

Abstract
After intracranial inoculation, neurovirulent mouse hepatitis virus (MHV) strains induce acute inflammation, demyelination, and axonal loss in the central nervous system. Prior studies using recombinant MHV strains that differ only in the spike gene, which encodes a glycoprotein involved in virus-host cell attachment, demonstrated that spike mediates anterograde axonal transport of virus to the spinal cord. A demyelinating MHV strain induces optic neuritis, but whether this is due to the retrograde axonal transport of viral particles to the retina or due to traumatic disruption of retinal ganglion cell axons during intracranial inoculation is not known. Using recombinant isogenic MHV strains, we examined the ability of recombinant MHV to induce optic neuritis by retrograde spread from the brain through the optic nerve into the eye after intracranial inoculation. Recombinant demyelinating MHV induced macrophage infiltration of optic nerves, demyelination, and axonal loss, whereas optic neuritis and axonal injury were minimal in mice infected with the nondemyelinating MHV strain that differs in the spike gene. Thus, optic neuritis was dependent on a spike glycoprotein-mediated mechanism of viral antigen transport along retinal ganglion cell axons. These data indicate that MHV spreads by retrograde axonal transport to the eye and that targeting spike protein interactions with axonal transport machinery is a potential therapeutic strategy for central nervous system viral infections and associated diseases.
Keywords: Axonal transport, Demyelination, Mouse hepatitis virus, Multiple sclerosis, Optic neuritis, Spike glycoprotein
Introduction
Neurotropic mouse hepatitis virus (MHV) infection in mice causes meningoencephalitis, myelitis, and demyelination, with relative axonal preservation. Recent studies have additionally demonstrated that neurotropic MHV strains can also induce axonal loss (1, 2); direct virus-mediated axonal damage can occur concurrently with and independently of demyelination (2). Thus, neurovirulent MHV strains provide useful tools for studying the neuroinflammation, demyelination, and axonal loss and as a virus-induced model of multiple sclerosis.
Recombinant MHV strains RSA59 (demyelinating strain; DM) and RSMHV2 (nondemyelinating strain; NDM) are isogenic except for the spike gene, which encodes the host attachment spike glycoprotein. Studies of these strains have elucidated mechanisms of axonal loss and demyelination (3). RSA59 and RSMHV2 both cause hepatitis, encephalitis, and meningitis after intracranial inoculation. However, they differ in their ability to induce macrophage infiltration and subsequent demyelination and axonal loss in spinal cord (2). There is a lack of viral antigen spread and subsequent inflammation extending into spinal cord white matter after intracranial infection with the NDM strain, whereas there is extensive macrophage-mediated white matter pathology secondary to DM strain infection. Thus, the spike protein plays a critical role in anterograde axonal transport of viral particles, an important mechanism mediating axonal damage and demyelination (2, 4). Because both strains cause encephalitis after transcranial inoculation, the differences in spike protein between DM and NDM strains do not impair viral entry; however, differential neural cell tropism may contribute to the mechanism of demyelination (2, 4, 5). Infection of brain neurons and oligodendrocytes occurs on inoculation with either strain, whereas in the spinal cord, oligodendrocyte infection is only seen with the DM strain. This is likely due to the route by which the virus gains access to white matter, that is, spinal cord infection does not occur as a result of direct trauma, whereas transcranial inoculation results in traumatic disruption of the brain gray-white matter interface. Viral particles that would require anterograde axonal transport from infected neurons to reach myelin are able to gain direct access to the myelin sheath and spread proximally to oligodendrocyte cytoplasm.
Anterograde axonal transport and spread of virus from neurons to oligodendrocytes have been documented, but retrograde axonal spread of virus from nerve ending to neural cell body also needs to be considered. Earlier studies suggested that MHV strains may spread via retrograde axonal transport (6, 7), but the molecular mechanisms mediating such transport are not well defined. In the optic nerve, the parental demyelinating strain MHV-A59 causes inflammation, demyelination, and axonal loss (ie, optic neuritis), in contrast to the nondemyelinating MHV-2 strain (8). Whether MHV-induced optic neuritis is dependent on retrograde axonal transport of viral particles or is due to local traumatic disruption of the intracranial portion of retinal ganglion cell (RGC) axons during inoculation is not known. Moreover, the immune response in MHV recombinant strain optic neuritis has not been well characterized. Here, we compared the incidence and phenotype of optic neuritis after inoculation with RSA59 and RSMHV2 and assessed the ability of spike protein to facilitate retrograde axonal transport and induce optic neuritis.
Materials and Methods
Viruses
Recombinant isogenic DM strain of MHV (RSA59) and NDM strain (RSMHV2) have been described in previous studies (4, 9). RSA59 and RSMHV2 strains of MHV are isogenic except for the spike gene, which encodes an envelope glycoprotein that mediates many biological properties of MHV including viral attachment to host cells and virus-cell and cell-cell fusion (10). These recombinant strains also express enhanced green fluorescence protein (EGFP) (4, 9).
