

Abstract
We have developed a statistical regression modeling approach to discover genes that are differentially expressed between two predefined sample groups in DNA microarray experiments. Our model is based on well-defined assumptions, uses rigorous and well-characterized statistical measures, and accounts for the heterogeneity and genomic complexity of the data. In contrast to cluster analysis, which attempts to define groups of genes and/or samples that share common overall expression profiles, our modeling approach uses known sample group membership to focus on expression profiles of individual genes in a sensitive and robust manner. Further, this approach can be used to test statistical hypotheses about gene expression. To demonstrate this methodology, we compared the expression profiles of 11 acute myeloid leukemia (AML) and 27 acute lymphoblastic leukemia (ALL) samples from a previous study (Golub et al. 1999) and found 141 genes differentially expressed between AML and ALL with a 1% significance at the genomic level. Using this modeling approach to compare different sample groups within the AML samples, we identified a group of genes whose expression profiles correlated with that of thrombopoietin and found that genes whose expression associated with AML treatment outcome lie in recurrent chromosomal locations. Our results are compared with those obtained using t-tests or Wilcoxon rank sum statistics.
The development of oligonucleotide microarray technologies allows scientists to monitor the mRNA transcript levels of thousands of genes in a single experiment. Indeed, several groups have already begun to simultaneously examine the expression profiles of entire genomes for organisms such as yeast whose complete DNA sequences are known (Lashkari et al. 1997; Chu et al. 1998; Spellman et al. 1998; Ferea et al. 1999). This power of examination and discovery moves well beyond the traditional experimental approach of focusing on one gene at a time. Nevertheless, the tremendous amount of data that can be obtained from microarray studies presents a challenge for data analysis (Brent 2000).
At present, the most commonly used computational approach for analyzing microarray data is cluster analysis. Cluster analysis groups genes or samples into “clusters” based on similar expression profiles and provides clues to the function or regulation of genes or similarity of samples via shared cluster membership (Tamayo et al. 1999; Tavazoie et al. 1999; Gaasterland and Bekiranov 2000). Several clustering methods have been usefully applied to analyzing genome-wide expression data and can be classified largely into three categories. The tree-based approach uses distance measures between genes such as correlation coefficients to group genes into a hierarchical tree (Eisen et al. 1998). The second category clusters genes so that within-cluster variation is minimized and between-cluster variation is maximized (Tamayo et al. 1999; Tavazoie et al. 1999). The third category groups genes into blocks, in which the correlation is maximized and between which the correlation is minimized (Ben-Dor et al. 1999).
The power of cluster analysis for microarray studies lies in discovering gene transcripts or samples that show similar expression profiles. Examples include identification of transcripts that appear to be coregulated over a time course (Chu et al. 1998; Spellman et al. 1998), or uncovering previously unknown sample groupings (Alon et al. 1999; Alizadeh et al. 2000). However, identification of “like” groups is not necessarily the objective in a microarray study. For example, microarrays present a high-throughput method to discover genes that are differentially expressed between predefined sample groups, such as normal versus cancerous tissues (Alon et al. 1999; Coller et al. 2000). Cluster analysis is not a sensitive method for this type of study because it focuses on group similarities, not differences within each individual gene. Furthermore, clustering algorithms such as those listed above are also unable to take advantage of preexisting knowledge of the data, such as the sample groupings.
The technique that has been most commonly applied for group comparisons from microarray studies is to simply look for genes with a twofold or higher difference between the mean intensities for each group (DeRisi et al. 1997). However, relative mean comparisons fail to account for sample variation, may require ad hoc data manipulation (e.g., to avoid divide-by-zero errors), and ignore the fact that differences in expression level of <100% can exert meaningful biological effects. Indeed, scientists would rarely use similar criteria when focusing their analysis on a single gene, such as comparing a panel of Northern blots or enzymatic assays between healthy and cancer tissue samples.
Classic statistical approaches used for detecting differences between two groups include the parametric t-test and the nonparametric Wilcoxon rank sum (Snedecor and Cochran 1980). Recently, the t-test was used to compare expression profiles in microarray experiments (Arfin et al. 2000; Tanaka et al. 2000). One must bear in mind three important issues when applying such standard statistical tests to microarray data analysis. First, the t-test assumes normality and constant variance for every gene across all samples. These assumptions are certainly inappropriate for a subset of genes despite any given transformation. Second, these tests cannot take advantage of the genomic data when correcting for heterogeneity between samples. Third, it is essential to correct for the high false-positive rate resulting from multiple comparisons. Otherwise, if a typical P-value of 0.05 were used to signify differential expression for individual genes between two groups, one would expect to find 50 positives for every 1000 genes under examination, even though none of these genes are differentially expressed.
In this manuscript, we introduce a well-founded and robust statistical procedure that compares the expression profiles of individual genes between two sample groups while taking into consideration the complexity of the genomic data. This methodology makes no distributional assumptions about the data and accounts for high false-positive error rate resulting from multiple comparisons. To demonstrate the statistical modeling technique, we examined expression profiles from 38 leukemia patients, 27 of whom were diagnosed with acute lymphoblastic leukemia (ALL) and 11 of whom were diagnosed with acute myeloid leukemia (AML) (Golub et al. 1999). Our results are compared with those obtained with the t-test or Wilcoxon rank sum. The findings show that our statistical modeling approach provides a sensitive and robust means to extract relevant information from DNA microarrays.
RESULTS
Methodology
The first step in our statistical analysis of oligonucleotide-array expression profiles is preprocessing and/or transformation of the data. In the present work this includes removal of the spiked oligonucleotide controls. The second step is to estimate correction factors for sample-specific heterogeneity, as well as for chip-specific heterogeneity, and to use these factors to normalize the data. The final step is to perform a regression analysis to estimate the relevant model parameters (equation 1 in Methods) for each gene transcript using robust statistical techniques. The results are ranked by the absolute value of the Z-score for each transcript. The higher the Z-score, the greater the confidence level that the corresponding gene is differentially expressed between the two groups.
Our methodology is implemented in a software program. Interested investigaors may contact L.P.Z. for details.
Multiple Comparisons
At issue when performing a large number of statistical tests is the high occurrence rate of false positives resulting from the multiple comparisons. To address this concern, we propose to raise the statistical threshold for declaring a transcript differentially expressed to ensure that the significance level is applicable on the genomic scale. A conservative choice to adjust the significance is the Bonferroni's correction, which divides the desired significance, for example, 1% or P-value = 0.01, by the total number of statistical tests performed. In this work, we calculated the significance value (i.e., P-value) for each probe set using a modified Bonferroni's correction as proposed by Hochberg (Hochberg 1988) (see Methods for details).
Applying Bonferroni's correction to data from Affymetrix Hu6800 GeneChip oligonucleotide arrays, which contain 7070 noncontrol probe sets for 6817 individual genes, the adjusted significance level for each probe set is 0.01/7070. Assuming that the Z-score follows the normal distribution, the corresponding 1% significance threshold at the genomic level is a Z-score of 4.8. Alternatively, one may adjust the significance by the total number of genes rather than the total number of probe sets. However, different probe sets for the same gene may yield dissimilar results, and either level of correction results in a rounded Z-score of 4.8 at the 1% significance level.
Leukemia Study
A previous study examined mRNA expression profiles from 38 leukemia patients (27 ALL and 11 AML) to develop an expression-based classification method for acute leukemia (Golub et al. 1999). Affymetrix Hu6800 GeneChips were used in the study. The data set from this study was ideal for illustrating our modeling technique as it contains a large number of patients and has been well characterized (Golub et al. 1999). Furthermore, there is a great deal of literature concerning leukemia from which we can assess the validity of our findings.
Our statistical modeling approach identified 141 probe sets that were differentially expressed between AML and ALL with a Z-score of 4.8 or higher. Twenty-four of these were detected at higher levels in AML and the remainder were expressed preferentially in ALL. Tables 1 and 2 list the top 25 differentially expressed probe sets in either sample group. These tables also include the corresponding P-values and ordering of the statistics given to each probe set by t-tests with either equal or unequal variance, and by the Wilcoxon rank sum. As expected, the ranked significance given to each gene by any of the statistical tests did not appear to correlate with either relative or absolute mean expression level differences. Tables 1 and 2 show that parametric t-tests under equal variances yielded rather different test statistics and ordering than our modeling approach. In contrast, the ordering of the probe sets by t-tests performed assuming unequal variances was very similar to that obtained in our regression analysis. Although t-tests are efficient under the assumption of equal variances, the results of this analysis appeared very sensitive to this assumption. In cases of discrepancies between t-tests with unequal variances and Z-scores, the latter are considered to be more robust because the assumptions of homogeneous variances within groups and normality made by the t-test may be violated. Note that the differences of P-values between the two statistics are associated with distributions; the t-distribution with heavy tails gives more conservative values than the asymptotic normal distribution we used to translate Z-scores to P-values. The Wilcoxon rank sum failed to identify any genes as differentially expressed at the 1% significance level. These findings are not surprising because nonparametric statistics may be too robust to yield any significant results.
