

Abstract
Nitric oxide (NO) synthesized by neuronal NO synthase (nNOS) has long been implicated in brain plasticity. However, it is unclear how this short-lived mediator contributes to the long-term molecular changes underlying neuroplasticity, which typically require activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase (ERK) signaling pathway and gene expression. To address this issue, we used a neuroplasticity model based on treatment of neuronal cultures with bicuculline and a model of experience-dependent plasticity in the barrel cortex. In neuronal cultures, NOS inhibition attenuated the bicuculline-induced activation of ERK and the expression of c-Fos, Egr-1, Arc, and brain-derived neurotrophic factor (BDNF), proteins essential for neuroplasticity. Furthermore, inhibition of the NO target soluble guanylyl cyclase or of the cGMP effector kinase protein kinase G (PKG) reduced both ERK activation and plasticity-related protein expression. NOS inhibition did not affect phosphorylation of cAMP response element-binding protein (CREB), a well-established ERK nuclear target, but it attenuated the nuclear accumulation of the CREB coactivator TORC1 and suppressed the activation of Elk-1, another transcription factor target of ERK. Consistent with these in vitro observations, induction of c-Fos, Egr-1, and BDNF was attenuated in the D1 cortical barrel of nNOS−/− mice subjected to single whisker experience. These results establish nNOS-derived NO as a key factor in the expression of proteins involved in neuroplasticity, an effect mediated through cGMP, PKG, and ERK signaling. These actions of NO do not depend on CREB phosphorylation but may involve TORC1 and Elk-1. Our data unveil a previously unrecognized link between neuronal NO and the molecular machinery responsible for the sustained synaptic changes underlying neuroplasticity.
Introduction
The NMDA receptor (NMDAR) is central to the structural and functional synaptic changes underlying neuroplasticity (Citri and Malenka, 2008). Ca2+ influx through NMDAR activates multiple signaling cascades that convey synaptic information to the cell nucleus for new gene expression (Flavell and Greenberg, 2008). Among these signaling pathways, the extracellular signal-regulated kinase (ERK) cascade is required for different forms of synaptic plasticity, such as long-term potentiation (LTP) and long-term depression (LTD) (Rosenblum et al., 2002; Thiels et al., 2002), and is linked to activity-dependent changes in dendritic structure (Wu et al., 2001b). ERK signaling constitutes a major link between NMDAR activation and new protein synthesis required for long-term synaptic modifications (Thomas and Huganir, 2004). Furthermore, ERK is involved in the phosphorylation of the transcription factors cAMP response element-binding protein (CREB) and Elk-1, which drive the expression of key plasticity-related genes (Flavell and Greenberg, 2008). CREB can also be regulated in a phosphorylation-independent manner via the coactivator transducer of regulated CREB activity (TORC), which is required for long-term plasticity (Conkright et al., 2003; Zhou et al., 2006; Sasaki et al., 2011).
Among the most extensively studied plasticity-related proteins are the transcription factors c-Fos and Egr-1, which drive the expression of delayed-onset effector genes and are thought to initiate the complex genomic response underlying long-lasting synaptic changes (Loebrich and Nedivi, 2009). Other proteins, such as activity-regulated cytoskeleton-associated protein (Arc) and brain-derived neurotrophic factor (BDNF), are thought to directly modulate the number and structure of dendrites and synapses (Bramham et al., 2008; Waterhouse and Xu, 2009). However, the signaling pathways linking NMDAR activation to ERK and gene expression in the context of neuroplasticity have not been elucidated.
The neuronal isoform of nitric oxide (NO) synthase (nNOS) is activated in response to Ca2+/calmodulin to produce the diffusible second-messenger NO (Bredt and Snyder, 1990). nNOS is strategically positioned near NMDAR to generate NO in response to incoming Ca2+ (Brenman et al., 1996). Although NO has been implicated in neuroplasticity (O'Dell et al., 1991; Schuman and Madison, 1991; Lu et al., 1999), it is unclear how this short-lived molecule results in the underlying, long-lasting cellular changes. In particular, a direct link between nNOS-derived NO and the expression of specific proteins linked to neuroplasticity has not been provided.
Here, we used well-established models of neuroplasticity in cortical neurons and in the mouse whisker barrel cortex to investigate the role of nNOS-derived NO in the expression of proteins involved in neuroplasticity. We found that neuronal NO is critical for the full expression of neuroplasticity-associated proteins both in vitro and in vivo. This effect of NO involves activation of cGMP–protein kinase G (PKG) and ERK signaling. NO is not involved in CREB phosphorylation but contributes to nuclear accumulation of the CREB coactivator TORC1 and to Elk-1 activation. The findings provide evidence that NO plays an important role in driving the long-term molecular changes underlying neuroplasticity by linking NMDAR signaling to downstream gene expression programs.
Materials and Methods
Mice.
Male mice (7–10 weeks old) were used with approval of the Institutional Animal Care and Use Committee of Weill Cornell Medical College. Studies were conducted in nNOS−/− mice in C57BL/6 background and wild-type C57BL/6 age-matched controls (nNOS+/+) obtained from in-house colonies.
Primary cortical neuronal cultures.
Mixed primary neocortical cultures were prepared from embryonic days 16–17 mice and established in Neurobasal medium (Invitrogen) supplemented with B27 and l-glutamine, as described previously (Zhou et al., 2005; Kawano et al., 2006). Cultures were stimulated with 50 μm bicuculline methiodide (Tocris Bioscience) at 11–12 d in vitro. The following agents were applied for 30 min before bicuculline treatment: l-NAME (N-ω-nitro-l-arginine methyl ester) (2 mm) from Tocris Bioscience; PD98059 [2-(2-amino-3-methyoxyphenyl)-4H-1-benzopyran-4-one] (50 μm) from Cell Signaling Technology); MK-801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate] (10 μm) and MnTBAP [Mn (III) tetrakis(4-benzoic acid) porphyrin] (200 μm) from Sigma-Aldrich; and TRIM [1-(2-trifluoromethylphenyl)imidazole] (100 μm), ODQ (1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one) (100 μm), and KT5823 [(9S,10R,12R)-2,3,9,10,11,12-hexahydro-10-methoxy-2,9-dimethyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg:3′,2′,1′-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylic acid methyl ester] (5 μm) from Enzo Life Sciences. Agents were applied at effective concentrations as established in previous studies from our laboratory or from the literature (Chaban et al., 2001; Wu et al., 2001a; Chen et al., 2005; Kawano et al., 2006; Riccio et al., 2006; Park et al., 2008; Girouard et al., 2009).
Western blot analysis.
