2O, 159 mg (0.710 mmol) Mg(ClO4)2, 307 µL (1.76 mmol) i-Pr2NEt, and 14.1 mL MeCN and an irradiation time of 1.5 hours. Purification by flash column chromatography (20:1 hexanes:Et2O) afforded 58 mg cycloadduct (0.20 mmol, 58%). Experiment 2: 101 mg (0.354 mmol) bis(enone), 14 mg (0.018 mmol) Ru(bpy)3Cl2·6H2O, 158 mg (0.708 mmol) Mg(ClO4)2, 307 µL (1.76 mmol) i-Pr2NEt, and 14.1 mL MeCN. Isolated 58 mg cycloadduct (0.20 mmol, 58%) as a colorless oil. IR (thin film): 2927, 1674, 1448, 1268 cm−1. 1H NMR: (500 MHz, CDCl3) δ8.07 (ddd, J = 7.8, 1.2, 1.0 Hz, 2H), 7.57 (tt, J = 7.8, 1.2 Hz, 1H), 7.46 (ddd, J = 7.8, 7.8, 1.0 Hz, 2H), 4.70 (d, J = 10.4 Hz, 1H), 4.42 (s, 1H), 2.27 (sept, J = 6.8 Hz, 1H), 1.95 (dddd, J = 10.5, 10.5, 1.8, 1.8 Hz, 1H), 1.80-1.59 (m, 4H), 1.47 (dddd, J = 13.0, 3.0, 3.0, 3.0 Hz, 1H), 1.38-1.16 (m, 2H), 1.24-0.92 (m, 8H); 13C NMR: (125 MHz, CDCl3) δ197.5, 157.6, 135.7, 133.3, 129.4, 128.4, 98.4, 83.2, 41.7, 37.9, 32.9, 32.0, 27.6, 26.0, 26.0, 20.5. HRMS (EI) calculcated for [C19H24O2]+ requires m/z 284.1771, found 284.1780.
3.3 Diastereoselective functionalization of cycloadduct 4
3.3.1 (12, Scheme 3)
To an oven-dried 10 mL round bottom flask equipped with a magnetic stir bar was added cycloadduct 4 (100. mg, 0.314 mmol), p-TsOH·H2O (6 mg, 0.03 mmol), and methanol (3.2 mL). The reaction was allowed to stir at room temperature for 3.5 h and then concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (8:1 hexanes:EtOAc) to afford the product (104 mg, 0.297 mmol, 95%) as a white solid (m.p. 91–97 °C). IR (thin film): 3059, 2924, 2854, 1679 cm−1. 1H NMR: (500 MHz, CDCl3) δ8.21 (ddd, J = 7.5, 1.0, 1.0 Hz, 2H), 7.61 (tt, J = 7.5, 1.0 Hz, 1H), 7.51 (t, J = 7.8 Hz, 2H), 7.49 (d, J = 7.3 Hz, 2H), 7.34 (td, J = 7.4, 1.0 Hz, 2H), 7.28 (tt, J = 7.3, 1.0 Hz, 1H), 4.73 (d, J = 10.6 Hz, 1H), 3.05 (s, 3H), 2.09 (dd, J = 13.6, 3.6 Hz, 1H), 1.94 (qt, J = 11.8, 3.5 Hz, 1H), 1.70 (m, 4H), 1.51 (t, J = 13.0 Hz, 1H), 1.44 (dt, J = 12.8, 2.2 Hz, 1H), 1.35 (qt, J = 12.7, 3.5 Hz, 1H), 1.23 (qt, J = 12.8, 3.5 Hz, 1H), 1.07 (qd, J = 12.2, 3.5 Hz, 1H), 1.00 (qd, J = 12.2, 3.5 Hz, 1H); 13C NMR: (125 MHz, CDCl3) δ197.8, 142.0, 136.2, 133.3, 129.4, 128.5, 128.2, 127.8, 125.9, 100.8, 78.5, 49.5, 44.8, 43.2, 36.0, 32.8, 27.4, 25.9, 25.7;. HRMS (EI) calculated for [C23H26O3 -MeO]+ requires m/z 319.1693, found 319.1689.