Table 1.
Top 25 Genes More Highly Expressed in AML Than in ALL
| Gene description | Probe | Difference | S.E. | Z-score | P-value1 | Fold diff2 | t-test3 | t rank4 | ut-test5 | ut rank6 | W. r.s.7 | W rank8 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fumarylacetoacetate | M55150 | 978.04 | 101.41 | 9.64 | <0.0001 | 2.21 | <0.0001 | 1 | <0.0001 | 1 | 0.0146 | 2 |
| Neuromedin B | M21551 | 216.62 | 33.81 | 6.41 | <0.0001 | 1.94 | 0.1952 | 64 | 0.0025 | 2 | 0.0432 | 7 |
| Leukotriene C4 synthase | U50136 | 1551.78 | 253.94 | 6.11 | <0.0001 | 2.57 | <0.0001 | 2 | 0.0082 | 4 | 0.0584 | 11 |
| CDC25A Cell division cycle 25A | M81933 | 178.74 | 29.31 | 6.10 | <0.0001 | 3.60 | 0.0510 | 38 | 0.0070 | 3 | 0.0678 | 14 |
| Thrombospondin 1 | U12471 | 128.52 | 21.94 | 5.86 | <0.0001 | 1.83 | 0.0570 | 39 | 0.0145 | 5 | 0.3275 | 33 |
| Zyxin | X95735 | 2587.14 | 445.32 | 5.81 | <0.0001 | 7.98 | <0.0001 | 3 | 0.0199 | 6 | 0.0124 | 1 |
| LYN V-yes-1 Yamaguchi sarcoma viral related oncogene homolog | M16038 | 1337.77 | 231.45 | 5.78 | <0.0001 | 4.49 | <0.0001 | 4 | 0.0216 | 7 | 0.0503 | 10 |
| Interferon-gamma inducing factor (IGIF) | D49950 | 167.60 | 29.25 | 5.73 | <0.0001 | 3.18 | 0.0004 | 7 | 0.0241 | 8 | 0.6428 | 50 |
| ATP6C Vacuolar H+ ATPase proton channel subunit | M62762 | 1686.88 | 300.07 | 5.62 | 0.0001 | 2.33 | 0.0024 | 13 | 0.0322 | 9 | 0.4925 | 43 |
| Metargidin | U41767 | 481.47 | 86.18 | 5.59 | 0.0002 | 1.50 | 0.0049 | 19 | 0.0353 | 10 | 0.1220 | 16 |
| Leptin receptor | Y12670 | 859.65 | 154.73 | 5.56 | 0.0002 | 3.10 | <0.0001 | 5 | 0.0414 | 12 | 0.0233 | 3 |
| Ferritin, light polypeptide | M11147 | 7928.14 | 1428.69 | 5.55 | 0.0002 | 1.97 | 0.0799 | 44 | 0.0368 | 11 | 0.8361 | 55 |
| HoxA9 | U82759 | 602.71 | 110.33 | 5.46 | 0.0003 | 3.48 | 0.0017 | 12 | 0.0521 | 13 | 0.1409 | 18 |
| NAB50 | U63289 | 121.53 | 23.29 | 5.22 | 0.0013 | n.a. | 0.3464 | 79 | 0.0955 | 14 | 1.0000 | >58 |
| Calnexin | D50310 | 1982.20 | 385.66 | 5.14 | 0.0019 | 1.47 | 0.6143 | 92 | 0.1184 | 15 | 1.0000 | >58 |
| PLCB2 Phospholipase C, beta 2 | M95678 | 1281.19 | 249.67 | 5.13 | 0.0020 | 1.97 | 0.0147 | 28 | 0.1347 | 16 | 0.4924 | 41 |
| Polyadenylate binding protein II | Z48501 | 4474.14 | 879.99 | 5.08 | 0.0026 | 1.50 | 0.5856 | 90 | 0.1404 | 17 | 1.0000 | >58 |
| GTP-binding protein (RAB31) | U59877 | 398.95 | 79.87 | 5.00 | 0.0041 | n.a. | 1.0000 | >105 | 0.1772 | 18 | 1.0000 | >58 |
| Chloride channel (putative) 2163bp | Z30644 | 760.30 | 154.08 | 4.93 | 0.0056 | 1.69 | 0.2998 | 76 | 0.2253 | 19 | 1.0000 | >58 |
| Proteogylcan 1, secretory granule | X17042 | 5188.71 | 1060.82 | 4.89 | 0.0070 | 3.82 | 0.0129 | 27 | 0.2738 | 20 | 0.8359 | 53 |
| PPIase, mitochondrial | M80254 | 377.99 | 77.41 | 4.88 | 0.0073 | 10.75 | 0.0015 | 10 | 0.2883 | 22 | 0.4925 | 42 |
| CD33 | M23197 | 579.93 | 119.10 | 4.87 | 0.0078 | 4.25 | 0.0002 | 6 | 0.3060 | 25 | 0.0503 | 9 |
| Activated leucocyte cell adhesion molecule | L38608 | 119.96 | 24.71 | 4.85 | 0.0084 | 2.51 | 0.0488 | 37 | 0.2984 | 23 | 1.0000 | >58 |
| FCGR2B Fc fragment of IgG, low affinity IIb, receptor for (CD32) | X62573 | 257.44 | 53.18 | 4.84 | 0.0090 | 1.47 | 1.0000 | >105 | 0.2835 | 21 | 1.0000 | >58 |
| Interkeukin-8 | Y00787 | 8645.98 | 1802.75 | 4.80 | 0.0112 | 9.63 | 0.0015 | 11 | 0.3032 | 24 | 0.2158 | 27 |
Dataset from Golub et al. (1999).
P-value computed from Z-score using a modified Bonferroni's correction.
(n.a.) The mean of one of the groups < = zero.
P-value obtained from t-test with equivalent variances using a modified Bonferroni's correction.
Relative ranking by significance values obtained from t-test with equivalent variances. >105 indicates that the gene was not ranked because the P-value was 1.0.
P-value obtained from t-test with unequal variances using a modified Bonferroni's correction.
Relative ranking by significance values obtained from t-test with unequal variances.
P-value obtained from Wilcoxon rank sum using a modified Bonferroni's correction.
Relative ranking by Wilcoxon significance values. >58 indicates that the gene was not ranked because the P-value was 1.0.
Table 2.