Lysates were processed in SDS sample buffer, followed by SDS-PAGE and transferred to polyvinylidene difluoride membranes (Millipore). Membranes were incubated with primary antibodies against the following proteins: c-Fos (1:200), Egr-1 (1:250), Arc (1:200), BDNF (1:250), and Sp1 (1:200) from Santa Cruz Biotechnology; phospho-ERK1/2 (1:2000), ERK1/2 (1:2000), phospho-TrkA/B (1:1000), TrkB (1:1000), phospho-CREB (1:1000), TORC1 (1:1000), and phospho-Elk-1 (1:1000) from Cell Signaling Technology; and β-actin (1:10,000) from Sigma. Blots were digitally exposed using a Kodak Molecular Imaging Station. Densitometric analysis was performed using NIH ImageJ software. Band density values, normalized to loading controls, were then converted to relative percentage values for each blot and averaged across independent experiments.
Subcellular fractionation.
Cortical neurons were processed using the Proteoextract Subcellular Proteome Extraction kit (Calbiochem), following the instructions of the manufacturer for adherent cells. Briefly, cultures were rinsed with ice-cold wash buffer two times for 5 min, followed by incubation in extract buffer I for 10 min on ice and under gentle shaking. Supernatants were collected as fraction 1 (cytosolic proteins) and then extract buffer II was added for 30 min. The supernatant yielded membrane proteins (fraction 2). To obtain fraction 3 (nuclear proteins), extract buffer 3 was added for 10 min, and the supernatants were collected in 1.5 ml of microcentrifuge tubes and spun at 8,600 rpm for 10 min at 4°C to remove debris. Supernatants containing the nuclear fraction were collected.
Immunofluorescence.
Cultures were briefly rinsed with ice-cold PBS and fixed with 4% paraformaldehyde (PFA) for 20 min. Neurons were rinsed and then permeabilized with 0.1% Triton X-100 in PBS for 20 min at room temperature. After 30 min blocking step in 5% normal donkey serum in PBS, neurons were labeled with primary antibody against TORC1 (1:200; Bethyl Laboratories) overnight at 4°C. FITC-conjugated donkey anti-rabbit secondary antibodies (Jackson ImmunoResearch) were used at 1:200. Images were acquired using a Leica confocal microscope.
Single whisker experience.
Mice were subjected to unilateral single whisker experience (SWE), as done by others (Glazewski et al., 1996; Barth et al., 2000), in which all large mystacial whiskers on the right side of the snout (A1–A4, B1–B4, C1–C5, D2–D5, E1–E5, α, β, γ, and δ), except for a single whisker (D1), are carefully removed under isoflurane anesthesia. During recovery, animals were returned to their home cages for 16 h and were then deeply anesthetized (pentobarbital, 100 mg/kg, i.p.) and perfused intracardially with ice-cold 4% paraformaldehyde in PBS. Brains were harvested and postfixed in 4% PFA and sunk in 30% sucrose at 4°C for immunohistochemical processing. The hemisphere contralateral to the spared D1 whisker was marked for subsequent identification of the active barrel in free-floating sections. Brains used for in situ hybridization (ISH) were harvested after 6 h SWE, fresh frozen on dry ice, and sectioned at 20 μm thickness on a Leica cryostat. Sections were thaw mounted on Superfrost Plus Slides (Thermo Fisher Scientific).
Immunohistochemistry.
Free-floating coronal sections (40 μm thick) were obtained using a sliding microtome. All consecutive sections spanning 1 mm rostrocaudally (approximately between bregma −1.15 and −2.15), containing the barrel cortex, were collected. For each immunohistochemical run, sections from wild-type and knock-out brains were processed simultaneously, using the same reagents and conditions. After incubation in blocking solution [0.5% bovine serum albumin (BSA) in PBS] for 30 min at room temperature, sections were labeled overnight at 4°C with primary rabbit polyclonal antibodies against c-Fos (1:10,000, Ab-5; Calbiochem) and Egr-1 (1:15,000, C-19; Santa Cruz Biotechnology) in 0.1% BSA in PBS plus 0.25% Triton X-100. After 30 min incubation in biotinylated goat anti-rabbit secondary antibodies (1:400; Vector Laboratories), sections were incubated in avidin–biotin complex for 30 min (Vector Laboratories) and 3,3′-diaminobenzidine with H2O2 for 5 min. Sections were mounted on gelatin-coated slides, air dried, dehydrated and defatted by ethanol and xylenes series, and coverslipped with DPX mounting medium (Aldrich Chemical Co.).
In situ hybridization.
Antisense oligonucleotides matching BDNF exon IX (5′-TTTATCTGCCGCTGTGACCCACTCGCTACAGCAGATAAA-3′) were kindly provided by Dr. E. M. Waters (Rockefeller University, New York, NY) and labeled with [33P]dATP. Control sense sequences were used to confirm hybridization specificity. The ISH procedure was done as described previously by Hunter et al. (2006). Air-dried slides were exposed to Kodak MR autoradiography films for 10 d.
Image analysis.
Images were acquired from coded slides in blinded manner on a Nikon Eclipse 80i microscope with a QImaging Micropublisher 5.0 RTV digital camera, using IPLab software. Same illumination levels and exposure times were set to acquire images at 10× magnification from all consecutive sections containing c-Fos or Egr-1 immunoreactivity in the D1 barrel. Images were acquired from the D1 barrel contralateral to the single spared D1 whisker (“experimental D1 barrel”) and from the D1 barrel contralateral to the undeprived whiskers (“control D1 barrel”). Previous assessments of c-Fos induction in D1 barrel after 16 h SWE enable location of this structure according to anatomical landmarks. Digitized images of the experimental and control D1 for each section were set to eight-bit grayscale and inverted. Average pixel density for regions lacking immunolabeling was determined within each captured image and subtracted from that image using NIH ImageJ software (Gammie and Nelson, 2001; Matys et al., 2004; Dallaporta et al., 2007). Images were set at a fixed optical density threshold value for each run, and a region of interest (ROI) box was drawn around the experimental D1 barrel and applied to the image of its corresponding control D1 barrel. NIH ImageJ particle analysis tool was used to measure the mean area of immunoreactivity within that ROI (Gammie and Nelson, 2001; Matys et al., 2004; Dallaporta et al., 2007), thereby avoiding automated counting of overlapping c-Fos or Egr-1-like nuclei as one particle (Dallaporta et al., 2007). The immunoreactive area for the experimental D1 was normalized to the immunoreactive area for the control D1 barrel within each section to control for individual differences in basal expression levels of these proteins. Values were expressed as the mean induction of immunoreactivity across all measured sections in each brain. For BDNF ISH, images were processed using MCID software. Relative optical density was measured bilaterally, within the experimental and control D1 barrel columns, whereas background density from a region lacking hybridization (corpus callosum) was subtracted.