3.3.2 (13, Scheme 3)
A 1.5 dram vial was charged with potassium osmate (VI) dihydrate (2 mg, 0.005 mmol) and sealed with a teflon cap. To the vial was added cycloadduct 4 (100. mg, 0.314 mmol), N-methylmorpholine N-oxide (54 mg, 0.46 mmol), citric acid (60 mg, 0.31 mmol) and a magnetic stir bar, followed by t-butanol (0.93 mL) and distilled water (0.31 mL). The reaction was then resealed and allowed to stir at room temperature for 23 h. Sodium sulfite (40 mg, 0.32 mmol) was then added and the reaction was stirred another 30 minutes. The reaction was diluted with water and extracted twice with EtOAc. The combined organic extracts were dried over sodium sulfate, filtered and concentrated in vacuo to a white solid. The solid was purified by flash column chromatography on silica gel (10:1 toluene:acetone) then recrystallized in hexanes and ethyl acetate to afford the product (65 mg, 0.18 mmol, 60%) as a white crystalline solid (m.p. 153–154 °C). IR (thin film): 3447, 2931, 2856, 1684 cm−1. 1H NMR: (500 MHz, Acetone-d6) δ8.14 (dt, J = 8.5, 1.5 Hz, 2H), 7.61 (dd, J = 8.5, 1.5 Hz, 1H), 7.60 (qt, J = 6.7, 1.3 Hz, 2H), 7.48 (tt, J = 7.8, 1.5 Hz, 2H), 7.29-7.21 (m, 3H), 5.63 (s, 1H (OH)), 5.13 (d, J = 10.4 Hz, 1H), 3.40 (d, J = 9.3 Hz, 1H (OH)), 3.29 (t, J = 9.3 Hz, 1H), 2.24 (m, 1H), 1.98 (qd, J = 11.0, 3.2 Hz, 1H), 1.86 (qd, J = 11.0, 3.2 Hz, 1H), 1.81 (m, 1H), 1.72 (m, 1H), 1.57 (m, 1H), 1.38-1.24 (m, 2H), 1.13-0.94 (m, 2H); 13C NMR: (125 MHz, Acetone-d6) δ196.1, 143.8, 136.4, 133.1, 129.2, 128.5, 127.5, 127.2, 126.6, 98.7, 76.7, 74.8, 42.7, 41.7, 28.4, 27.2, 25.7, 25.2. HRMS (EI) calculated for [C22H24O4 + Na]+ requires m/z 375.1567, found 375.1583.
3.3.3 (14, Scheme 3)
To an oven-dried 500 mL pressure flask equipped with a magnetic stir bar was added 10% palladium on carbon (62 mg) followed by cycloadduct 4 (100. mg, 0.314 mmol) in dichloromethane (8.0 mL) under an atmosphere of nitrogen. The flask was fitted with a regulator and filled with H2 and evacuated twice, then filled with 20 psi of H2. The reaction was allowed to stir at room temperature for 3 h, and then the excess H2 was vented. The reaction was filtered through Celite and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (8:1 hexanes:EtOAc) to afford the product (70 mg, 0.22 mmol, 69%) as a clear oil. IR (thin film): 3553, 3432, 2926, 2852 cm−1. 1H NMR: (500 MHz, CDCl3) δ 7.46 (dt, J = 7.0, 1.5 Hz, 2H), 7.39-7.26 (m, 8H), 4.80 (dd, J = 9.4, 3.2 Hz, 1H), 4.49 (dd, J = 11.3, 2.2 Hz, 1H), 3.70 (dd, J = 10.1, 3.3 Hz, 1H), 3.40 (d, J = 9.6 Hz, 1H, (OH)), 1.80 (dddd, J = 12.4, 2.8, 2.8, 2.8 Hz, 1H), 1.73 (m, 1H), 1.67 (tt, J = 12.4, 2.8 Hz, 2H), 1.56 (dddd, J = 12.9, 2.8, 2.8, 2.8 Hz, 1H), 1.40 (tt, J = 11.7, 3.3 Hz, 1H), 1.27 (qt, J = 13.0, 3.7 Hz, 1H), 1.16 (q, J = 12.3 Hz, 1H), 1.12 (qt, J = 13.0, 3.7 Hz, 1H), 1.00 (qd, J = 12.1, 3.3 Hz, 1H), 0.89 (qd, J = 12.8, 3.7 Hz, 1H), 0.76 (qd, J = 10.2, 3.2 Hz, 1H); 13C NMR: (125 MHz, CDCl3) δ143.2, 140.6, 128.3, 128.0, 127.9, 127.5, 127.3, 125.8, 84.5, 80.1, 73.8, 42.4, 41.7, 41.1, 32.7, 27.2, 25.9, 25.6. HRMS (EI) calculated for [C22H26O2 + Na]+ requires m/z 345.1825, found 345.1835.