Mice
MHV-free, C57BL/6 (B6) mice (Jackson Laboratory, Bar Harbor, ME) were inoculated intracranially at 4 weeks of age with 50% LD50 dose of RSA59 strain (20,000 plaque forming units) or RSMHV2 (100 plaque forming units), as described previously (4). Mice were monitored daily for signs of disease. Mock-infected controls were inoculated similarly but with an uninfected cell lysate at a comparable dilution. Animals were killed (3-5 mice per group) at day 3, days 5 to 7, and day 30 post inoculation (pi). All experimental procedures adhered to guidelines of, and were approved by, the Institutional Animal Care and Use Committee.
Histopathological Analysis
Mice were killed at days 5 to 7 pi (peak of inflammation) or at day 30 pi (during chronic demyelination) and were perfused transcardially with phosphate-buffered saline followed by phosphate-buffered saline containing 4% paraformaldehyde. Brain, spinal cord, eyes, and optic nerve tissues were collected, postfixed in 4% paraformaldehyde overnight, and embedded in paraffin; sections were then stained with hematoxylin and eosin (H&E) to evaluate inflammation and Luxol fast blue to evaluate demyelination. Experiments were repeated at least 3 times with 3 to 5 mice. Areas of demyelination and inflammation were quantified as previously described (8, 11). All slides were coded and read in a blinded fashion. To confirm expected virulence of the strains used, livers from the infected mice were embedded in paraffin, sectioned at 5 μm, and stained with H&E (5, 12). The degree of optic nerve inflammation was scored by 2 blinded investigators on a 0- (no inflammation) to 4-point (severe inflammation) scale, as described (8, 11), during the time of peak inflammation (days 5-7 pi). Any amount of inflammation (score of 1-4) was considered positive for optic neuritis.
Immunohistochemical Analysis
Serial sections from optic nerves were stained by the avidin-biotin-immunoperoxidase technique (Vector Laboratories, Burlingame, CA) using 3, 3′ diaminobenzidine as substrate, and antibodies against lymphocytic cell markers, antiviral nucleocapsid antiserum, or the axonal marker antineurofilament antiserum as primary immunoglobulin G antibodies. The sources and dilution of primary antibodies are listed in the Table. Control slides from mock-infected mice were incubated in parallel.
TABLE.
Antibodies Used for Immunohistochemistry

Detection of Viral Antigen
Fixed sections of optic nerve, brain, and eyes from mice at 3 or 6 days pi were stained by the previously described immunohistochemical methods using antiviral nucleocapsid antiserum. To visualize viral antigen by EGFP expression directly, frozen sections from infected mice were examined by fluorescence microscopy. Viral antigen was also assayed by Western blotting using anti-hepatitis virus nonstructural protein 9 monoclonal antibody (Rockland, Inc., Gilbertsville, PA), according to the manufacturer's instructions. Briefly, protein was extracted from the optic nerve and retina using RIPA buffer (Sigma, St. Louis, MO) in the presence of a complete protease inhibitor cocktail (Pierce, Rockford, IL) at 4°C. Protein concentration was determined with a Micro BCA Protein Assay Kit (Pierce). Protein (30 μg) was electrophoresed on 4% to 15% SDS-PAGE, transferred to cellulose membrane, blocked, and probed with 1:1000 dilution of primary antibody. Expression of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was determined as a loading control using anti-GAPDH (Sigma) diluted 1:10,000. After incubation with horseradish peroxidase-conjugated secondary antibodies, signals were developed with enhanced chemiluminescence agent (GE Healthcare, Buckinghamshire, UK), and intensity was determined using the NIH Image J program.
Evaluation of Axonal Loss
Longitudinal optic nerve sections were stained with antineurofilament antibody, and areas of axonal staining were quantified as described (11). Briefly, 3 photographs were taken at 40× magnification of each stained nerve at predefined locations (one each of the proximal, central, and distal portion of the nerve) covering a total area of 38,500 μm2 of each nerve. The amount of tissue within this area that stained positively for neurofilament was calculated using ImagePro Plus 6.0 (Media Cybernetics, Silver Spring, MD) software. Data shown represent the cumulative area of positive staining/nerve.
Statistics
Comparisons of optic neuritis incidence, demyelination, and axonal density were analyzed by one-way analysis of variance followed by Tukey multiple comparison test using GraphPad Prism 5.0 (GraphPad Software, San Diego, CA). Data represent mean (SD) percentage of eyes that developed optic nerve inflammation or demyelination or the mean (SD) density of axonal staining.
Results
Pathology of RSA59- and RSMHV2-Infected Mice
As in prior studies, RSA59-infected mice showed meningitis, encephalitis, myelitis, and concurrent axonal loss and demyelination as early as day 5 pi with an increase at day 30, whereas RSMHV2 showed only meningitis, encephalitis, and myelitis with no significant demyelination or axonal loss (data not shown) (2). The livers of both strains showed moderate to severe hepatitis (data not shown), thereby confirming virulence.