Data analysis.
Data are expressed as mean ± SEM. Statistical analyses were performed using GraphPad Prism software. Two-group comparisons were analyzed by Student's t test. Multiple comparisons were evaluated by ANOVA and Tukey's post hoc test, when appropriate. Statistical significance was considered for p < 0.05.
Results
Nitric oxide activates neuronal ERK signaling
To determine whether nNOS-derived NO plays a role in neuroplasticity associated gene expression, we examined whether ERK signaling is triggered by synaptic NMDAR activation, using a well-established in vitro model of neuroplasticity (Hardingham et al., 2002; Arnold et al., 2005). Primary cortical neuronal cultures were stimulated with the GABAA receptor antagonist bicuculline, which suppresses tonic GABAergic inhibition and triggers synaptically evoked bursts of action potentials (Arnold et al., 2005). This synchronous bursting depends on calcium influx through synaptic NMDAR and constitutes a form of neuronal network plasticity (Hardingham et al., 2002; Arnold et al., 2005). The dual phosphorylation of the ERK cascade downstream effectors p44/p42 mitogen-activated protein (MAP) kinase (ERK1/2) was examined by Western blot after 5 min of bicuculline treatment. Bicuculline (50 μm) resulted in a robust increase in phospho-ERK1/2, an effect suppressed by the NMDAR antagonist MK-801(10 μm), confirming that ERK signaling relies on active NMDAR (Fig. 1A). To determine whether NO contributes to the NMDAR-dependent activation of ERK, we pretreated cultures with the nonselective NOS inhibitor l-NAME (2 mm) or the nNOS inhibitor TRIM (100 μm). The bicuculline-evoked increase in phospho-ERK1/2 levels was attenuated by either l-NAME or TRIM (Fig. 1A). These results suggest that nNOS-derived NO is involved in the activation of the ERK pathway after a neuroplasticity-inducing stimulus.
Figure 1.

NO activates neuronal ERK signaling. A, Left, Western blots showing increased ERK1/2 phosphorylation in cortical neurons treated with bicuculline for 5 min (Bic; 50 μm), whereas pretreatment with l-NAME (2 mm) or TRIM (100 μm) reduces phospho-ERK1/2. The NMDAR antagonist MK-801 (MK; 10 μm) significantly attenuates the increase in ERK1/2 phosphorylation. The MEK inhibitor PD98059 (PD; 50 μm) effectively blocks ERK1/2 phosphorylation. Right, Densitometric analysis of phospho-ERK2 (normalized to total ERK2), expressed as a percentage of the induction obtained with bicuculline (% Bic Induction). Veh, Vehicle. B, Western blots showing bicuculline (Bic)-induced increase in c-Fos, Egr-1, Arc (1 h), and BDNF (8 h). Pretreatment with PD98059 (PD; 50 μm) blocks the increase in plasticity-related proteins after bicuculline. *p < 0.05 from vehicle (Veh), #p < 0.05 from bicuculline; ANOVA and Tukey's test; n = 4–5 per group.
The full expression of plasticity-related proteins induced by bicuculline depends on nNOS-derived NO
Our data indicating that NO is involved in the activation of ERK raises the possibility that NO contributes to the expression of key proteins associated with neuroplasticity. First, we determined whether the ERK pathway is specifically involved in the expression of neuroplasticity-associated proteins. To this end, we analyzed the levels of the transcription factors c-Fos and Egr-1 and synaptic effector proteins Arc and BDNF after bicuculline. Bicuculline treatment increased the levels of c-Fos, Egr-1, Arc (1 h), and BDNF (8 h) (Fig. 1B). Pretreatment with the MAP kinase kinase 1 (MEK1) inhibitor PD98059 (50 μm), which inhibited ERK1/2 phosphorylation (Fig. 1A), blocked the bicuculline-induced expression of all four proteins (Fig. 1B). The bicuculline-evoked increase in protein expression also relied on NMDAR activation, because it was reduced by MK-801 (Fig. 2A). Next, we investigated whether NO is involved in the expression of plasticity-related proteins. NOS inhibition with either l-NAME or TRIM attenuated bicuculline-induced expression of c-Fos, Egr-1, Arc, and BDNF (Fig. 2B,C), indicating that nNOS-derived NO is required for the full induction of key proteins associated with neuroplasticity. In addition, we found that NOS inhibitors suppress the bicuculline-induced phosphorylation of the BDNF receptor TrkB (Fig. 2C). Because TrkB is autophosphorylated and activated when bound by BDNF (Huang and Reichardt, 2003), this observation suggests that NO may be necessary for BDNF signaling.
Figure 2.

The full expression of plasticity-related proteins induced by bicuculline (Bic) depends on nNOS-derived NO. A, Western blots showing that expression of c-Fos, Egr-1, Arc (1 h bicuculline), and BDNF (8 h) is reduced by MK-801 (MK). B, Neurons pretreated with l-NAME or TRIM display attenuated levels of c-Fos, Egr-1, and Arc. C, Western blot showing a similar effect of NOS inhibition on the levels of BDNF (left) and phosphorylated TrkB (p-TrkB; normalized to total TrkB, right) after 8 h bicuculline. *p < 0.05 from vehicle (Veh), #p < 0.05 from bicuculline; ANOVA and Tukey's test; n = 5 per group.