3.4 Stereochemical determinations
NOE correlations were used to verify the relative stereochemistry of acetal 12 and dihydroxylation product 13, as well as the regiochemistry of the cycloadduct in Table 2, entry 11. The structure of 14 was determined unambiguously by single-crystal X-ray diffraction of the corresponding p-nitrobenzoate ester.xii The structures of other compounds were assigned by analogy.
Acknowledgments
We thank Dr. Ilia Guzei for performing the crystallographic determination of the structure of 14. We are indebted to the ACS PRF (49817-ND1), NIH (GM095666), Sloan Foundation, Beckman Foundation, and Research Corporation for research funding. The NMR facilities at UW–Madison are funded by the NSF (CHE-9208463, CHE-9629688) and NIH (RR08389-01)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- i.For recent reviews, see: Tietze LF, Kettschau G. Top. Curr. Chem. 1997;189:1–120. Tietze LF, Kettschau G, Gewert JA, Schuffenhauer A. Curr. Org. Chem. 1998;2:19–62. Boger DL, Weinreb SM. In: Hetero-Diels–Alder Methodology in Organic Synthesis. Wasserman HH, editor. San Diego: Academic Press; 1987. Jørgensen KA. Eur. J. Org. Chem. 2004:2093–2102. Pellissier H. Tetrahedron. 2009;65:2839–2877.
- ii.For seminal examples, see: Thompson CF, Jamison TF, Jacobsen EN. J. Am. Chem. Soc. 2000;122:10482–10483. Liu P, Jacobsen EJ. J. Am. Chem. Soc. 2001;123:10772–10773. doi: 10.1021/ja016893s. Chavez DE, Jacobsen EN. Angew. Chem., Int. Ed. 2001;40:3667–3670. doi: 10.1002/1521-3773(20011001)40:19<3667::aid-anie3667>3.0.co;2-6. Paterson I, De Savi C, Tudge M. Org. Lett. 2001;3:3149–3152. doi: 10.1021/ol010150u. Evans DA, Starr JT. Angew. Chem., Int. Ed. 2002;41:1787–1790. doi: 10.1002/1521-3773(20020517)41:10<1787::aid-anie1787>3.0.co;2-v. Paterson I, Tudge M. Angew. Chem., Int. Ed. 2003;42:343–347. doi: 10.1002/anie.200390112. Voight EA, Seradj H, Roethle PA, Burke SD. Org. Lett. 2004;6:4045–4048. doi: 10.1021/ol0483044. Tietze LF, Rackelmann N, Müller I. Chem.—Eur. J. 2004;10:2722–2731. doi: 10.1002/chem.200306039. Lucas BS, Luther LM, Burke SD. J. Org. Chem. 2005;70:3757–3760. doi: 10.1021/jo050034v. Majumder U, Cox JM, Johnson HWB, Rainier JD. Chem.—Eur. J. 2006;12:1736–1746. doi: 10.1002/chem.200500993. Louis I, Hungerford NL, Humphries EJ, McLeod MD. Org. Lett. 2006;8:1117–1120. doi: 10.1021/ol053092b. Lucas BS, Gopalsamuthiram V, Burke SD. Angew. Chem., Int. Ed. 2007;46:769–772. doi: 10.1002/anie.200603656. Ghosh AK, Gong G. Org. Lett. 2007;9:1437–1440. doi: 10.1021/ol0701013. Bonazzi S, Güttinger S, Zemp I, Kutay U, Gademann K. Angew. Chem., Int. Ed. 2007;46:8707–8710. doi: 10.1002/anie.200703134. Dilger AK, Gopalsamuthiram V, Burke SD. J. Am. Chem. Soc. 2007;129:16273–16277. doi: 10.1021/ja077336u.