Optic Neuritis
As expected, mice infected with parental demyelinating strain MHV-A59 at days 5 to 7 pi had optic nerve inflammation (Fig. 1C), whereas optic nerves from mice infected with parental nondemyelinating strain MHV-2 did not (Fig. 1B); their optic nerves appeared similar to those of mock-infected mice (Fig. 1A) (8). The inability of MHV-2 to induce optic neuritis was expected because MHV-2 does not infect the brain parenchyma and is unable to induce encephalitis (13, 14). By contrast, the RSMHV-2 strain induces encephalitis (2, 4, 5). RSA59 induced optic neuritis similar to the parental MHV-A59 strain at days 5 to 7 pi (Fig. 1F), whereas most eyes of RSMHV2-infected mice exhibited no optic nerve inflammation (Fig. 1D); however, there was mild inflammation in a few optic nerves (Fig. 1E).
FIGURE 1.

Comparative histopathology of demyelinating and nondemyelinating mouse hepatitis virus (MHV)-infected mouse optic nerve sections during acute infection. (A-F) Longitudinal sections of optic nerve from mock- (A), MHV-2- (B), MHV-A59- (C), RSMHV2- (D, E), and RSA59 (F)-infected mice were stained with H&E. There is inflammation (arrows) in an MHV-A59-infected mouse optic nerve on day 5 post inoculation (pi) (C) and in the RSA59-infected mouse optic nerve (F). In contrast, almost no RSMHV2-infected mice developed optic neuritis on day 5 pi (D), as in the parental MHV-2 strain (B). Optic nerves in a few RSMHV2-infected mice had only mild inflammation (E). (G) The incidence of MHV-A59-induced optic neuritis was significantly higher than that induced by MHV-2. (H) The incidence of RSA59-induced optic neuritis was also significantly higher than that induced by RSMHV2. *, p < 0.001. Mean (SD) percentage of eyes with optic neuritis in 4 experiments (n = 6-10 eyes/group/experiment; scored by blinded investigators) is shown. Original magnification: (A-F) ×40.
MHV-A59 induced optic neuritis in a mean of 55.6% of optic nerves in 4 experiments, whereas MHV-2 induced optic neuritis in only 3.7% of optic nerves (Fig. 1G); this result is similar to the incidence in prior studies (8). RSA59-infected mice developed optic neuritis with a high incidence (68.5%) similar to that in MHV-A59-infected mice, and significantly higher than the incidence in RSMHV-2 infected mice (25.1%) (Fig. 1H).
Serial sections from RSA59- and RSMHV2-infected mice were stained with anti-CD45 (leukocyte common antigen; LCA), anti-Iba-1 (microglia/macrophage marker), anti-CD3 (T-cell marker), or anti-CD19 (B-cell marker) (Table). LCA staining confirmed the presence of infiltrating inflammatory cells in optic nerves from RSA59-infected mice, whereas few LCA-positive cells were found in those of RSMHV2-infected mice (not shown). Among the LCA-positive inflammatory cells, the majority in the RSA59-infected mice were Iba-1+ microglia/macrophages (Fig. 2C). The optic nerves of some control mice (Fig. 2) and RSMHV2-infected mice (Fig. 2B) also had scattered Iba1+ cells that likely represent resident microglia, but there were far fewer than in the RSA59-infected samples. This result was confirmed by 2 blinded investigators. Significantly more optic nerves from RSA59-infected mice had increased Iba1+ cells versus nerves from RSMHV2-infected mice (14/23 total nerves from 4 experiments versus 4/20, respectively; p = 0.0124 by Fisher exact test).
FIGURE 2.

Characterization of inflammatory cells in optic nerves of RSA59- and RSMHV-2- infected mice. (A-E) Longitudinal sections (5 μm thick) of optic nerve isolated 5 to 6 days post inoculation from mock-infected (A) and RSA59- (C-E) and RSMHV2 (B)-infected mice were immunostained with antibodies to Iba-1 (A-C), CD3 (D), or CD19 (E). Only a few Iba1-positive microglia/macrophages are present in optic nerve from control (A) and RSMHV2-infected mice (B). In RSA59-infected mice, most inflammatory cells are Iba-1-positive (C); few or no CD3+ T cells (D) or CD19+ B cells (E) are present. (F) A section of normal spleen from a mock-infected mouse was used as positive control for CD3. T cells are seen in a blood vessel and within the parenchyma. Original magnification: (A-E) ×40; (F) ×64.
Few or no CD3-positive T cells were present in optic nerves of RSA59-infected mice (Fig. 2D) and no CD19-stained B cells were observed (Fig. 2E). Spleen sections from mock-infected mice stained with either anti-CD3 (Fig. 2F) or anti-CD19 antibodies (not shown) served as positive controls. Thus, there was an increase of Iba-1-positive cells and few CD3-positive cells in optic nerves of RSA59-infected mice.
Optic nerves from RSA59-infected mice had areas of demyelination detected by Luxol fast blue staining both at day 7 and day 30 pi (Fig. 3), whereas no demyelinating plaque was observed in day 7 pi RSMHV2 mouse optic nerve and little or no demyelination was observed at day 30 pi. These data are consistent with the previous result obtained from parental demyelinating strain MHV-A59 and nondemyelinating strain MHV-2 (8).
FIGURE 3.