nNOS-derived NO is involved in the induction of c-Fos, Egr-1, and BDNF in the whisker barrel cortex after single-whisker experience
To determine whether nNOS-derived NO is critical for gene expression associated with neuroplasticity in vivo, we used a model of experience-dependent plasticity in the whisker barrel cortex (Glazewski and Fox, 1996; Barth et al., 2000; Clem et al., 2008). In this SWE model, mice are deprived of all but one whisker on one side of the face and then allowed to naturally explore their environment. In adult mice, a period of 16–24 h of SWE evokes NMDAR-dependent potentiation at synapses within the barrel corresponding to the “active” spared whisker (Barth et al., 2000; Clem et al., 2008). Because SWE-induced plasticity occurs specifically in the active barrel column, this provides a unique opportunity to assess the expression of genes and proteins within a site undergoing plasticity (Barth et al., 2000, 2004; Clem et al., 2008). We used this model to determine whether nNOS-derived NO is involved in plasticity-related protein expression by comparing the induction of c-Fos and Egr-1 in nNOS+/+ and nNOS−/− mice 16 h after the removal of all whiskers unilaterally, except for D1. Using immunohistochemistry and light microscopic analysis, we assessed c-Fos or Egr-1 immunoreactivity within the D1 barrel corresponding to the spared whisker (termed experimental D1) and in the control D1 barrel (corresponding to the undeprived whiskers) for each brain section containing c-Fos or Egr-1 induction in the D1 barrel. We found an increase of c-Fos immunoreactivity in the experimental D1 over the control D1 barrel in nNOS+/+ mice, but the increase was attenuated in nNOS−/− (Fig. 3A). Similarly, the increase in Egr-1 immunoreactivity induced by SWE was attenuated in nNOS−/− mice (Fig. 3B). Upregulation of BDNF mRNA was previously demonstrated after 6 h of artificial whisker stimulation (Rocamora et al., 1996). Therefore, using in situ hybridization, we compared induction of BDNF mRNA in nNOS+/+ and nNOS−/− after 6 h SWE. We found robust BDNF induction in the experimental D1 of nNOS+/+ mice after SWE (Fig. 3C). However, the BDNF induction observed in nNOS−/− mice was attenuated compared with nNOS+/+ mice (Fig. 3C). These data suggest a role for nNOS-derived NO in the gene and protein expression linked to experience-dependent plasticity.
Figure 3.
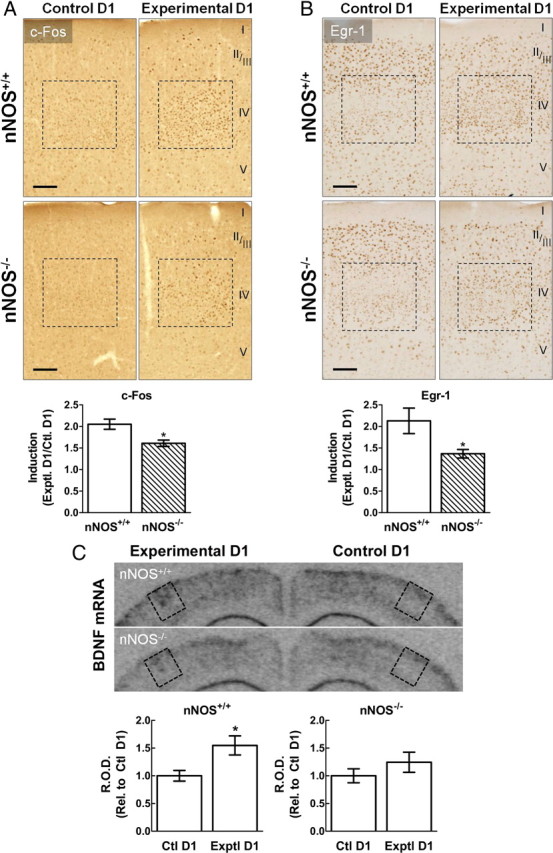
In vivo induction of c-Fos, Egr-1, and BDNF in barrel cortex after SWE is attenuated in the absence of nNOS-derived NO. A, Top, Representative images depicting c-Fos immunoreactivity within control D1 barrels (corresponding to the undeprived whisker pad) and experimental D1 barrels (corresponding to the single spared whisker) in both nNOS+/+ and nNOS−/− mice. Cortical layers are labeled on the right. Upregulation of c-Fos immunolabeling is observed within experimental D1 compared with control D1 in both nNOS+/+ and nNOS−/− mice. However, c-Fos induction is attenuated in nNOS−/− mice. Bottom, Quantification of c-Fos immunoreactivity after 16 h SWE, expressed as a ratio of experimental D1 over control D1 labeling. B, Induction of Egr-1 immunoreactivity after 16 h SWE is also less prominent in nNOS−/− mice. Scale bars, 100 μm. C, In situ hybridization against BDNF reveals that 6 h SWE leads to induction of BDNF mRNA in the experimental D1 barrel, an effect that is attenuated in nNOS−/− mice. R.O.D., Relative optical density. *p < 0.05; t test; n = 5–6 per group.
ERK activation involves the NO targets soluble guanylyl cyclase and PKG
Next, we used the bicuculline model to examine the signaling pathways by which NO activates ERK. Because many of the biological effects of NO are mediated by activation of soluble guanylyl cyclase (sGC) and its synthesis of the second-messenger cGMP (Hofmann et al., 2006; Francis et al., 2010), we tested whether sGC was involved in ERK activation induced by bicuculline. Pretreatment of cultures with the sGC inhibitor ODQ (100 μm) attenuated ERK1/2 phosphorylation after bicuculline (Fig. 4A). Furthermore, KT5823 (5 μm), an inhibitor of PKG, the main target of cGMP (Hofmann et al., 2006), also attenuated ERK1/2 phosphorylation (Fig. 4A), suggesting that PKG is involved in ERK activation. NO may also exert its effects via the signaling molecule peroxynitrite, the product of the reaction of NO with the free radical superoxide (Liaudet et al., 2009). Therefore, we examined whether superoxide was involved in ERK activation. In contrast to the effects of ODQ and KT5823, the cell-permeable reactive oxygen species (ROS) scavenger MnTBAP (200 μm) did not affect ERK1/2 phosphorylation (Fig. 4B). Collectively, these findings implicate cGMP and PKG as the major NO effectors in ERK activation.
Figure 4.

ERK activation involves the NO targets sGC and PKG. A, Inhibition of sGC with ODQ (100 μm) or of PKG with KT5823 (KT; 5 μm) leads to attenuated ERK1/2 phosphorylation after 5 min bicuculline (Bic). B, ERK1/2 phosphorylation evoked by 5 min bicuculline is not affected by pretreatment with the ROS scavenger MnTBAP (TBAP; 200 μm). *p < 0.05 from vehicle (Veh), #p < 0.05 from bicuculline; ANOVA and Tukey's test; n = 5 per group.
Inhibition of sGC and PKG attenuates the expression of plasticity-related proteins induced by bicuculline
Because ERK activation involves cGMP and PKG, we examined whether these mediators also contribute to the expression of neuroplasticity-associated proteins. The sGC inhibitor ODQ attenuated the induction of c-Fos, Egr-1, Arc, and BDNF after bicuculline (Fig. 5A). Likewise, the PKG inhibitor KT5823 attenuated the bicuculline-induced increase in all four proteins (Fig. 5B). The degree of reduction in all four proteins obtained after sGC or PKG inhibition was comparable with that observed after NOS inhibition (Fig. 2B,C). In contrast, MnTBAP had no effect on protein levels after bicuculline (Fig. 5C). These findings implicate NO, cGMP, and PKG in the expression of plasticity-related proteins.
Figure 5.