Top 25 Genes More Highly Expressed in ALL Than in AML
| Gene description | Probe | Difference | S.E. | Z-score | P-value1 | Fold diff2 | t-test3 | t rank4 | ut-test5 | ut rank6 | W. r.s.7 | W rank8 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C-myb | U22376 | −3183.79 | 429.30 | −7.42 | <0.0001 | 5.39 | 0.1827 | 7 | <0.0001 | 1 | 0.0371 | 2 |
| p48 | X74262 | −1115.97 | 152.35 | −7.33 | <0.0001 | 5.24 | 0.2268 | 8 | <0.0001 | 2 | 0.0911 | 6 |
| Proteasome iota chain | X59417 | −3331.23 | 481.79 | −6.91 | <0.0001 | 3.94 | 0.3777 | 12 | <0.0001 | 3 | 0.3756 | 23 |
| Myosin light chain (alkali) | M31211 | −408.99 | 59.50 | −6.87 | <0.0001 | 3.57 | 0.1584 | 5 | <0.0001 | 4 | 0.2854 | 19 |
| Macmarcks | HG1612– | −2512.37 | 372.76 | −6.74 | <0.0001 | 2.87 | 0.2555 | 9 | <0.0001 | 5 | 0.0432 | 3 |
| HT1612 | ||||||||||||
| Transcription factor 3 (E2A) | M65214 | −471.84 | 70.04 | −6.74 | <0.0001 | 1.56 | 0.1759 | 6 | <0.0001 | 6 | 0.8348 | >59 |
| Inducible protein | L47738 | −1055.39 | 159.50 | −6.62 | <0.0001 | 6.64 | 0.7782 | 23 | 0.0012 | 7 | 0.0584 | 5 |
| MB-1 (CD79b) | U05259 | −3399.02 | 523.02 | −6.50 | <0.0001 | 12.26 | 1.0000 | >28 | 0.0016 | 8 | 1.0000 | >59 |
| Crystallin zeta (quinone reductase) | L13278 | −118.93 | 18.33 | −6.49 | <0.0001 | 27.36 | 0.4366 | 16 | 0.0018 | 9 | 0.5629 | 37 |
| Transcriptional activator hSNF2b | D26156 | −766.23 | 118.24 | −6.48 | <0.0001 | 2.35 | 0.3940 | 13 | 0.0019 | 10 | 0.4304 | 18 |
| Acyl-Coenzyme A dehydrogenase, C-4 to C-12 straight chain | M91432 | −669.68 | 104.25 | −6.42 | <0.0001 | 4.29 | 1.0000 | >28 | 0.0020 | 11 | 0.1626 | 11 |
| Oncoprotein 18 | M31303 | −2013.15 | 314.83 | −6.39 | <0.0001 | 2.12 | 0.0309 | 2 | 0.0029 | 13 | 0.1874 | 13 |
| Thymopoietin beta | U09087 | −125.08 | 19.56 | −6.39 | <0.0001 | 3.43 | 0.8446 | 26 | 0.0023 | 12 | 0.3277 | 22 |
| Cyclin D3 | M92287 | −3025.10 | 484.75 | −6.24 | <0.0001 | 3.86 | 1.0000 | >28 | 0.0035 | 14 | 0.0911 | 7 |
| Serine kinase SRPK2 | U88666 | −105.40 | 16.98 | −6.21 | <0.0001 | 2.08 | 0.4101 | 15 | 0.0044 | 17 | 0.7333 | 44 |
| Transcription factor 3 (E2A) | M31523 | −1044.46 | 169.32 | −6.17 | <0.0001 | 4.38 | 1.0000 | >28 | 0.0043 | 15 | 0.0371 | 1 |
| Adenosine triphosphatase, calcium | Z69881 | −1809.52 | 293.38 | −6.17 | <0.0001 | 7.26 | 1.0000 | >28 | 0.0043 | 16 | 0.6423 | 39 |
| IEF SSP 9502 | L07758 | −278.58 | 45.28 | −6.15 | <0.0001 | 2.06 | 0.5292 | 19 | 0.0052 | 18 | 0.2483 | 18 |
| Minichromosome maintenance deficient 3 | D38073 | −598.54 | 97.78 | −6.12 | <0.0001 | 2.85 | 0.8160 | 24 | 0.0055 | 20 | 0.4921 | 29 |
| Cytoplasmic dynein light chain 1 | U32944 | −1349.78 | 221.68 | −6.09 | <0.0001 | 5.16 | 1.0000 | >28 | 0.0054 | 19 | 1.0000 | >59 |
| Aldehyde reductase 1 | X15414 | −818.57 | 135.98 | −6.02 | <0.0001 | 2.26 | 0.0312 | 20 | 0.0090 | 24 | 0.4305 | 27 |
| Spectrin, alpha, non-erythrocytic 1 (alpha-fodrin) | J05243 | −732.24 | 122.22 | −5.99 | <0.0001 | 6.26 | 1.0000 | >28 | 0.0078 | 21 | 0.1626 | 12 |
| Rabaptin-5 | Y08612 | −220.83 | 36.90 | −5.98 | <0.0001 | 2.20 | 1.0000 | >28 | 0.0079 | 23 | 1.0000 | >59 |
| Topoisomerase (DNA) II beta | Z15115 | −2927.58 | 490.37 | −5.97 | <0.0001 | 3.20 | 1.0000 | >28 | 0.0079 | 22 | 0.0678 | 5 |
| HKR-T1 | S50223 | −287.95 | 48.38 | −5.95 | <0.0001 | 5.68 | 0.5208 | 18 | 0.0097 | 25 | 1.0000 | >59 |
Dataset from Golub et al. (1999).
P-value computed from Z-score using a modified Bonferroni's correction.
P-value obtained from t-test with equivalent variances using a modified Bonferroni's correction.
Relative ranking by significance values obtained from t-test with equivalent variances. >28 indicates that the gene was not ranked because the P-value was 1.0.
P-value obtained from t-test with unequal variances using a modified Bonferroni's correction.
Relative ranking by significance values obtained from t-test with unequal variances.
P-value obtained from Wilcoxon rank sum using a modified Bonferroni's correction.
Relative ranking by Wilcoxon significance values. >59 indicates that the gene was not ranked because the P-value was 1.0.
We next applied the statistical modeling method to examine expression profiles within subgroups of the 11 AML patients. Thrombopoietin (TPO) is the major cytokine responsible for the transition of myeloid progenitors into megakaryocytes (Caen et al. 1999), but also plays a more general role in the differentiation of hematopoietic stem cells into all types of progenitors (Kaushansky 1999). Furthermore, TPO is known to be expressed in a number of AML cell lines (Graf et al. 1996). We noticed a sharp delineation of TPO expression profiles between patients 28, 30, 32, 34, 36, and 38 versus patients 29, 31, 33, 35, and 37 and therefore compared these patient groups using our statistical modeling technique. This approach identified eight transcripts with a Z-score >4.8, with TPO itself yielding the highest ranking (Table 3). In contrast, neither t-tests nor Wilcoxon rank sum identified any gene with a genomic significance level of 1% (Table 3). Of the 15 highest ranking mRNAs from our analysis, three of the corresponding gene products are known to be influenced by or interact directly with TPO, two have not been characterized heavily but are highly homologous to proteins that interact with TPO, and eight others are involved in myeloid hematopoiesis. Although we have no evidence for any biological significance of the patient groups used in this comparison other than TPO transcript level, we noted that the groupings appear to fall along the lines of samples with high or low percentage of blasts (see http://www.genome.wi.mit.edu/MPR). Interestingly, TPO can stimulate the proliferation of AML blasts (Motoji et al. 1996; Luo et al. 2000).
Table 3.
Top 15 Genes Whose Expression Profiles Correlated with Differential Expression of TPO Among 11 AML Samples
| Gene description | Probe | Z-score | P-value1 | t-test2 | W. r.s.3 | Relation to TPO and/or hematopoiesis |
|---|---|---|---|---|---|---|
| Thrombopoietin (TPO) | L36051 | −9.39 | <0.0001 | 0.0971 | 1.000 | TPO supports megakaryopoiesis, most important regulator of platelet production (Caen et al. 1999). |
| Jagged 1 | U73936 | 6.26 | <0.0001 | 1.0000 | 1.0000 | Jagged 1 signaling through notch 1 plays a role in hematopoiesis (Schroeder and Just 2000). |
| Carboxypeptidase (MAX.1) | J04970 | −5.81 | <0.0001 | 1.0000 | 1.0000 | MAX.1 associated with monocyte to macrophage differentiation and is expressed in AML cells (Rehli et al. 1995). |
| Dynamin 1 | L07807 | −5.27 | 0.0010 | 1.0000 | 1.0000 | Dynamin 1 induced by Grb2 when monocytes stimulated with M-CSF (Kharbanda et al. 1995). |
| Neutrophil gelatinase B-associated lipocalin (NGAL) | X99133 | 5.24 | 0.0011 | 1.0000 | 1.0000 | NGAL mainly expressed in myeloid cells (Bundgaard et al. 1994), NGAL specific granules are marker for neutrophil maturation (Le Cabec et al. 1997). |
| Beta 1 integrin D | U33880 | −5.12 | 0.0021 | 1.0000 | 1.0000 | TPO up-regulates adhesion of hematopoetic progenitors to fibronectin through activation of integrin alpha4beta1 and alpha5beta1 (Gotoh et al. 1997). |
| Sp4 transcription factor | X68561 | −5.06 | 0.0030 | 1.0000 | 1.0000 | Sp4 not well characterized but closely related to Sp1 (Suske 1999), TPO activates several Sp1-dependent genes during megakaryopoiesis (Wang et al. 1999). |
| Prothrombin | M17262 | 4.86 | 0.0082 | 1.0000 | 1.0000 | Thrombin cleaves TPO to various isoforms (Kato et al. 1997), thrombin and TPO may co-regulate myeloid differentiation (van Willigen et al. 2000). |
| Wilms tumor 1 | X69950 | 4.65 | 0.0236 | 1.0000 | 1.0000 | WT1 inhibits differentiation of myeloid progenitor cells (Tsuboi et al. 1999). |
| LIM-homeobox domain (hLH-2) | U11701 | −4.61 | 0.0284 | 1.0000 | 1.0000 | hLH-2 has a role in control of cell fate decision and/or hematopoietic proliferation (Pinto do et al. 1998). |
| Mitochondrial creatine kinase | J04469 | −4.61 | 0.0284 | 1.0000 | 1.0000 | ? |
| Thrombospondin 2 (TSP2) | HG896– HT896 | 4.61 | 0.0286 | 1.0000 | 1.0000 | TSP2 inhibits tumor growth and angiogenesis (Streit et al. 1999), close relative thrombospondin 1 is negative regulator of megakaryopoiesis (Chen et al. 1997; Touhami et al. 1997). |
| Lysyl hydroxylase 2 (PLOD2) | U84573 | 4.49 | 0.0507 | 1.0000 | 1.0000 | ? |
| Serotonin receptor | M83181 | 4.33 | 0.1039 | 1.0000 | 1.0000 | Serotonin secretion stimulated by TPO (FontenayRoupie et al. 1998). |
| Karyopherin beta 3 | U72761 | 4.30 | 0.1192 | 1.0000 | 1.0000 | ? |
Dataset from Golub et al. (1999).