Demyelination in optic nerves of RSA59- and RSMHV-2-infected mice. (A-F) Longitudinal sections of optic nerves from mock- (A), RSA59- (D-F), and RSMHV2 (B, C)-infected mice at days 6 and 30 post inoculation (pi) stained with Luxol fast blue. There is normal myelin staining in the control optic nerve (A). Almost all of the optic nerves in RSMHV2-infected mice had normal myelin at day 6 (B) and at day 30 (C) pi. During acute infection at day 6 pi, the optic nerves in most RSA59-infected mice maintained normal myelin (D); a few developed focal areas of demyelination (E). During chronic infection stage at day 30 pi, most RSA59-infected mouse optic nerves showed areas of demyelination (F). (G, H) Incidence of demyelination at the acute (G) and chronic time points (H). There was a significantly higher incidence of demyelination induced by RSMHV2 in chronically infected mice (H). *, p = 0.0004. Original magnification: (A-F) ×40.
Axonal Loss in RSA59-Infected But Not RSMHV2-Infected Optic Nerves
No axonal loss was identified at 5 to 7 days pi in optic nerves of RSA59- or RSMHV2-infected mice (Figs. 4A-C). At day 30 pi, RSMHV2-infected mice continued to show no axonal loss (Fig. 4D), whereas optic nerves from RSA59-infected mice showed regions of mildly reduced axonal staining, with focal areas of axon loss intermixed with areas of normal axons (Figs. 4E, F). Quantification of the area of axonal staining across all sections of optic nerves of RSA59-infected mice demonstrated a small but significant decrease compared with optic nerves of either RSMHV2-infected or mock-infected control mice (p = 0.0306) (Fig. 4G).
FIGURE 4.
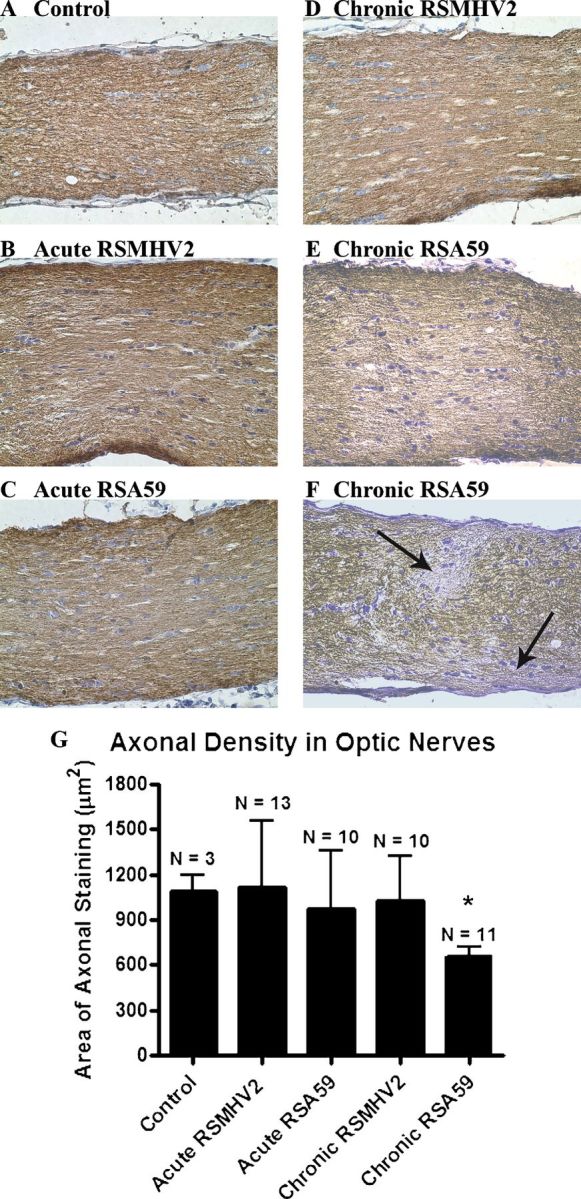
Axon loss in optic nerves of RSA59-infected mice. (A-F) Normal axonal staining is present in a control optic nerve (A), as well as optic nerves from RSMHV2- (B), and RSA5- (C) infected mice at day 6 post inoculation (pi) and at day 30 pi in a RSMHV2-infected mouse (D). Chronic RSA59 infection at 30 days pi resulted in regions of mild diffuse decrease in axonal staining (E) and focal areas with loss of neurofilament staining (arrows) (F) in some mice. (G) The average area of neurofilament-positive axonal staining detected at day 30 pi was significantly lower in optic nerves from RSA59-infected mice than in optic nerves from control, uninfected mice, or RSMHV2-infected mice. *, p = 0.0306. Sections were stained with antineurofilament antibody. Original magnification: (A-F) ×40.