Inhibition of sGC and PKG attenuates the expression of plasticity-related proteins induced by bicuculline. A, ODQ attenuates the expression of c-Fos, Egr-1, Arc (1 h), and BDNF (8 h) after bicuculline (Bic)-induced synaptic activity. B, KT5823 (KT) also attenuates the induction of all four plasticity-related proteins. C, MnTBAP (TBAP) has no effect on the induction of c-Fos, Egr-1, Arc, or BDNF. *p < 0.05 from vehicle (Veh), #p < 0.05 from bicuculline; ANOVA and Tukey's test; n = 5 per group.
NO contributes to nuclear accumulation of the CREB coactivator TORC1 and to Elk-1 phosphorylation
We then examined the role of NO in the activation of the nuclear targets of ERK: CREB and Elk-1. First, we tested whether ERK is involved in phosphorylation of CREB at Ser-133, a key event in CREB-mediated transcription (Lonze and Ginty, 2002; Alberini, 2009). Indeed, the MEK inhibitor PD98059 attenuated the increase in phospho-CREB after 5 min bicuculline (Fig. 6A). To determine whether NO contributes to CREB phosphorylation, we tested the effect of NOS inhibition on phospho-CREB levels after bicuculline. The bicuculline-evoked increase in phospho-CREB was not affected by either l-NAME or TRIM (Fig. 6B), suggesting that NO is not involved in CREB phosphorylation. Although CREB phosphorylation is key for CREB activity, it is not sufficient to drive CREB-dependent gene expression (Alberini, 2009), raising the possibility that NO may activate CREB through a different mechanism. The TORC protein family has emerged as a critical Ser-133-independent means of CREB activation (Conkright et al., 2003). TORC translocation into the nucleus is an essential step in CREB-mediated transcription (Bittinger et al., 2004). We examined TORC1 immunofluorescence in cortical cultures after 30 min bicuculline and found an increase in TORC1 labeling associated with the nucleus (Fig. 6C). This was confirmed using subcellular fractionation, which revealed increased TORC1 levels in the nuclear fraction after 30 min bicuculline (Fig. 6D). This effect was reduced by PD98059, suggesting an involvement of ERK in TORC1 regulation (Fig. 6D). To test the involvement of NO, cultures were pretreated with l-NAME or TRIM. Nuclear TORC levels after bicuculline were attenuated after NOS inhibition, pointing to a role of NO in TORC1 nuclear accumulation (Fig. 6E).
Figure 6.

NO contributes to nuclear accumulation of CREB coactivator TORC1 and to Elk-1 phosphorylation. A, CREB phosphorylation on Ser-133 (p-CREB) is increased after 5 min bicuculline (Bic) and attenuated by PD98059 (PD). B, The bicuculline-evoked increase in p-CREB is not affected by l-NAME or TRIM. C, Increased TORC1 immunofluorescent labeling in the nuclei of cortical neurons treated with bicuculline for 30 min, as shown by the arrowheads. TO-PRO-3 was used to stain and identify cell nuclei. D, E, TORC1 levels are increased in the nuclear fraction of cortical neuron lysates after 30 min bicuculline. This increase is attenuated by PD98059 (D) and by either l-NAME or TRIM (E). The nuclear protein Sp1 was used as loading control. F, G, Elk-1 phosphorylation on Ser-383 (p-Elk-1) induced by 5 min bicuculline is blocked by PD98059 (F) and attenuated by NOS inhibition (G). *p < 0.05 from vehicle (Veh), #p < 0.05 from bicuculline; ANOVA and Tukey's test; n = 5 per group.
Active Elk-1 interacts with target DNA and the serum response factor, driving serum response element (SRE)-dependent transcription (Buchwalter et al., 2004). Elk-1 is activated by direct phosphorylation by ERK1/2 (Buchwalter et al., 2004), but whether Elk-1 activation involves NO is unknown. We examined phosphorylation of Elk-1 on Ser-383, the major residue enabling ternary complex formation and transactivation (Gille et al., 1995). We found that phospho-Elk-1 levels were increased after 5 min bicuculline in an ERK-dependent manner, because PD98059 prevented Elk-1 phosphorylation (Fig. 6F). Pretreatment with NOS inhibitors attenuated phospho-Elk-1 levels after bicuculline, suggesting an involvement of NO in Elk-1 activation (Fig. 6G).
Discussion
NO is known to play an important role in NMDAR-dependent neuroplasticity (Garthwaite, 2008). However, it is unclear how this short-lived molecule contributes to the underlying long-term synaptic modifications. Considering that persistent changes in neuronal function and structure typically require new gene expression (Flavell and Greenberg, 2008), we tested the hypothesis that nNOS-derived NO is involved in the expression of proteins critical to neuroplasticity. We found that neuronal NO is required for the full expression of c-Fos, Egr-1, Arc, and BDNF in cortical cultures after bicuculline-evoked synaptic activity. Moreover, we found that NO not only participates in BDNF expression but is also required for activation of the BDNF receptor TrkB. In vivo, we found that nNOS-derived NO contributes to the induction of c-Fos, Egr-1, and BDNF in the affected barrel after single whisker experience, supporting our in vitro findings. The signaling pathways underlying this effect involve cGMP, PKG, and ERK. In addition, we identified a role for NO in the activation of transcriptional regulators Elk-1 and TORC1. By implicating NO and its signaling targets as a key link between NMDAR and protein synthesis, these new observations provide a more complete understanding of the mechanisms by which neuronal NO leads to the long-term modifications associated with neuroplasticity.
The ERK signaling cascade is a major link between synaptic stimuli and gene expression (Thomas and Huganir, 2004). We found that activation of this pathway involves NO. A previous study in hippocampal neurons implicated NO in the activation of p21Ras, an upstream effector of the ERK cascade (Yun et al., 1998). Although this finding established a potential link between NO and ERK, its relevance to neuroplasticity is unclear because the stimulation method used activates both synaptic and extrasynaptic NMDAR, which have opposing roles in the activation of ERK (Ivanov et al., 2006). In the present study, we used the bicuculline model, which activates only the synaptic NMDAR population (Hardingham et al., 2002; Arnold et al., 2005), and found that NOS inhibition attenuates ERK activation. These observations, in concert with previous evidence for involvement of NO in p21Ras activation (Yun et al., 1998), implicate ERK signaling as a key target for the regulation of gene expression by NO.