P-value computed from Z-score using a modified Bonferroni's correction.
P-value obtained from t-test with unequal variances using a modified Bonferroni's correction. When equal variances were assumed, the P-value for TPO was 0.1105 and the P-values for the other genes did not change.
P-value obtained from Wilcoxon rank sum using a modified Bonferroni's correction.
We next examined the association of gene expression with the success or failure of treatment. Among the 11 AML patients, 6 patients did not respond to treatment (patients 28–33) and five patients survived (patients 34–38) (see www.genome.wi.mit.edu/MPR [Golub et al. 1999]). The 25 transcripts with the highest Z-scores from the comparison of these groups are listed in Table 4, five of which had a Z-score greater than 4.8. As above, neither t-tests nor Wilcoxon rank sum identified any genes as differentially expressed between these groups at a 1% significance level (Table 4). We examined the chromosomal locations of the corresponding genes because chromosomal abnormalities are prevalent in leukemia and often have prognostic implications (El-Rifai et al. 1997; Rowley 2000). Almost all of the genes listed in Table 4 lie in regions that have been identified previously to contain abnormalities in AML or other forms of leukemia. Furthermore, three of the genes are encoded within 5q11–31, four are in the 2q region, two are within 1q32–26, and two others are found at 6p12–p11 (Table 4). The identification of five “mini-clusters” of chromosomal locales in the top 25 genes from a random pool of 6800+ genes is striking. Of note, the region 5q11–31 is frequently lost in AML and known to influence prognosis (Shipley et al. 1996; El-Rifai et al. 1997; Van den Berghe and Michaux 1997). Furthermore, Set (Li et al. 1996) and HoxA9 (Lawrence et al. 1999) are known to play a role in AML progression, and COL4A4 (Verfaillie et al. 1992), thioredoxin (Nilsson et al. 2000; Soderberg et al. 2000), caspase-8 (Pervaiz et al. 1999), integrin beta5 (Feng et al. 1999), α-tubulin (Hirose and Takiguchi 1995), and SPS2 (Soderberg et al. 2000) may well contribute to the disease. Although it should be kept in mind that clinical outcome is influenced by a number of nongenetic factors, including patient age, time of diagnosis, and treatment protocol, the above findings are promising for the discovery of prognostic indicators using genome-wide microarray analysis.
Table 4.
Top 25 Genes Differentially Expressed between AML Patients Who Lived or Died After Treatment
| Gene description | Probe | Z-score | P-value1 | t-test2 | W. r.s.3 | Locus4 | Locus anomalies observed in AML |
|---|---|---|---|---|---|---|---|
| Alpha IV collagen | D17391 | −5.96 | <0.0001 | 1.0000 | 1.0000 | 2q35-q37 | Rearrangement (Berger et al. 1991), ring formation (Whang-Peng et al. 1987) |
| Integrin beta-5 subunit | X53002 | 5.24 | 0.0011 | 1.0000 | 1.0000 | 3 (q22?) | Inversion, translocation (Testoni et al. 1999) |
| Pyrroline-5-carboxylate synthetase | X94453 | 5.00 | 0.0041 | 1.0000 | 1.0000 | 10q24.3 | Translocation in CML (Aguiar et al. 1997), hotspot for translocations in ALL (Kagan et al. 1989; Salvati et al. 1999) |
| Alpha-tubulin | X01703 | 4.96 | 0.0051 | 1.0000 | 1.0000 | 2q | ?, Rearrangement (Berger et al. 1991), ring formation (Whang-Peng et al. 1987) |
| KIAA0076 | D38548 | −4.84 | 0.0092 | 1.0000 | 1.0000 | 6 (p12-21?) | Rearrangement (Raynaud et al. 1994; Haase et al. 1995) |
| Cockayne syndrome complementation group A | U28413 | 4.74 | 0.0147 | 1.0000 | 1.0000 | 5 (q13?) | 5q11-31 frequently lost in AML (Shipley et al. 1996; Van den Berghe and Michaux 1997) |
| Set | M93651 | 4.73 | 0.0158 | 1.0000 | 1.0000 | 9q34 | Translocation, may create Set-Can fusion (von Lindern et al. 1992) |
| KIAA0172 | D79994 | −4.63 | 0.0254 | 1.0000 | 1.0000 | 9 (p?) | 9p abnormalities are common in leukemia and other cancers (Ragione and Iolascon 1997) |
| Selenophosphate synthetase 2 (SPS2) | U43286 | 4.61 | 0.0285 | 1.0000 | 1.0000 | (16p or 10q)? | Inversions and translocations are common in 16p (Marlton et al. 1995; Mancini et al. 2000), CML/ALL translocations identified in 10q (Kagan et al. 1989; Aguiar et al. 1997; Salvati et al. 1999) |
| Centromere protein E (312kD) | Z15005 | −4.60 | 0.0293 | 1.0000 | 1.0000 | 4q24-q25 | 4q25 translocation in ALL (Nowell et al. 1986) |
| Thioredoxin | X77584 | 4.54 | 0.0392 | 1.0000 | 1.0000 | 9q31 | Loss (Shipley et al. 1996) |
| PIG-B | D42138 | −4.53 | 0.0409 | 1.0000 | 1.0000 | 15q21-q22 | 15q observed deleted or translocated (Gogineni et al. 1997; Grimwade et al. 1997) |
| Survival motor neuron protein SMN | U80017 | 4.32 | 0.1088 | 1.0000 | 1.0000 | 5q13 | 5q11-31 frequently lost in AML (Shipley et al. 1996; Van den Berghe and Michaux 1997) |
| Caspase-8 | X98176 | −4.22 | 0.1755 | 1.0000 | 1.0000 | 2q33-q34 | Duplication in non-Hodgkin's lymphoma (Bajalica-Lagercrantz et al. 1996), ring formation (Whang-Peng et al. 1987) |
| Bullous pemphigoid antigen | M69225 | −4.21 | 0.1795 | 1.0000 | 1.0000 | 6p12-p11 | Rearrangement (Raynaud et al. 1994; Haase et al. 1995) |
| Sp2 transcription factor | D28588 | −4.18 | 0.2020 | 1.0000 | 1.0000 | 17q21.3-q22 | Translocation spot (Melnick et al. 1999), isochromosomes on 17q common (Fioretos et al. 1999) |
| Biglycan | J04599 | −4.17 | 0.2119 | 1.0000 | 1.0000 | Xq28 | Translocation found in AML (Weis et al. 1985), common in ALL (Stern 1996) |
| 26S proteasome-associated pad1 homolog (POH1) | U86782 | 4.16 | 0.2226 | 1.0000 | 1.0000 | 2 (q24-32?) | Duplication in non-Hodgkin's lymphoma (Bajalica-Lagercrantz et al. 1996), ring formation (Whang-Peng et al. 1987) |
| Homeobox-like | L32606 | 4.14 | 0.2427 | 1.0000 | 1.0000 | ? | ? |
| Pre-mRNA splicing factor SRP75 | L14076 | −4.13 | 0.2582 | 1.0000 | 1.0000 | 1 (p32-36?) | 1p32 and 1p36 both involved in translocations (Selypes and Laszlo 1987; Shimizu et al. 2000) |
| Autoantigen PM-SCL | X66113 | −4.12 | 0.2614 | 1.0000 | 1.0000 | 1p36 | Translocation (Shimizu et al. 2000) |
| Bactericidal/permeability-increasing protein | J04739 | 4.09 | 0.3030 | 1.0000 | 1.0000 | 20q11.23-q12 | Deletion (commonly deleted in MDS) (Fracchiolla et al. 1998) |
| HoxA9 | U82759 | 4.04 | 0.3836 | 1.0000 | 1.0000 | 7p15-p14 | Inversion (Stanley et al. 1997) |
| Matrin 3 | M63483 | 4.03 | 0.3940 | 1.0000 | 1.0000 |
Dataset from Golub et al. (1999).
P-value computed from Z-score using a modified Bonferroni's correction.
P-value obtained from tPvalue for TPO was 0.1105 and the Pvalues for the otehr genes did not change.3P-value obtained from Wilcoxon rank sum using a modified Bonferroni's correction.
Chromosomal locus determined by survey of NCBI LocusLink (http://www.ncbi.nlm.nih.gov/LocusLink). Purative loci are shown in parentheses.