Viral Antigen in RSA59-Infected Optic Nerves and Brain Regions Containing RGC Projections
The optic nerve inflammation, demyelination, and axonal loss observed after RSA59 infection, but not after RSMHV2 infection, demonstrate that the MHV-A59 spike protein is required for the induction of optic neuritis. This suggests that the spike protein may mediate retrograde axonal transport of the virus, allowing it to travel from brain regions containing RGC axonal projections along the optic nerve. Viral antigen was consistently detected within thalamic neurons in RSA59-infected mouse brains (Figs. 5C, D), as well as in some RSMHV2-infected brains (Fig. 5B). RSA59 viral antigen was also detected in the superior colliculi in some animals (data not shown). Light diffuse staining detected in optic nerves from RSA59-infected mice (Fig. 5G), but not RSMHV2-infected mice (Fig. 5F), suggests that only the RSA59 viral antigen is transported to the optic nerves. To further investigate this, the optic nerves were isolated 3 or 6 days pi and frozen sections were examined for the presence of EGFP. Viral antigen-positive EGFP signal was observed in optic nerves from RSA59-infected mice (Figs. 5I, J) but not RSMHV2-infected mice (Fig. 5H). The punctate EGFP signal in optic nerve sections observed by fluorescent microscopy in RSA59-infected mice (Fig. 5J) further suggests axonal transport through the optic nerve and was seen as early as 3 days pi.
FIGURE 5.

Viral antigen distribution during acute CNS infection assessed by immunohistochemistry (A-G) and immunofluorescence (H-J). Thalamus from a control mouse shows no viral antigen staining (A). Viral antigen-positive cells in the thalamus of RSMHV2- (B) and RSA59- (C) infected mice at 3 days post inoculation (pi). Higher magnification (D) of the RSA59-positive thalamic neurons shown in C. No viral antigen is detected by immunohistochemistry in optic nerves of control (E) or RSMHV2 (F)-infected mice at 6 days pi. In contrast, there is mild diffuse brown staining of viral antigen in the optic nerve of an RSA59-infected mouse at 6 days pi. Enhanced green fluorescence protein (EGFP) viral signal is absent in a cross section of optic nerve from an RSMHV2-infected mouse (H). EGFP signal is detected in cross section (I) and longitudinal section (J) of optic nerve from RSA59-infected mice. Nuclei, counterstained with DAPI, are shown in blue (H, I). The boxed area in J, shown at higher magnification in the upper left inset, demonstrates punctate staining observed within retinal ganglion cell axons. Original magnifications: (A-C) ×4; (D-J) ×40; (J, inset) ×100.
Viral Antigen Spreads to RGCs After RSA59 Infection
To determine whether viral antigen is transported all the way to the RGC cell bodies, whole eyes were isolated and sectioned. Viral antigen immunostaining of retinal sections demonstrated no viral antigen in cells of the RGC layer of RSMHV2-infected mice (Fig. 6A), whereas RSA59 viral antigen was detected in some cells within the RGC layer (Figs. 6B, C) at day 6 pi. Overall, viral antigen staining was detected in 60% of eyes from RSA59-infected mice, with 15 serial cross sections examined from each of 5 eyes. The timing of viral antigen spread and relative levels of antigen in retina and optic nerve were determined in protein extracts isolated at days 3 and 6 pi. Western blot analysis demonstrated presence of viral antigen in retinal tissue from RSA59-infected mice at day 6 pi but not at day 3 and no viral antigen was detected in RSMHV2-infected mice (Fig. 6D). Viral antigen was detected in protein extracts from optic nerves of RSA59-infected mice earlier than it was detected in retina. Antigen was detected on Western blots at both days 3 and 6 pi (Fig. 6E). These results suggest that the viral antigen was able to enter the RGC axons and was transported retrograde to the cell bodies within the eye.
FIGURE 6.

RSA59, but not RSMHV2, spreads from the CNS into the retina. (A-C) No viral antigen staining is present in the retina of an RSMHV2-infected mouse at 6 days post inoculation (pi). Scattered viral antigen-positive cells are found in the retinal ganglion cell layer of the retina of an RSA59-infected mouse (B). Higher magnification view of the boxed area in B shows viral antigen-positive cells (C). (D) Western blot shows the presence of a 13-kb band representing viral antigen in retinal protein extracts from 3 representative RSA59-infected mice at day 6 pi. In contrast, little or no viral antigen is present at day 3 pi with RSA59, and as late as day 6 pi with RSMHV2. (E) The average amount of viral antigen present in protein extracts of optic nerve and retina is shown as the ratio of optical density of MHV nsp9 antibody binding normalized against the binding of glyceraldehyde 3-phosphate dehydrogenase (GAPDH). RSA59 is not detectable in retina until day 6 pi, whereas in optic nerve, it is detected at equally high levels at both days 3 and 6 pi.
Discussion
RSA59 induces optic neuritis at comparable levels of severity and incidence as that seen in mice infected with its parental strain MHV-A59 (8), whereas RSMHV2 has a limited ability to induce optic nerve inflammation. Accordingly, nerves from RSA59-infected mice at the chronic stage showed significantly decreased axonal density compared with nerves from RSMHV2-infected mice. This differential ability of MHV strains to induce optic neuritis and axonal injury is dependent on spike glycoprotein mediated retrograde transport of viral antigen along RGC axons. Prior studies demonstrated that RSA59 and RSMHV2 both can cause meningitis and encephalitis, but RSMHV2 did not induce subsequent demyelination and axonal loss in spinal cord. Our current results demonstrate that in addition to demyelination and axonal loss, RSMHV2 also has limited ability to cause significant optic nerve inflammation and RGC infection. However, RSMHV2 did induce some optic nerve inflammation, unlike the parental strain MHV2. This difference may be due to the ability of RSMHV2 to enter neurons in the brain parenchyma and replicate, as demonstrated previously (2, 4, 5) and shown again here, leading to a higher viral load that may allow some diffusion of the virus at an undetectable level. Alternatively, the higher viral load might trigger a more diffuse central nervous system (CNS) inflammatory response associated with mild, nonspecific inflammation in some optic nerves. MHV2, on the other hand, does not enter the brain parenchyma, and infection with it results in meningitis without inducing encephalitis (3).