We found that NO contributes to ERK activation and plasticity-related protein expression via cGMP and PKG. cGMP mediates many of the biological effects of NO, and, in neurons, this cyclic nucleotide is involved in NO-dependent forms of synaptic plasticity in hippocampus and other brain regions (Boulton et al., 1995; Kleppisch and Feil, 2009). Moreover, elevations in cGMP levels by inhibition of cGMP-hydrolyzing phosphodiesterases have been shown to reverse deficits in LTP and LTD in neurodegenerative disease models (Puzzo et al., 2009; Picconi et al., 2011). Because both nNOS and the α2β1 isoform of sGC are anchored to the postsynaptic membrane through their interaction with postsynaptic density protein-95 (Brenman et al., 1996; Russwurm et al., 2001), postsynaptic sGC can be rapidly activated by NO, making it a likely mediator of NMDAR–nNOS signaling (Russwurm et al., 2001). PKG is a major target of cGMP in neurons and is involved in the activation of CREB and other transcription factors (Lu et al., 1999; Kleppisch and Feil, 2009). Our experiments in cortical cultures demonstrate a role for cGMP and PKG in neuronal ERK activation and in the expression of plasticity-related proteins. Therefore, our data provide evidence for a pathway linking activation of NMDAR at the synapse to ERK signaling, partly through NO/cGMP/PKG.
NO could also react with superoxide to form peroxynitrite, which can modulate various cellular signaling pathways (Liaudet et al., 2009). However, we found no effect on either ERK activation or protein expression using the ROS scavenger MnTBAP, suggesting that an NO-superoxide reaction is unlikely to mediate these effects. Another mechanism by which NO could exert its effects on gene expression is via S-nitrosylation of target proteins, such as transcription factors or histone deacetylases (HDAC) (Contestabile, 2008; Nott and Riccio, 2009). NO was shown recently to be involved in CREB binding to target DNA sequences after stimulation with exogenous BDNF (Riccio et al., 2006). This effect did not depend on cGMP/PKG or ERK but on S-nitrosylation of HDAC2, facilitating CREB–DNA binding (Riccio et al., 2006; Nott et al., 2008). In contrast, our findings suggest that, in response to synaptic NMDAR activation, cGMP, PKG, and ERK contribute to the expression of key plasticity-related proteins. This raises the interesting possibility that NO, when produced after distinct extracellular stimuli, can initiate different signaling pathways leading to gene expression. These pathways could work in parallel to converge on transcriptional regulation or could occur sequentially, whereby NMDAR–NO leads to BDNF expression, which then results in NO production and S-nitrosylation of nuclear proteins. However, additional work is required to define these possible relationships.
We also studied the involvement of NO in the activation of well-known ERK nuclear targets CREB and Elk-1. Phosphorylation of CREB at Ser-133 is considered a prerequisite for CREB activation (Alberini, 2009). However, in agreement with Riccio et al. (2006), we found that NO is not required for CREB phosphorylation. Instead, our results indicate that NO contributes to activity-dependent TORC1 nuclear accumulation. Because this is considered to be a necessary step for CREB activation (Bittinger et al., 2004), our findings suggest a novel role for NO and ERK in the regulation of TORC1/CREB-dependent gene expression. Furthermore, our data reveal an involvement of NO in the phosphorylation of Elk-1, pointing to a new role for NO in Elk-1 activation, which is a main mechanism leading to SRE-dependent gene expression (Buchwalter et al., 2004). Together, our results suggest that NO contributes to gene expression through the regulation of different transcription factor targets of the ERK signaling pathway.
In conclusion, we found that nNOS-derived NO contributes to the induction of proteins involved in synaptic changes in neuroplasticity models in vitro and in vivo (Fig. 7). The signaling pathway responsible for this effect involves cGMP, PKG, and ERK. Moreover, the effect of NO on protein expression does not involve CREB phosphorylation but may be linked to the CREB coactivator TORC1 and the transcription factor Elk-1. These findings provide new evidence that NO, a short-lived mediator, participates in the long-term molecular changes underlying neuroplasticity, in part by regulating the expression of critical proteins governing synaptic structure and function.
Figure 7.

Hypothetical model of the role of neuronal NO in neuroplasticity-associated protein expression. Ca2+ influx through NMDAR on the postsynaptic membrane activates nNOS. The resulting NO is required for ERK activation and for the full expression of plasticity-related proteins, an effect primarily mediated by cGMP and PKG. In addition, NO contributes to Elk-1 but not CREB phosphorylation. However, NO may activate CREB by modulating nuclear accumulation of its essential coactivator TORC1. Although NO-independent pathways may also be activated by NMDAR, NO exerts significant control over gene expression. These effects of NO may occur postsynaptically, as depicted in the schematic, but can also take place in neighboring or synaptically apposed neurons, as NO diffuses across the plasma membrane.
Footnotes
The present work was supported by National Institutes of Health Grants R36 AG035067 (E.F.G.) and R01 NS37853 (C.I.). We thank Jasmine Tan for valuable technical assistance with primary cultures and animal husbandry, Dr. Josef Anrather for helpful discussion and comments, and Dr. Elizabeth M. Waters for providing the BDNF oligoprobes and for assistance with the in situ hybridization.