DISCUSSION
The Z-scores we propose for testing differences of mean expression levels between two groups are connected closely with classical t-tests or Wilcoxon rank sum statistics, but it is important to realize that there are subtle differences between these tests. The t-test requires that expression levels be normally distributed and homogeneous within groups, and may also require equal variances between the groups. In contrast, the estimating equation technique we used to calculate Z-scores does not require any distributional assumptions or homogeneity of variances (see Methods for details). In practice, Z-scores are expected to be similar to t-test statistics, particularly those calculated assuming unequal variances, when the distribution of expression levels can be approximated by the normal distribution. When these assumptions are violated, Z-scores will differ from t-statistics and will be more reliable for making statistical inferences. On the other hand, the Wilcoxon statistic for two-group comparisons is nonparametric and thus robust. However, its power is reduced, which could be of concern in light of small sample sizes in typical array studies. Indeed, the Wilcoxon test did not detect any genes as differentially expressed between AML and ALL at the 1% genomic significance level (Tables 1 and 2, data not shown). Finally, there is no obvious method, besides ad hoc corrections of the expression values, to adjust for heterogeneity among samples when using the t-test or Wilcoxon statistics. The regression paradigm we propose provides a natural correction for heterogeneity using all expression values.
It is important to note that we analyzed the leukemia data without applying any questionable filtering methods to the Affymetrix data. For example, we did not subtract a background noise level from the data, rescale any values other than to correct for between-chip heterogeneity, or remove genes based on fluorescent signal intensities or Affymetrix present/absent calls. These filtering techniques may be required to make the strongest associations when clustering data or when calculating fold changes in means. However, ad hoc filtering could remove potential genes of interest, especially those with modest expression levels, and therefore reduce the power of discovery. For example, the difference of only a few transcripts to zero transcripts per cell may become undetectable after applying filtering techniques, but could nevertheless have a very real biological significance or present a considerable opportunity to target a cell specifically for therapeutic treatment. To illustrate this point, we note that TPO was called absent by the Affymetrix software in every sample in the leukemia data set. Nevertheless, by dichotomizing the AML samples along the lines of TPO expression values, we were able to uncover a group of proteins that interact directly with TPO or perform similar cellular functions.
Another distinct advantage of statistical modeling is that these tests take advantage of the random variations (i.e., “noise”) in the data. For example, the mean expression level of activation-induced C-type lectin (AICL) was threefold higher in AML than ALL, and the absolute mean difference was substantial at 826 units. Considering that AICL is expressed in a variety of hematopoietic-derived cell lines (Hamann et al. 1997), one might reasonably conclude that AICL was indeed overexpressed in AML based on this evidence. However, our modeling approach gave AICL a Z-score of 0.91. This apparent discrepancy is explained by the fact that one of the AICL samples in the AML set had an intensity value more than fivefold higher than any other. Excluding just this one sample, the relative and absolute mean differences for AICL between AML and ALL were 1.3-fold and −94 ± 216, respectively. Clearly, simple comparisons of fold changes are insufficient for drawing proper conclusions.
Our modeling approach can be extended. First, we can incorporate nonlinear models or apply other transformations to the observed expression levels to account for nonlinearity in fluorescent intensity. Second, the model (equation 1 in Methods) can be extended naturally to incorporate additional covariates. For example, in a clinical study of multiple patients, one may be interested in assessing the association of expression profiles with several clinical variables. Third, one may extend the model (equation 1) by incorporating nonparametric smoothing function for a continuous covariate, for example, in the assessment of nonlinear dose-response relationship. Fourth, as our knowledge accumulates about the genetic regulatory circuitry of multiple genes, we may be able to formulate a functional relationship among genes, via postulating a “high-level” model for regression coefficients α(π) = (α1,α2,…,αJ) and β(π) = (β1,β2,…,βJ), in which π could be a common set of parameters characterizing the entire genetic regulatory circuitry. One may then test how well such a genetic circuitry model fits the data using estimating equations.
The main limitation of the current approach is associated with the calculation of P-values. As noted earlier, a Z-score of 4.8 is chosen to ensure that the genome-wide significance is controlled at 1% for the Affymetrix 6800 GeneChips. However, the calculation of the corresponding P-value relies on the asymptotic normal distribution for Z-scores. With small to modest sample sizes this normality may be questionable, and such a threshold value is overly conservative. Currently, we are developing simulation-based methods to evaluate the exact significance level. It is also important to note that for the purpose of discovery science with small sample sizes, the Z-score 4.8 threshold value should be treated as a tentative guideline. In the context of testing associations with a specific candidate gene, the accepted threshold value to ensure the false-error rate of 1% for a single gene is a Z-score of 2.58. Finally, we note that the Bonferroni's correction or modifications thereof do not take into account covariation of gene expression levels, resulting in conservative estimates for the P-values. Our future research will improve on Bonferroni's correction by acknowledging expression dependencies among genes.
The capability of simultaneously assessing the expression of thousands of gene transcripts provides an opportunity of monitoring cellular activity at the genomic level. We can therefore begin addressing complex pathways of basic physiology and disease etiology, the foundation of functional genomics. The development of the statistical method described here provides a tool for researchers to pursue functional genomics systematically and rigorously. Modeling can also be used to aid the design of efficient and robust functional genomic studies, and to develop methods that estimate sample sizes and powers required for expression studies. The use of rigorous statistical tools will help functional genomic studies yield much-needed information in understanding human biology and pathology.
METHODS
Leukemia Study
The Affymetrix 6800 GeneChip oligonucleotide arrays contain a combined total of 7070 oligonucleotide probe sets (excluding controls) for 6817 individual genes. Investigators at the Massachusetts Institute of Technology gathered blood samples from 38 leukemia patients (27 ALL and 11 AML) and used Affymetrix Hu6800 GeneChip oligonucleotide arrays to assess gene expression profiles for each patient (Golub et al. 1999). We used the training data set exclusively in this work. Experimental protocols used to perform the microarray analysis and the data values obtained are available to the public at (http://waldo.wi.mit.edu/MPR/pubs.html).
Regression Model
An array of gene expression profiles may be conceptualized as a vector of outcomes. Let Yk = (Y1k, Y1k,…, YJk)‘ denote the array, where Yjk denotes the expression of the jth gene in the kth sample (j = 1,2,…,J; k = 1,2,…, K). Let xk denote a covariate associating with each kth sample. For example, xk = 1 for the presence of a marker gene and xk = 0 for its absence. We propose a regression model for the expression level of the jth gene in the kth sample:
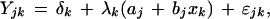 |
1 |
in which (aj, bj) are gene-specific regression coefficients, (δk,λk) are the sample-specific additive and multiplicative heterogeneity factors, respectively, and ɛjk is a random variable reflecting variation due to sources other than the one identified by the known covariate and the systematic heterogeneity between samples. Because xk is binary, aj measures the mean expression level of the jth gene in normal samples (xk = 0), and bj measures the difference of averaged expression levels of the jth gene between the two sample groups.
The heterogeneity factors, (δk,λk), are introduced to account for variations in preparing multiple mRNA samples. Such corrections have been well conceived in comparing two samples. Under the null hypothesis of no overall differential expression between these two samples, one can adjust this heterogeneity by normalizing the sample data to fall on the diagonal line, a common technique (Wodicka et al. 1997). An intercept may also be estimated to ensure the numerical stability. If the intercept is different from zero, the diagonal line is adjusted to compensate. Formalizing this correction, one may assume that typical genome-wide expression patterns are stable, and hence may use a linear model, μjk = δk + λkaj, to characterize average expression values for every gene in every sample. These heterogeneity factors are then estimated via the weighted least square method (Carroll and Ruppert 1988). Estimated heterogeneity factors are used to adjust the observed expression level as 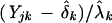 , and corrected expression values are then used for further analysis under the above model (equation 1).
, and corrected expression values are then used for further analysis under the above model (equation 1).
The random variation, ɛjk, is used to depict variations due to all unknown sources. Specifically, this variation may be associated with sampling preparations, cross-hybridization of genes, or other anomalies on microarrays. The stochastic distribution of these random variations is typically unknown and is unlikely to follow any familiar distributions, such as the normal distribution. Hence, no distribution assumption is made.
Analytic Strategy
The first step in the statistical analysis of oligonucleotide-array expression profiles is preprocessing of the data, which includes elimination of control genes and transformation of the data (e.g., logarithmic transformation) as desired. The second step is to examine heterogeneity among samples by estimating additive and multiplicative heterogeneity factors, (δk,λk). The estimate is obtained via minimizing the weighted least square, ∑j,k (Yjk − δk − λkaj)2w , where the summation is over all genes and samples (Carroll and Ruppert 1988). The weight is chosen so that the contribution of every gene is standardized between 0 and 1. Consequently, the above weighted least square equals the number of genes when samples are homogeneous. The estimated parameters (δk,λk) are used to correct the data. Because we do not impose distributional assumptions about residuals, the third step is to use the weighted least square (Huber 1967) to estimate gene-specific parameters (aj, bj) in the model (equation 1). The corresponding robust standard errors for each gene are calculated using estimating equation theory (Godambe 1960; Liang and Zeger 1986; Prentice and Zhao 1991). Z-scores for each gene are computed as the ratio of mean difference between the two groups for each gene, bj, over the standard error for the corresponding gene, S.E.j.