- iii.(a) Desimoni G, Tacconi G. Chem. Rev. 1975;75:651–692. [Google Scholar]; (b) Schmidt JA, Jorgensen WL. J. Org. Chem. 1983;48:3923–3941. [Google Scholar]; (c) Jun J-G, Shin DG, Lee CK, Sin KS. Bull. Korean Chem. Soc. 1990;11:307–309. [Google Scholar]
- iv.a) Ischay MA, Anzovino ME, Du J, Yoon TP. J. Am. Chem. Soc. 2008;130:12886–12887. doi: 10.1021/ja805387f. [DOI] [PubMed] [Google Scholar]; (b) Du J, Yoon TP. J. Am. Chem. Soc. 2009;131:14604–14605. doi: 10.1021/ja903732v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ischay MA, Lu Z, Yoon TP. J. Am. Chem. Soc. 2010;132:8572–8574. doi: 10.1021/ja103934y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lu Z, Shen M, Yoon TP. J. Am. Chem. Soc. 2011 doi: 10.1021/ja107849y. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- v.(a) Nicewicz D, MacMillan DWC. Science. 2008;322:70–80. doi: 10.1126/science.1161976. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nagib DA, Scott ME, MacMillan DWC. J. Am. Chem. Soc. 2009;131:10875–10877. doi: 10.1021/ja9053338. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Narayanam JMR, Tucker JW, Stephenson CRJ. J. Am. Chem. Soc. 2009;131:8756–8757. doi: 10.1021/ja9033582. [DOI] [PubMed] [Google Scholar]; (d) Koike T, Akita M. Chem. Lett. 2009;38:166–167. [Google Scholar]; (e) Tucker JW, Narayanam JMR, Krabbe SW, Stephenson CRJ. Org. Lett. 2010;12:368–371. doi: 10.1021/ol902703k. [DOI] [PubMed] [Google Scholar]; (f) Condie AG, González-Gómez JC, Stephenson CRJ. J. Am. Chem. Soc. 2010;132:1464–1465. doi: 10.1021/ja909145y. [DOI] [PubMed] [Google Scholar]; (g) Tucker JW, Nguyen JD, Narayanam JMR, Krabbe SW, Stephenson CRJ. Chem. Commun. 2010;46:4985–4987. doi: 10.1039/c0cc00981d. [DOI] [PubMed] [Google Scholar]; (h) Furst L, Matsuura BS, Narayanam JMR, Tucker JW, Stephenson CRJ. Org. Lett. 2010;12:3104–3107. doi: 10.1021/ol101146f. [DOI] [PubMed] [Google Scholar]; (i) Andrews RS, Becker JJ, Gagné MR. Angew. Chem. Int. Ed. 2010:7274–7276. doi: 10.1002/anie.201004311. [DOI] [PubMed] [Google Scholar]; (j) Shih H-W, Vander Wal MN, Grange RL, MacMillan DWC. J. Am. Chem. Soc. 2010;132:13600–13603. doi: 10.1021/ja106593m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Rueping M, Vila C, Koenigs RM, Poscharny K, Fabry DC. Chem. Commun. 2011;47:2360–2362. doi: 10.1039/c0cc04539j. [DOI] [PubMed] [Google Scholar]; (l) Maji T, Karmakar A, Reiser O. J. Org. Chem. 2011;76:736–739. doi: 10.1021/jo102239x. [DOI] [PubMed] [Google Scholar]
- vi.For reviews on recent developments in transition metal photoredox catalysis in organic synthesis, see: Zeitler K. Angew. Chem. Int. Ed. 2009;48:9785–9789. doi: 10.1002/anie.200904056. Narayanam JMR, Stephenson CRJ. Chem. Soc. Rev. 2010 Yoon TP, Ischay MA, Du J. Nature Chem. 2010;2:527–532. doi: 10.1038/nchem.687.
- vii.(a) Roh Y, Jang HY, Lynch V, Bauld NL, Krische MJ. Org. Lett. 2002;4:611–613. doi: 10.1021/ol0172065. [DOI] [PubMed] [Google Scholar]; (b) Yang J, Felton GAN, Bauld NL, Krische MJ. J. Am. Chem. Soc. 2004;126:1634–1635. doi: 10.1021/ja030543j. [DOI] [PubMed] [Google Scholar]; (c) Felton GAN, Bauld NL. Tetrahedron Lett. 2004;45:8465–8469. [Google Scholar]; (d) Felton GAN, Bauld NL. Tetrahedron. 2004;60:10999–11010. [Google Scholar]
- viii.The origin of this effect is not clear at this time, but given the polar nature of the intermediates and the step-wise mechanism proposed for radical anion cycloadditions, the hydrophobic effect proposed by Breslow for the rate acceration of thermal Diels–Alder reactions in water is most likely not operational: Breslow R. Acc. Chem. Res. 1991;24:159–164.
- ix.Fournier F, Fournier M. Can. J. Chem. 1986;64:881–890. [Google Scholar]
- x.VanRheenen V, Kelly RC, Cha DY. Tetrahedron Lett. 1976;17:1973–1976. [Google Scholar]
- xi.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Organometallics. 1996;15:1518–1520. [Google Scholar]
- xii.CCDC 807947 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.