References
- Alberini CM. Transcription factors in long-term memory and synaptic plasticity. Physiol Rev. 2009;89:121–145. doi: 10.1152/physrev.00017.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold FJ, Hofmann F, Bengtson CP, Wittmann M, Vanhoutte P, Bading H. Microelectrode array recordings of cultured hippocampal networks reveal a simple model for transcription and protein synthesis-dependent plasticity. J Physiol. 2005;564:3–19. doi: 10.1113/jphysiol.2004.077446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth AL, McKenna M, Glazewski S, Hill P, Impey S, Storm D, Fox K. Upregulation of cAMP response element-mediated gene expression during experience-dependent plasticity in adult neocortex. J Neurosci. 2000;20:4206–4216. doi: 10.1523/JNEUROSCI.20-11-04206.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth AL, Gerkin RC, Dean KL. Alteration of neuronal firing properties after in vivo experience in a FosGFP transgenic mouse. J Neurosci. 2004;24:6466–6475. doi: 10.1523/JNEUROSCI.4737-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittinger MA, McWhinnie E, Meltzer J, Iourgenko V, Latario B, Liu X, Chen CH, Song C, Garza D, Labow M. Activation of cAMP response element-mediated gene expression by regulated nuclear transport of TORC proteins. Curr Biol. 2004;14:2156–2161. doi: 10.1016/j.cub.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Boulton CL, Southam E, Garthwaite J. Nitric oxide-dependent long-term potentiation is blocked by a specific inhibitor of soluble guanylyl cyclase. Neuroscience. 1995;69:699–703. doi: 10.1016/0306-4522(95)00349-n. [DOI] [PubMed] [Google Scholar]
- Bramham CR, Worley PF, Moore MJ, Guzowski JF. The immediate early gene arc/arg3.1: regulation, mechanisms, and function. J Neurosci. 2008;28:11760–11767. doi: 10.1523/JNEUROSCI.3864-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredt DS, Snyder SH. Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc Natl Acad Sci U S A. 1990;87:682–685. doi: 10.1073/pnas.87.2.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenman JE, Chao DS, Gee SH, McGee AW, Craven SE, Santillano DR, Wu Z, Huang F, Xia H, Peters MF, Froehner SC, Bredt DS. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and α1-syntrophin mediated by PDZ domains. Cell. 1996;84:757–767. doi: 10.1016/s0092-8674(00)81053-3. [DOI] [PubMed] [Google Scholar]
- Buchwalter G, Gross C, Wasylyk B. Ets ternary complex transcription factors. Gene. 2004;324:1–14. doi: 10.1016/j.gene.2003.09.028. [DOI] [PubMed] [Google Scholar]
- Chaban VV, McRoberts JA, Ennes HS, Mayer EA. Nitric oxide synthase inhibitors enhance mechanosensitive Ca2+ influx in cultured dorsal root ganglion neurons. Brain Res. 2001;903:74–85. doi: 10.1016/s0006-8993(01)02407-6. [DOI] [PubMed] [Google Scholar]
- Chen Y, Wang PY, Ghosh A. Regulation of cortical dendrite development by Rap1 signaling. Mol Cell Neurosci. 2005;28:215–228. doi: 10.1016/j.mcn.2004.08.012. [DOI] [PubMed] [Google Scholar]
- Citri A, Malenka RC. Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology. 2008;33:18–41. doi: 10.1038/sj.npp.1301559. [DOI] [PubMed] [Google Scholar]
- Clem RL, Celikel T, Barth AL. Ongoing in vivo experience triggers synaptic metaplasticity in the neocortex. Science. 2008;319:101–104. doi: 10.1126/science.1143808. [DOI] [PubMed] [Google Scholar]
- Conkright MD, Canettieri G, Screaton R, Guzman E, Miraglia L, Hogenesch JB, Montminy M. TORCs: transducers of regulated CREB activity. Mol Cell. 2003;12:413–423. doi: 10.1016/j.molcel.2003.08.013. [DOI] [PubMed] [Google Scholar]
- Contestabile A. Regulation of transcription factors by nitric oxide in neurons and in neural-derived tumor cells. Prog Neurobiol. 2008;84:317–328. doi: 10.1016/j.pneurobio.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Dallaporta M, Pecchi E, Jacques C, Berenbaum F, Jean A, Thirion S, Troadec JD. c-Fos immunoreactivity induced by intraperitoneal LPS administration is reduced in the brain of mice lacking the microsomal prostaglandin E synthase-1 (mPGES-1) Brain Behav Immun. 2007;21:1109–1121. doi: 10.1016/j.bbi.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Flavell SW, Greenberg ME. Signaling mechanisms linking neuronal activity to gene expression and plasticity of the nervous system. Annu Rev Neurosci. 2008;31:563–590. doi: 10.1146/annurev.neuro.31.060407.125631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis SH, Busch JL, Corbin JD, Sibley D. cGMP-Dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol Rev. 2010;62:525–563. doi: 10.1124/pr.110.002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gammie SC, Nelson RJ. cFOS and pCREB activation and maternal aggression in mice. Brain Res. 2001;898:232–241. doi: 10.1016/s0006-8993(01)02189-8. [DOI] [PubMed] [Google Scholar]
- Garthwaite J. Concepts of neural nitric oxide-mediated transmission. Eur J Neurosci. 2008;27:2783–2802. doi: 10.1111/j.1460-9568.2008.06285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gille H, Kortenjann M, Thomae O, Moomaw C, Slaughter C, Cobb MH, Shaw PE. ERK phosphorylation potentiates Elk-1-mediated ternary complex formation and transactivation. EMBO J. 1995;14:951–962. doi: 10.1002/j.1460-2075.1995.tb07076.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girouard H, Wang G, Gallo EF, Anrather J, Zhou P, Pickel VM, Iadecola C. NMDA receptor activation increases free radical production through nitric oxide and NOX2. J Neurosci. 2009;29:2545–2552. doi: 10.1523/JNEUROSCI.0133-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glazewski S, Fox K. Time course of experience-dependent synaptic potentiation and depression in barrel cortex of adolescent rats. J Neurophysiol. 1996;75:1714–1729. doi: 10.1152/jn.1996.75.4.1714. [DOI] [PubMed] [Google Scholar]
- Glazewski S, Chen CM, Silva A, Fox K. Requirement for alpha-CaMKII in experience-dependent plasticity of the barrel cortex. Science. 1996;272:421–423. doi: 10.1126/science.272.5260.421. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Hofmann F, Feil R, Kleppisch T, Schlossmann J. Function of cGMP-dependent protein kinases as revealed by gene deletion. Physiol Rev. 2006;86:1–23. doi: 10.1152/physrev.00015.2005. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Hunter RG, Jones D, Vicentic A, Hue G, Rye D, Kuhar MJ. Regulation of CART mRNA in the rat nucleus accumbens via D3 dopamine receptors. Neuropharmacology. 2006;50:858–864. doi: 10.1016/j.neuropharm.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Ivanov A, Pellegrino C, Rama S, Dumalska I, Salyha Y, Ben-Ari Y, Medina I. Opposing role of synaptic and extrasynaptic NMDA receptors in regulation of the extracellular signal-regulated kinases (ERK) activity in cultured rat hippocampal neurons. J Physiol. 2006;572:789–798. doi: 10.1113/jphysiol.2006.105510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano T, Anrather J, Zhou P, Park L, Wang G, Frys KA, Kunz A, Cho S, Orio M, Iadecola C. Prostaglandin E2 EP1 receptors: downstream effectors of COX-2 neurotoxicity. Nat Med. 2006;12:225–229. doi: 10.1038/nm1362. [DOI] [PubMed] [Google Scholar]