, where the summation is over all genes and samples (Carroll and Ruppert 1988). The weight is chosen so that the contribution of every gene is standardized between 0 and 1. Consequently, the above weighted least square equals the number of genes when samples are homogeneous. The estimated parameters (δk,λk) are used to correct the data. Because we do not impose distributional assumptions about residuals, the third step is to use the weighted least square (Huber 1967) to estimate gene-specific parameters (aj, bj) in the model (equation 1). The corresponding robust standard errors for each gene are calculated using estimating equation theory (Godambe 1960; Liang and Zeger 1986; Prentice and Zhao 1991). Z-scores for each gene are computed as the ratio of mean difference between the two groups for each gene, bj, over the standard error for the corresponding gene, S.E.j.
Statistical Significance and Multiple Comparisons
To measure the significance of the findings, we translated Z-scores into P-values under asymptotic normality. To address the multiple comparison issue, we adjusted the threshold for declaring genes differentially expressed using a modified Bonferroni’s correction proposed by Hochberg (1988). The Hochberg stepdown method divides the P-values by the total number of comparisons with equal or lesser test statistics; for 7070 probe sets, the 1% genomic significance level for the probe set with the highest test statistic is 0.01/7070, the genomic significance threshold for the probe set with the second highest test statistic is 0.01/7069, etc.
t-test and Wilcoxon Rank Sum Test
The t-test and Wilcoxon rank sum test were performed after correcting the data for heterogeneity using our regression approach. t-tests were performed assuming both equal and unequal variances between the sample groups. The functions used were those built into MATLAB (MathWorks). The P-values derived from these tests were adjusted using the modified Bonferroni's correction described above.
Acknowledgments
We thank Tracy Bergemann, Chun Cheng, Robert Eisenman, and Jerry Radich for comments on this manuscript. We also thank T.R. Golub and colleagues at MIT for making their excellent AML/ALL data set (Golub et al. 1999) available in the public domain. This work was supported by National Institute of Health grants HG02283, GM58897, and CA53996.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL lzhao@fhcrc.org; FAX (206) 667-2437.
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.165101.
REFERENCES
- Aguiar RC, Chase A, Oscier DG, Carapeti M, Goldman JM, Cross NC. Characterization of a t(10;12)(q24;p13) in a case of CML in transformation. Genes Chromosomes Cancer. 1997;20:408–411. [PubMed] [Google Scholar]
- Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- Alon U, Barkai N, Notterman DA, Gish K, Ybarra S, Mack D, Levine A J. Broad patterns of gene expression revealed by clustering analysis of tumor and normal colon tissues probed by oligonucleotide arrays. Proc Natl Acad Sci. 1999;96:6745–6750. doi: 10.1073/pnas.96.12.6745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arfin SM, Long AD, Ito ET, Tolleri L, Riehle MM, Paegle ES, Hatfield GW. Global gene expression profiling in Esherichia coli K12: The effects of integration host factor. J Biol Chem. 2000;275:29672–29684. doi: 10.1074/jbc.M002247200. [DOI] [PubMed] [Google Scholar]
- Bajalica-Lagercrantz S, Tingaard Pedersen N, Sorensen AG, Nordenskjold M. Duplication of 2q31-qter as a sole aberration in a case of non-Hodgkin's lymphoma. Cancer Genet Cytogenet. 1996;90:102–105. doi: 10.1016/s0165-4608(96)00092-1. [DOI] [PubMed] [Google Scholar]
- Ben-Dor A, Shamir R, Yakhini Z. Clustering gene expression patterns. J Comput Biol. 1999;6:281–297. doi: 10.1089/106652799318274. [DOI] [PubMed] [Google Scholar]
- Berger R, Le Coniat M, Derre J, Vecchione D, Jonveaux P. Cytogenetic studies in acute promyelocytic leukemia: A survey of secondary chromosomal abnormalities. Genes Chromosomes Cancer. 1991;3:332–337. doi: 10.1002/gcc.2870030503. [DOI] [PubMed] [Google Scholar]
- Brent R. Genomic biology. Cell. 2000;100:169–183. doi: 10.1016/s0092-8674(00)81693-1. [DOI] [PubMed] [Google Scholar]
- Bundgaard JR, Sengelov H, Borregaard N, Kjeldsen L. Molecular cloning and expression of a cDNA encoding NGAL: A lipocalin expressed in human neutrophils. Biochem Biophys Res Commun. 1994;202:1468–1475. doi: 10.1006/bbrc.1994.2096. [DOI] [PubMed] [Google Scholar]
- Caen JP, Han ZC, Bellucci S, Alemany M. Regulation of megakaryocytopoiesis. Haemostasis. 1999;29:27–40. doi: 10.1159/000022458. [DOI] [PubMed] [Google Scholar]
- Carroll RJ, Ruppert D. Transformation and weighting in regression. London: Chapman and Hall; 1988. [Google Scholar]
- Chen YZ, Incardona F, Legrand C, Momeux L, Caen J, Han ZC. Thrombospondin, a negative modulator of megakaryocytopoiesis. J Lab Clin Med. 1997;129:231–238. doi: 10.1016/s0022-2143(97)90144-x. [DOI] [PubMed] [Google Scholar]
- Chu S, DeRisi J, Eisen M, Mulholland J, Botstein D, Brown PO, Herskowitz I. The transcriptional program of sporulation in budding yeast. Science. 1998;282:699–705. doi: 10.1126/science.282.5389.699. [DOI] [PubMed] [Google Scholar]
- Coller HA, Grandori C, Tamayo P, Colbert T, Lander ES, Eisenman RN, Golub TR. Expression analysis with oligonucleotide microarrays reveals that MYC regulates genes involved in growth, cell cycle, signaling, and adhesion. Proc Natl Acad Sci. 2000;97:3260–3265. doi: 10.1073/pnas.97.7.3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRisi JL, Iyer VR, Brown PO. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science. 1997;278:680–686. doi: 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Rifai W, Elonen E, Larramendy M, Ruutu T, Knuutila S. Chromosomal breakpoints and changes in DNA copy number in refractory acute myeloid leukemia. Leukemia. 1997;11:958–963. doi: 10.1038/sj.leu.2400706. [DOI] [PubMed] [Google Scholar]
- Feng X, Teitelbaum SL, Quiroz ME, Towler DA, Ross FP. Cloning of the murine β5 integrin subunit promoter. Identification of a novel sequence mediating granulocyte-macrophage colony-stimulating factor-dependent repression of β5 integrin gene transcription. J Biol Chem. 1999;274:1366–1374. doi: 10.1074/jbc.274.3.1366. [DOI] [PubMed] [Google Scholar]
- Ferea TL, Botstein D, Brown PO, Rosenzweig RF. Systematic changes in gene expression patterns following adaptive evolution in yeast. Proc Natl Acad Sci. 1999;96:9721–9726. doi: 10.1073/pnas.96.17.9721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fioretos T, Strombeck B, Sandberg T, Johansson B, Billstrom R, Borg A, Nilsson PG, Van Den Berghe H, Hagemeijer A, Mitelman F, et al. Isochromosome 17q in blast crisis of chronic myeloid leukemia and in other hematologic malignancies is the result of clustered breakpoints in 17p11 and is not associated with coding TP53 mutations. Blood. 1999;94:225–232. [PubMed] [Google Scholar]
- Fontenay-Roupie M, Huret G, Loza JP, Adda R, Melle J, Maclouf J, Dreyfus F, Levy-Toledano S. Thrombopoietin activates human platelets and induces tyrosine phosphorylation of p80/85 cortactin. Thromb Haemost. 1998;79:195–201. [PubMed] [Google Scholar]
- Fracchiolla NS, Colombo G, Finelli P, Maiolo AT, Neri A. EHT, a new member of the MTG8/ETO gene family, maps on 20q11 region and is deleted in acute myeloid leukemias. Blood. 1998;92:3481–3484. [PubMed] [Google Scholar]
- Gaasterland T, Bekiranov S. Making the most of microarray data. Nat Genet. 2000;24:204–206. doi: 10.1038/73392. [DOI] [PubMed] [Google Scholar]