Prior studies demonstrated that one mechanism limiting the ability of the NDM strain to induce demyelination and axonal loss in the spinal cord involves impaired interneuronal spread of viral particles and defective translocation of viral antigen from gray matter to white matter (2). Evaluation of axonal loss and demyelination in the spinal cord demonstrated that DM MHV infection begins in the neuronal cell body, propagates to the axon, and subsequently induces axonal degeneration and demyelination. The propagation of viral antigen from gray to white matter is dependent on anterograde axonal transport of virus particle mediated by the spike protein (2, 4). Spike protein-mediated axonal transport as an underlying mechanism is further supported by the current studies and demonstrates that it also plays a role in mediating retrograde axonal transport. It is especially intriguing that MHV uses similar molecular mechanisms to interact with cellular axonal transport machinery, suggesting that targeted disruption of this protein function might prevent all pathogenic spread of the virus.
Although it has been reported previously that neurotropic MHV strains can spread within neurons in a retrograde direction, the molecular interactions mediating this spread have not been fully examined (6, 7). In cultured neurons, viral interaction with the microtubule network in neuronal processes has been shown to be important; based on cross-interaction of antimicrotubule antibodies and the nucleocapsid protein (N), it is possible that such interactions might be involved in axonal transport mechanisms (15). However, in view of the fact that the DM and NDM strains used in the present study differ only in spike protein and contain identical N proteins, our data suggest more of a role for the spike protein.
How the DM strain of MHV initially infects neurons requires further study. One possible explanation could be that traumatic disruption to the nerve endings in the brain allows access of the virus through the damaged axolemma; or the virus may be capable of directly infecting intact axons either in the brain, or after diffusing into the optic nerve in the cerebrospinal fluid. Alternatively, virus may be engulfed into the axons at synaptic terminals. Although direct infection after diffusion in cerebrospinal fluid could potentially explain the presence of viral antigen in optic nerves, the fact that antigen is detected in RGC bodies in the retina demonstrates that retrograde transport does occur. Although the presence of viral antigen in RGCs alone cannot exclude hematogenous spread as an alternate mechanism from retrograde transport, the timing of its spread (ie, detected only at day 6 pi in the retina versus day 3 pi within optic nerves and within liver via hematogenous spread), the lack of hematogenous spread to the CNS after systemic infection with equivalent doses of MHV-A59 (6), and the presence of viral antigen in areas of the brain containing RGC projections all suggest that retrograde transport is the more likely mechanism of viral spread. Figure 7 shows a diagram of the optic nerve pathway across which viral antigen is likely transported.
FIGURE 7.
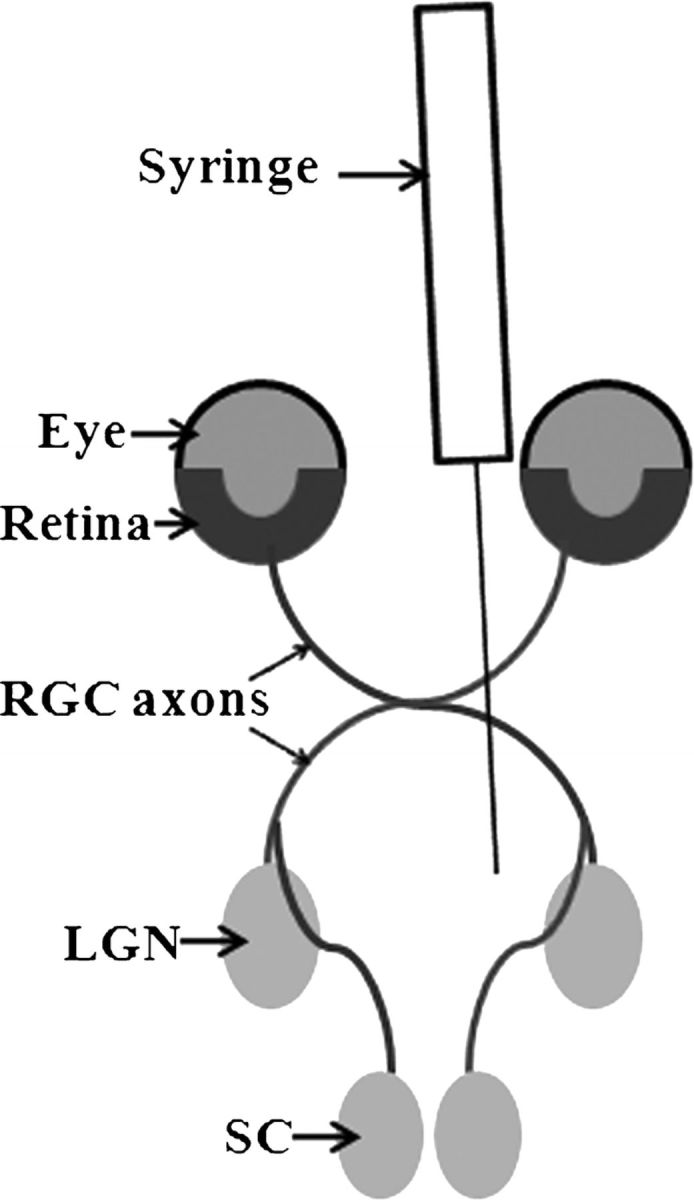
Schematic diagram demonstrating the likely route of MHV transport into the retina. Virus is injected directly into the brain by syringe needle through the skull posterior to the orbits. Virus infects neurons in regions containing retinal ganglion cell (RGC) axonal projections, including the lateral geniculate nucleus (LGN) within the thalamus. Viral antigen is then transported retrograde along RGC axons to the RGC cell bodies in the retina. SC, superior colliculus.