- Kleppisch T, Feil R. cGMP Signalling in the mammalian brain: role in synaptic plasticity and behaviour. In: Schmidt HHHW, Hofmann F, Stasch JP, editors. cGMP: generators, effectors and therapeutic implications. Springer: Berlin; 2009. pp. 549–579. [DOI] [PubMed] [Google Scholar]
- Liaudet L, Vassalli G, Pacher P. Role of peroxynitrite in the redox regulation of cell signal transduction pathways. Front Biosci. 2009;14:4809–4814. doi: 10.2741/3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loebrich S, Nedivi E. The function of activity-regulated genes in the nervous system. Physiol Rev. 2009;89:1079–1103. doi: 10.1152/physrev.00013.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002;35:605–623. doi: 10.1016/s0896-6273(02)00828-0. [DOI] [PubMed] [Google Scholar]
- Lu YF, Kandel ER, Hawkins RD. Nitric oxide signaling contributes to late-phase LTP and CREB phosphorylation in the hippocampus. J Neurosci. 1999;19:10250–10261. doi: 10.1523/JNEUROSCI.19-23-10250.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matys T, Pawlak R, Matys E, Pavlides C, McEwen BS, Strickland S. Tissue plasminogen activator promotes the effects of corticotropin-releasing factor on the amygdala and anxiety-like behavior. Proc Natl Acad Sci U S A. 2004;101:16345–16350. doi: 10.1073/pnas.0407355101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nott A, Riccio A. Nitric oxide-mediated epigenetic mechanisms in developing neurons. Cell Cycle. 2009;8:725–730. doi: 10.4161/cc.8.5.7805. [DOI] [PubMed] [Google Scholar]
- Nott A, Watson PM, Robinson JD, Crepaldi L, Riccio A. S-nitrosylation of histone deacetylase 2 induces chromatin remodelling in neurons. Nature. 2008;455:411–415. doi: 10.1038/nature07238. [DOI] [PubMed] [Google Scholar]
- O'Dell TJ, Hawkins RD, Kandel ER, Arancio O. Tests of the roles of two diffusible substances in long-term potentiation: evidence for nitric oxide as a possible early retrograde messenger. Proc Natl Acad Sci U S A. 1991;88:11285–11289. doi: 10.1073/pnas.88.24.11285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park L, Gallo EF, Anrather J, Wang G, Norris EH, Paul J, Strickland S, Iadecola C. Key role of tissue plasminogen activator in neurovascular coupling. Proc Natl Acad Sci U S A. 2008;105:1073–1078. doi: 10.1073/pnas.0708823105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picconi B, Bagetta V, Ghiglieri V, Paillè V, Di Filippo M, Pendolino V, Tozzi A, Giampà C, Fusco FR, Sgobio C, Calabresi P. Inhibition of phosphodiesterases rescues striatal long-term depression and reduces levodopa-induced dyskinesia. Brain. 2011;134:375–387. doi: 10.1093/brain/awq342. [DOI] [PubMed] [Google Scholar]
- Puzzo D, Staniszewski A, Deng SX, Privitera L, Leznik E, Liu S, Zhang H, Feng Y, Palmeri A, Landry DW, Arancio O. Phosphodiesterase 5 inhibition improves synaptic function, memory, and amyloid-β load in an Alzheimer's disease mouse model. J Neurosci. 2009;29:8075–8086. doi: 10.1523/JNEUROSCI.0864-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccio A, Alvania RS, Lonze BE, Ramanan N, Kim T, Huang Y, Dawson TM, Snyder SH, Ginty DD. A nitric oxide signaling pathway controls CREB-mediated gene expression in neurons. Mol Cell. 2006;21:283–294. doi: 10.1016/j.molcel.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Rocamora N, Welker E, Pascual M, Soriano E. Upregulation of BDNF mRNA expression in the barrel cortex of adult mice after sensory stimulation. J Neurosci. 1996;16:4411–4419. doi: 10.1523/JNEUROSCI.16-14-04411.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblum K, Futter M, Voss K, Erent M, Skehel PA, French P, Obosi L, Jones MW, Bliss TV. The role of extracellular regulated kinases I/II in late-phase long-term potentiation. J Neurosci. 2002;22:5432–5441. doi: 10.1523/JNEUROSCI.22-13-05432.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russwurm M, Wittau N, Koesling D. Guanylyl cyclase/PSD-95 interaction: targeting of the nitric oxide-sensitive alpha2beta1 guanylyl cyclase to synaptic membranes. J Biol Chem. 2001;276:44647–44652. doi: 10.1074/jbc.M105587200. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Takemori H, Yagita Y, Terasaki Y, Uebi T, Horike N, Takagi H, Susumu T, Teraoka H, Kusano K, Hatano O, Oyama N, Sugiyama Y, Sakoda S, Kitagawa K. SIK2 is a key regulator for neuronal survival after ischemia via TORC1-CREB. Neuron. 2011;69:106–119. doi: 10.1016/j.neuron.2010.12.004. [DOI] [PubMed] [Google Scholar]
- Schuman EM, Madison DV. A requirement for the intercellular messenger nitric oxide in long-term potentiation. Science. 1991;254:1503–1506. doi: 10.1126/science.1720572. [DOI] [PubMed] [Google Scholar]
- Thiels E, Kanterewicz BI, Norman ED, Trzaskos JM, Klann E. Long-term depression in the adult hippocampus in vivo involves activation of extracellular signal-regulated kinase and phosphorylation of Elk-1. J Neurosci. 2002;22:2054–2062. doi: 10.1523/JNEUROSCI.22-06-02054.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL. MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci. 2004;5:173–183. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- Waterhouse EG, Xu B. New insights into the role of brain-derived neurotrophic factor in synaptic plasticity. Mol Cell Neurosci. 2009;42:81–89. doi: 10.1016/j.mcn.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GY, Deisseroth K, Tsien RW. Activity-dependent CREB phosphorylation: convergence of a fast, sensitive calmodulin kinase pathway and a slow, less sensitive mitogen-activated protein kinase pathway. Proc Natl Acad Sci U S A. 2001a;98:2808–2813. doi: 10.1073/pnas.051634198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GY, Deisseroth K, Tsien RW. Spaced stimuli stabilize MAPK pathway activation and its effects on dendritic morphology. Nat Neurosci. 2001b;4:151–158. doi: 10.1038/83976. [DOI] [PubMed] [Google Scholar]
- Yun HY, Gonzalez-Zulueta M, Dawson VL, Dawson TM. Nitric oxide mediates N-methyl-d-aspartate receptor-induced activation of p21ras. Proc Natl Acad Sci U S A. 1998;95:5773–5778. doi: 10.1073/pnas.95.10.5773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, Qian L, Iadecola C. Nitric oxide inhibits caspase activation and apoptotic morphology but does not rescue neuronal death. J Cereb Blood Flow Metab. 2005;25:348–357. doi: 10.1038/sj.jcbfm.9600036. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Wu H, Li S, Chen Q, Cheng XW, Zheng J, Takemori H, Xiong ZQ. Requirement of TORC1 for late-phase long-term potentiation in the hippocampus. PLoS One. 2006;1:e16. doi: 10.1371/journal.pone.0000016. [DOI] [PMC free article] [PubMed] [Google Scholar]