- Godambe VP. An optimum property of regular maximum likelihood estimation. Annals Mathemat Stat. 1960;31:1208–1212. [Google Scholar]
- Gogineni SK, Shah HO, Chester M, Lin JH, Garrison M, Alidina A, Bayani E, Verma RS. Variant complex translocations involving chromosomes 1, 9, 9, 15 and 17 in acute promyelocytic leukemia without RAR alpha/PML gene fusion rearrangement. Leukemia. 1997;11:514–518. doi: 10.1038/sj.leu.2400610. [DOI] [PubMed] [Google Scholar]
- Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA, et al. Molecular classification of cancer: Class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- Gotoh A, Ritchie A, Takahira H, Broxmeyer HE. Thrombopoietin and erythropoietin activate inside-out signaling of integrin and enhance adhesion to immobilized fibronectin in human growth-factor-dependent hematopoietic cells. Ann Hematol. 1997;75:207–213. doi: 10.1007/s002770050344. [DOI] [PubMed] [Google Scholar]
- Graf G, Dehmel U, Drexler HG. Expression of thrombopoietin and thrombopoietin receptor MPL in human leukemia-lymphoma and solid tumor cell lines. Leuk Res. 1996;20:831–838. doi: 10.1016/s0145-2126(96)00057-4. [DOI] [PubMed] [Google Scholar]
- Grimwade D, Gorman P, Duprez E, Howe K, Langabeer S, Oliver F, Walker H, Culligan D, Waters J, Pomfret M, et al. Characterization of cryptic rearrangements and variant translocations in acute promyelocytic leukemia. Blood. 1997;90:4876–4885. [PubMed] [Google Scholar]
- Haase D, Feuring-Buske M, Konemann S, Fonatsch C, Troff C, Verbeek W, Pekrun A, Hiddemann W, Wormann B. Evidence for malignant transformation in acute myeloid leukemia at the level of early hematopoietic stem cells by cytogenetic analysis of CD34+ subpopulations. Blood. 1995;86:2906–2912. [PubMed] [Google Scholar]
- Hamann J, Montgomery KT, Lau S, Kucherlapati R, van Lier RA. AICL: A new activation-induced antigen encoded by the human NK gene complex. Immunogenetics. 1997;45:295–300. doi: 10.1007/s002510050208. [DOI] [PubMed] [Google Scholar]
- Hirose Y, Takiguchi T. Microtubule changes in hematologic malignant cells treated with paclitaxel and comparison with vincristine cytotoxicity. Blood Cells Mol Dis. 1995;21:119–130. doi: 10.1006/bcmd.1995.0014. [DOI] [PubMed] [Google Scholar]
- Hochberg Y. A sharper Bonferroni procedure for multiple test of significance. Biometrika. 1988;75:800–802. [Google Scholar]
- Huber PJ. Proceedings of the Fifth Berkeley Symposium in Mathematical Statistics and Probability. Berkeley: UC Press; 1967. The behavior of maximum likelihood estimates under nonstandard conditions. [Google Scholar]
- Kagan J, Finger LR, Letofsky J, Finan J, Nowell PC, Croce CM. Clustering of breakpoints on chromosome 10 in acute T-cell leukemias with the t(10;14) chromosome translocation. Proc Natl Acad Sci. 1989;86:4161–4165. doi: 10.1073/pnas.86.11.4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T, Oda A, Inagaki Y, Ohashi H, Matsumoto A, Ozaki K, Miyakawa Y, Watarai H, Fuju K, Kokubo A, et al. Thrombin cleaves recombinant human thrombopoietin: One of the proteolytic events that generates truncated forms of thrombopoietin. Proc Natl Acad Sci. 1997;94:4669–4674. doi: 10.1073/pnas.94.9.4669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushansky K. Thrombopoietin and hematopoietic stem cell development. Ann NY Acad Sci. 1999;872:314–319. doi: 10.1111/j.1749-6632.1999.tb08475.x. [DOI] [PubMed] [Google Scholar]
- Kharbanda S, Saleem A, Yuan Z, Emoto Y, Prasad KV, Kufe D. Stimulation of human monocytes with macrophage colony-stimulating factor induces a Grb2-mediated association of the focal adhesion kinase pp125FAK and dynamin. Proc Natl Acad Sci. 1995;92:6132–6136. doi: 10.1073/pnas.92.13.6132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashkari DA, DeRisi JL, McCusker JH, Namath AF, Gentile C, Hwang SY, Brown PO, Davis RW. Yeast microarrays for genome wide parallel genetic and gene expression analysis. Proc Natl Acad Sci. 1997;94:13057–13062. doi: 10.1073/pnas.94.24.13057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence HJ, Rozenfeld S, Cruz C, Matsukuma K, Kwong A, Komuves L, Buchberg AM, Largman C. Frequent co-expression of the HOXA9 and MEIS1 homeobox genes in human myeloid leukemias. Leukemia. 1999;13:1993–1999. doi: 10.1038/sj.leu.2401578. [DOI] [PubMed] [Google Scholar]
- Le Cabec V, Calafat J, Borregaard N. Sorting of the specific granule protein, NGAL, during granulocytic maturation of HL-60 cells. Blood. 1997;89:2113–2121. [PubMed] [Google Scholar]
- Li M, Makkinje A, Damuni Z. The myeloid leukemia-associated protein SET is a potent inhibitor of protein phosphatase 2A. J Biol Chem. 1996;271:11059–11062. doi: 10.1074/jbc.271.19.11059. [DOI] [PubMed] [Google Scholar]
- Liang KY, Zeger SL. Longitudinal data analysis using generalized linear models. Biometrika. 1986;73:13–22. [Google Scholar]
- Luo SS, Ogata K, Yokose N, Kato T, Dan K. Effect of thrombopoietin on proliferation of blasts from patients with myelodysplastic syndromes. Stem Cells. 2000;18:112–119. doi: 10.1634/stemcells.18-2-112. [DOI] [PubMed] [Google Scholar]
- Mancini M, Cedrone M, Diverio D, Emanuel B, Stul M, Vranckx H, Brama M, De Cuia MR, Nanni M, Fazi F, et al. Use of dual-color interphase FISH for the detection of inv(16) in acute myeloid leukemia at diagnosis, relapse and during follow-up: A study of 23 patients. Leukemia. 2000;14:364–368. doi: 10.1038/sj.leu.2401678. [DOI] [PubMed] [Google Scholar]
- Marlton P, Claxton DF, Liu P, Estey EH, Beran M, LeBeau M, Testa JR, Collins FS, Rowley JD, Siciliano MJ. Molecular characterization of 16p deletions associated with inversion 16 defines the critical fusion for leukemogenesis. Blood. 1995;85:772–779. [PubMed] [Google Scholar]
- Melnick A, Fruchtman S, Zelent A, Liu M, Huang Q, Boczkowska B, Calasanz M, Fernandez A, Licht JD, Najfeld V. Identification of novel chromosomal rearrangements in acute myelogenous leukemia involving loci on chromosome 2p23, 15q22 and 17q21. Leukemia. 1999;13:1534–1538. doi: 10.1038/sj.leu.2401513. [DOI] [PubMed] [Google Scholar]
- Motoji T, Takanashi M, Motomura S, Wang W H, Shiozaki H, Aoyama M, Mizoguchi H. Growth stimulatory effect of thrombopoietin on the blast cells of acute myelogenous leukaemia. Br J Haematol. 1996;94:513–516. doi: 10.1046/j.1365-2141.1996.d01-1832.x. [DOI] [PubMed] [Google Scholar]
- Nilsson J, Soderberg O, Nilsson K, Rosen A. Thioredoxin prolongs survival of B-type chronic lymphocytic leukemia cells. Blood. 2000;95:1420–1426. [PubMed] [Google Scholar]
- Nowell PC, Vonderheid EC, Besa E, Hoxie JA, Moreau L, Finan JB. The most common chromosome change in 86 chronic B cell or T cell tumors: A 14q32 translocation. Cancer Genet Cytogenet. 1986;19:219–227. doi: 10.1016/0165-4608(86)90050-6. [DOI] [PubMed] [Google Scholar]
- Pervaiz S, Seyed MA, Hirpara JL, Clement MV, Loh KW. Purified photoproducts of merocyanine 540 trigger cytochrome C release and caspase 8-dependent apoptosis in human leukemia and melanoma cells. Blood. 1999;93:4096–4108. [PubMed] [Google Scholar]
- Pinto do OP, Kolterud A, Carlsson L. Expression of the LIM-homeobox gene LH2 generates immortalized steel factor-dependent multipotent hematopoietic precursors. EMBO J. 1998;17:5744–5756. doi: 10.1093/emboj/17.19.5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prentice RL, Zhao LP. Estimating equations for parameters in means and covariances of multivariate discrete continuous responses. Biometrics. 1991;47:825–839. [PubMed] [Google Scholar]