Retrograde transport may follow fusion of the spike protein with axonal membranes followed by loss of most of the viral structural proteins and then binding of the nucleocapsid via one or more viral proteins to the retrograde molecular motors. Such a mechanism has been seen in other neurovirulent viruses that follow a retrograde direction of axonal transport (16, 17). Although it is not common that a single virus can use both directional transport mechanisms, this is not the first virus to demonstrate such properties. For example, herpesviruses can be transported rapidly along microtubules in the retrograde direction from the axon terminus to the dorsal root ganglion and then anterograde in the opposite direction (18, 19). Axonal transport is an important strategy used by several neurovirulent viruses, including herpes simplex, rabies, polio, influenza, and Borna disease viruses, which can be transported in axons either in anterograde or in retrograde direction (20-22). In some instances, viruses can be transported within axons for a long distance, yet this journey occurs within the cell. Therefore, the virus cannot be inactivated by neutralizing antibody during its transit and may spread in the CNS without inducing an antivirus immune response while it is within the cell. When viruses can spread only within axons and/or via direct cell-to-cell contact, they could potentially escape the attack of antiviral drugs or neutralizing antibodies. In the current studies, optic nerve inflammation induced by DM strain RSA59 consisted of predominantly macrophages/microglia, similar to immune responses in spinal cord (2) and without the marked T-cell infiltration seen in other demyelinating disease models (23-26). Iba-1 staining was diffuse and was observed in cell bodies as well as cell processes, as has been seen previously with this macrophage/microglia marker (27, 28). Although Iba-1 cannot distinguish between macrophages and microglia, we suspect most labeled cells represent infiltrating macrophages based on the overall increase in the number of cells observed. Macrophages have been shown to play both proinflammatory and anti-inflammatory roles in optic nerves (29), and we suggest that the DM strain viral antigen might be moving within axons to avoid being cleared by infiltrating macrophages.
It is known that some viruses make use of the microtubules and/or the actin cytoskeleton for axonal transport (30-32). Our studies have shown that a major mechanism of both retrograde and anterograde axonal transport of neurovirulent MHV is mediated by spike protein and further experiments will focus on identifying the molecular mechanisms by which the DM virus interacts with the axonal transport system and whether specific interventions targeting the transport system can delay or prevent the DM strain-induced axonal loss and demyelination. Analyzing the underlying principles of MHV axonal transport will be helpful in the design of viral vectors to be used in research, in human gene therapy, and in the identification of new antiviral therapies. Such therapies may also have the potential to prevent CNS demyelinating diseases that might be triggered initially by viral infection.
Acknowledgments
The authors thank the Du Pre Foundation for support of AG.