- Ragione FD, Iolascon A. Inactivation of cyclin-dependent kinase inhibitor genes and development of human acute leukemias. Leuk Lymphoma. 1997;25:23–35. doi: 10.3109/10428199709042493. [DOI] [PubMed] [Google Scholar]
- Raynaud SD, Brunet B, Chischportich M, Bayle J, Gratecos N, Pesce A, Dujardin P, Flandrin G, Ayraud N. Recurrent cytogenetic abnormalities observed in complete remission of acute myeloid leukemia do not necessarily mark preleukemic cells. Leukemia. 1994;8:245–249. [PubMed] [Google Scholar]
- Rehli M, Krause SW, Kreutz M, Andreesen R. Carboxypeptidase M is identical to the MAX.1 antigen and its expression is associated with monocyte to macrophage differentiation. J Biol Chem. 1995;270:15644–15649. doi: 10.1074/jbc.270.26.15644. [DOI] [PubMed] [Google Scholar]
- Rowley JD. Molecular genetics in acute leukemia. Leukemia. 2000;14:513–517. doi: 10.1038/sj.leu.2401600. [DOI] [PubMed] [Google Scholar]
- Salvati PD, Watt PM, Thomas WR, Kees UR. Molecular characterization of a complex chromosomal translocation breakpoint t(10;14) including the HOX11 oncogene locus. Leukemia. 1999;13:975–979. doi: 10.1038/sj.leu.2401421. [DOI] [PubMed] [Google Scholar]
- Schroeder T, Just U. Notch signalling via RBP-J promotes myeloid differentiation. EMBO J. 2000;19:2558–2568. doi: 10.1093/emboj/19.11.2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selypes A, Laszlo A. A new translocation t(1;4;11) in congenital acute nonlymphocytic leukemia (acute myeloblastic leukemia) Hum Genet. 1987;76:106–108. doi: 10.1007/BF00283060. [DOI] [PubMed] [Google Scholar]
- Shimizu S, Suzukawa K, Kodera T, Nagasawa T, Abe T, Taniwaki M, Yagasaki F, Tanaka H, Fujisawa S, Johansson B, et al. Identification of breakpoint cluster regions at 1p36.3 and 3q21 in hematologic malignancies with t(1;3)(p36;q21) Genes Chromosomes Cancer. 2000;27:229–238. doi: 10.1002/(sici)1098-2264(200003)27:3<229::aid-gcc2>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Shipley J, Weber-Hall S, Birdsall S. Loss of the chromosomal region 5q11-q31 in the myeloid cell line HL-60: Characterization by comparative genomic hybridization and fluorescence in situ hybridization. Genes Chromosomes Cancer. 1996;15:182–186. doi: 10.1002/(SICI)1098-2264(199603)15:3<182::AID-GCC7>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Snedecor GW, Cochran WG. Statistical methods. Ames, Iowa: The Iowa State University Press; 1980. [Google Scholar]
- Soderberg A, Sahaf B, Rosen A. Thioredoxin reductase, a redox-active selenoprotein, is secreted by normal and neoplastic cells: Presence in human plasma. Cancer Res. 2000;60:2281–2289. [PubMed] [Google Scholar]
- Spellman PT, Sherlock G, Zhang MQ, Iyer VR, Anders K, Eisen MB, Brown PO, Botstein D, Futcher B. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol Biol Cell. 1998;9:3273–3297. doi: 10.1091/mbc.9.12.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley WS, Burkett SS, Segel B, Quiery A, George B, Lobel J, Shah N. Constitutional inversion of chromosome 7 and hematologic cancers. Cancer Genet Cytogenet. 1997;96:46–49. doi: 10.1016/s0165-4608(96)00268-3. [DOI] [PubMed] [Google Scholar]
- Stern MH. Oncogenesis of T-cell prolymphocytic leukemia (editorial) Pathol Biol (Paris) 1996;44:689–693. [PubMed] [Google Scholar]
- Streit M, Riccardi L, Velasco P, Brown LF, Hawighorst T, Bornstein P, Detmar M. Thrombospondin-2: A potent endogenous inhibitor of tumor growth and angiogenesis. Proc Natl Acad Sci. 1999;96:14888–14893. doi: 10.1073/pnas.96.26.14888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suske G. The Sp-family of transcription factors. Gene. 1999;238:291–300. doi: 10.1016/s0378-1119(99)00357-1. [DOI] [PubMed] [Google Scholar]
- Tamayo P, Slonim D, Mesirov J, Zhu Q, Kitareewan S, Dmitrovsky E, Lander ES, Golub TR. Interpreting patterns of gene expression with self-organizing maps: Methods and application to hematopoietic differentiation. Proc Natl Acad Sci. 1999;96:2907–2912. doi: 10.1073/pnas.96.6.2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka TS, Jaradat SA, Lim MK, Kargul GJ, Wang X, Grahovac MJ, Pantano S, Sano Y, Piao Y, Nagaraja R, et al. Genome-wide expression profiling of mid-gestation placenta and embryo using a 15,000 mouse developmental cDNA microarray. Proc Natl Acad Sci. 2000;97:9127–9132. doi: 10.1073/pnas.97.16.9127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavazoie S, Hughes JD, Campbell MJ, Cho RJ, Church GM. Systematic determination of genetic network architecture. Nat Genet. 1999;22:281–285. doi: 10.1038/10343. [DOI] [PubMed] [Google Scholar]
- Testoni N, Borsaru G, Martinelli G, Carboni C, Ruggeri D, Ottaviani E, Pelliconi S, Ricci P, Pastano R, Visani G, et al. 3q21 and 3q26 cytogenetic abnormalities in acute myeloblastic leukemia: Biological and clinical features. Haematologica. 1999;84:690–694. [PubMed] [Google Scholar]
- Touhami M, Fauvel-Lafeve F, Da Silva N, Chomienne C, Legrand C. Induction of thrombospondin-1 by all-trans retinoic acid modulates growth and differentiation of HL-60 myeloid leukemia cells. Leukemia. 1997;11:2137–2142. doi: 10.1038/sj.leu.2400866. [DOI] [PubMed] [Google Scholar]
- Tsuboi A, Oka Y, Ogawa H, Elisseeva OA, Tamaki H, Oji Y, Kim EH, Soma T, Tatekawa T, Kawakami M, et al. Constitutive expression of the Wilms' tumor gene WT1 inhibits the differentiation of myeloid progenitor cells but promotes their proliferation in response to granulocyte-colony stimulating factor (G- CSF) Leuk Res. 1999;23:499–505. doi: 10.1016/s0145-2126(99)00037-5. [DOI] [PubMed] [Google Scholar]
- Van den Berghe H, Michaux L. 5q-, twenty-five years later: A synopsis. Cancer Genet Cytogenet. 1997;94:1–7. doi: 10.1016/s0165-4608(96)00350-0. [DOI] [PubMed] [Google Scholar]
- van Willigen G, Gorter G, Akkerman JW. Thrombopoietin increases platelet sensitivity to α-thrombin via activation of the ERK2-cPLA2 pathway. Thromb Haemost. 2000;83:610–616. [PubMed] [Google Scholar]
- Verfaillie CM, McCarthy JB, McGlave PB. Mechanisms underlying abnormal trafficking of malignant progenitors in chronic myelogenous leukemia. Decreased adhesion to stroma and fibronectin but increased adhesion to the basement membrane components laminin and collagen type IV. J Clin Invest. 1992;90:1232–1241. doi: 10.1172/JCI115985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Lindern M, van Baal S, Wiegant J, Raap A, Hagemeijer A, Grosveld G. Can, a putative oncogene associated with myeloid leukemogenesis, may be activated by fusion of its 3′ half to different genes: Characterization of the set gene. Mol Cell Biol. 1992;12:3346–3355. doi: 10.1128/mcb.12.8.3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Zhang Y, Lu J, Sun S, Ravid K. Mpl ligand enhances the transcription of the cyclin D3 gene: A potential role for Sp1 transcription factor. Blood. 1999;93:4208–4221. [PubMed] [Google Scholar]
- Weis J, DeVito V, Allen L, Linder D, Magenis E. Translocation X;10 in a case of congenital acute monocytic leukemia. Cancer Genet Cytogenet. 1985;16:357–364. doi: 10.1016/0165-4608(85)90245-6. [DOI] [PubMed] [Google Scholar]
- Whang-Peng J, Lee EC, Kao-Shan CS, Schechter G. Ring chromosome in a case of acute myelomonocytic leukemia: Its significance and a review of the literature. Hematol Pathol. 1987;1:57–65. [PubMed] [Google Scholar]
- Wodicka L, Dong H, Mittmann M, Ho MH, Lockhart DJ. Genome-wide expression monitoring in Saccharomyces cerevisiae. Nat Biotechnol. 1997;15:1359–1367. doi: 10.1038/nbt1297-1359. [DOI] [PubMed] [Google Scholar]