References
- 1. Dandekar AA, Wu GF, Pewe L. Axonal damage is T cell mediated and occurs concomitantly with demyelination in mice infected with a neurotropic coronavirus. J Virol 2001;75:6115–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Das Sarma J, Kenyon LC, Hingley ST. Mechanisms of primary axonal damage in a viral model of multiple sclerosis. J Neurosci 2009;29:10272–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Das Sarma J. A mechanism of virus-induced demyelination. Interdiscip Perspect Infect Dis 2010;2010:109239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Das Sarma J, Iacono K, Gard L. Demyelinating and nondemyelinating strains of mouse hepatitis virus differ in their neural cell tropism. J Virol 2008;82:5519–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Das Sarma J, Fu L, Tsai JC. Demyelination determinants map to the spike glycoprotein gene of coronavirus mouse hepatitis virus. J Virol 2000;74:9206–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lavi E, Gilden DH, Highkin MK. The organ tropism of mouse hepatitis virus A59 in mice is dependent on dose and route of inoculation. Lab Anim Sci 1986;36:130–35 [PubMed] [Google Scholar]
- 7. Perlman S, Evans G, Afifi A. Effect of olfactory bulb ablation on spread of a neurotropic coronavirus into the mouse brain. J Exp Med 1990;172:1127–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shindler KS, Kenyon LC, Dutt M. Experimental optic neuritis induced by a demyelinating strain of mouse hepatitis virus. J Virol 2008;82:8882–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Das Sarma J, Scheene E, Seo SH. Enhanced green fluorescent protein expression may be used to monitor murine coronavirus spread in vitro and in the mouse central nervous system. J Neurovirol 2002;8:381–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gallagher TM, Buchmeier MJ. Coronavirus spike proteins in viral entry and pathogenesis. Virology 2001;279:371–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shindler KS, Ventura E, Dutt M. Inflammatory demyelination induces axonal injury and retinal ganglion cell apoptosis in experimental optic neuritis. Exp Eye Res 2008;87:208–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Navas S, Seo SH, Chua MM. Murine coronavirus spike protein determines the ability of the virus to replicate in the liver and cause hepatitis. J Virol 2001;75:2452–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Das Sarma J, Fu L, Hingley ST. Sequence analysis of the S gene of recombinant MHV-2/A59 coronaviruses reveals three candidate mutations associated with demyelination and hepatitis. J Neurovirol 2001;7:432–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Das Sarma J, Fu L, Hingley ST. Mouse hepatitis virus type-2 infection in mice: An experimental model system of acute meningitis and hepatitis. Exp Mol Pathol 2001;71:1–12 [DOI] [PubMed] [Google Scholar]
- 15. Pasick JMM, Kalicharran K, Dales S. Distribution and trafficking of JHM coronavirus structural proteins and virions in primary neurons and the OBL-21 neuronal cell line. J Virol 1994;68:2915–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Klingen Y, Conzelmann KK, Finke S. Double-labeled rabies virus: Live tracking of enveloped virus transport. J Virol 2008;82:237–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McGraw HM, Friedman HM. Herpes simplex virus type 1 glycoprotein E mediates retrograde spread from epithelial cells to neurites. J Virol 2009;83:4791–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McGraw HM, Awasthi S, Wojcechowskyj JA. Anterograde spread of herpes simplex virus type 1 requires glycoprotein E and glycoprotein I but not Us9. J Virol 2009;83:8315–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Smith GA, Gross SP, Enquist LW. Herpesviruses use bidirectional fast-axonal transport to spread in sensory neurons. Proc Natl Acad Sci U S A 2001;98:3466–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cunningham AL, Diefenbach RJ, Miranda-Saksena M, et al. The cycle of human herpes simplex virus infection: Virus transport and immune control. J Infect Dis 2006;194(Suppl 1):S11–S18 [DOI] [PubMed] [Google Scholar]
- 21. Lancaster KZ, Pfeiffer JK. Limited trafficking of a neurotropic virus through inefficient retrograde axonal transport and the type I interferon response. PLoS Pathog 2010;6:e1000791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tsunoda I, Fujinami RS. Inside-out versus outside-in models for virus induced demyelination: Axonal damage triggering demyelination. Springer Semin Immunopathol 2002;24:105–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ben-Nun A, Cohen IR. Experimental autoimmune encephalomyelitis (EAE) mediated by T cell lines: Process of selection of lines and characterization of the cells. J Immunol 1982;129:303–8 [PubMed] [Google Scholar]
- 24. Huseby ES, Liggitt D, Brabb T. A pathogenic role for myelin-specific CD8(+) T cells in a model for multiple sclerosis. J Exp Med 2001;194:669–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ji Q, Perchellet A, Goverman JM. Viral infection triggers central nervous system autoimmunity via activation of CD8+ T cells expressing dual TCRs. Nat Immunol 2010;11:628–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jin YH, Kang B, Kim BS. Theiler's virus infection induces a predominant pathogenic CD4+ T cell response to RNA polymerase in susceptible SJL/J mice. J Virol 2009;83:10981–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ito D, Tanaka K, Suzuki S. Enhanced expression of Iba1, ionized calcium-binding adapter molecule 1, after transient focal cerebral ischemia in rat brain. Stroke 2001;32:1208–15 [DOI] [PubMed] [Google Scholar]
- 28. Simmons GW, Pong WW, Emnett RJ. Neurofibromatosis-1 heterozygosity increases microglia in a spatially and temporally restricted pattern relevant to mouse optic glioma formation and growth. J Neuropathol Exp Neurol 2011;70:51–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. van Rossum D, Hilbert S, Strassenburg S. Myelin-phagocytosing macrophages in isolated sciatic and optic nerves reveal a unique reactive phenotype. Glia 2008;56:271–83 [DOI] [PubMed] [Google Scholar]
- 30. Douglas MW, Diefenbach RJ, Homa FL. Herpes simplex virus type 1 capsid protein VP26 interacts with dynein light chains RP3 and Tctex1 and plays a role in retrograde cellular transport. J Biol Chem 2004;279:28522–30 [DOI] [PubMed] [Google Scholar]
- 31. Vaughan JC, Brandenburg B, Hogle JM. Rapid actin-dependent viral motility in live cells. Biophys J 2009;97:1647–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ward BM, Moss B. Visualization of intracellular movement of vaccinia virus virions containing a green fluorescent protein-B5R membrane protein chimera. J Virol 2001;75:4802–13 [DOI] [PMC free article] [PubMed] [Google Scholar]