

Abstract
Both oxidative stress and aggregation of the protein α-synuclein (aS) have been implicated as key factors in the etiology of Parkinson’s disease. Specifically, oxidative modifications to aS disrupt its binding to lipid membranes, an interaction considered critical to its native function. Here we seek to provide a mechanistic explanation for this phenomenon by investigating the effects of oxidative nitration of tyrosine residues on the structure of aS and its interaction with lipid membranes. Membrane binding is mediated by the first ~95 residues of aS. We find that nitration of the single tyrosine (Y39) in this domain disrupts binding due to electrostatic repulsion. Moreover, we observe that nitration of the three tyrosines (Y125/133/136) in the C-terminal domain is equally effective in perturbing binding, an intriguing result given that the C-terminus is not thought to interact directly with membranes. Our investigations show that tyrosine nitration results in a change of the conformational states populated by aS in solution, with the most prominent changes occurring in the C-terminal region. These results lead us to suggest that nitration of Y125/133/136 reduces the membrane binding affinity of aS through allosteric coupling by altering the ensemble of conformational states and depopulating those capable of membrane binding. While allostery is a well-established concept for structured proteins, it has only recently been discussed in the context of disordered proteins. We propose that allosteric regulation through modification of specific residues in, or ligand binding to, the C-terminus may even be a general mechanism for modulating aS function.
Introduction
There is growing evidence to support a central role for the protein α-Synuclein (aS) in the pathogenesis of Parkinson’s disease (PD). It is the primary component of Lewy bodies, cytoplasmic amyloid inclusions, which are associated with selective loss of dopaminergic neurons in the substantia nigra and are a hallmark of the disease. Although its precise role in the development of PD remains unclear, point mutations to aS (A30P, E46K, and A53T) and multiplication of the aS gene have both been linked to familial forms of PD1–4. The native functions of aS are also not well-understood but have been proposed to involve synaptic vesicle trafficking5,6, regulation of the synaptic vesicle pool7, and maintenance of neuronal plasticity8. These functions are likely to be mediated by associations between aS and cellular membranes, and its ability to bind lipid membranes is well-documented9–11. A potential link between membrane association and disease has been established by the finding A30P and E46K show altered membrane binding properties12,13. Further, posttranslational modifications to aS that are thought to be related to PD, such as serine phosphorylation14,15 and tyrosine nitration16, disrupt aS-membrane interactions.
While oxidatively modified proteins accumulate to some extent during normal aging, there is mounting evidence that oxidative injury to aS, specifically nitration of tyrosine residues, contributes directly to the pathology of PD17–22. aS contains four tyrosines, one in the N-terminal, membrane binding region at residue 39, and three near the C-terminus at residues 125, 133 and 136 (Figure 1), all of which are readily nitrated in the presence of oxidizing agents such as peroxinitrite, a common mediator of oxidative stress in vivo23. Nitrated aS (nit-aS) was detected in Lewy bodies from post-mortem brain tissue using an antibody against 3-nitrotyrosine17. Furthermore, nit-aS has been shown to be toxic to dopaminergic neurons in vitro and in vivo22, and in an oxidative cellular model of PD, an increase of 3-nitrotyrosine was observed19. In vitro, nitration of tyrosines can inhibit fibrillation and lead to the accumulation of stable oligomers, most likely by covalent cross-linking due to formation of di-tyrosine species23.
Figure 1.
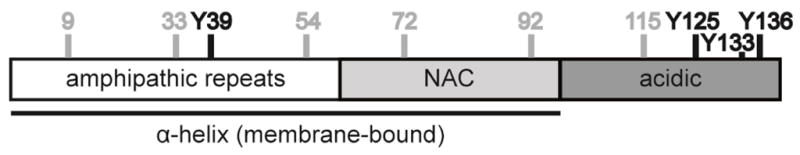
Schematic representation of aS showing modification sites. aS is divided into three regions: a positively charged N-terminal region, a hydrophobic central region (NAC region) forming the core of the β-sheet structures in amyloid fibrils, and an acidic C-terminus. The N-terminus and the NAC region form an amphipathic α-helix upon binding to lipid membranes. aS has four tyrosines which are indicated in black; the different modification sites for fluorescent labeling are indicated in gray.
In the present study we investigate the mechanism by which tyrosine nitration modulates binding of aS to model lipid membranes. We find that nitration of both the N-terminal and C-terminal tyrosines reduce the binding affinity of aS for negatively charged lipid vesicles. For the N-terminal tyrosine, we show that this decrease is primarily due to an increase in electrostatic repulsion between aS and the vesicle. In contrast, the reduction in binding affinity caused by nitration of the tyrosines in the C-terminus, which is not directly involved in membrane interactions, cannot be attributed to either electrostatic or hydrophobic effects. Rather, we find that nitration of the C-terminal tyrosines results in a compaction of the C-terminus in solution and when bound to vesicles. Our results lead us to propose a model whereby modifications to the C-terminus regulate binding through allosteric effects, which we suggest may be a more general mechanism for modulating interactions between aS and cellular membranes.
Materials and Methods
Protein preparation and nitration
aS was expressed and purified from a T7-7 plasmid in E. coli BL21 cells as described previously24,25 and lyophilized for storage at −20°C. To prevent the possible misincorporation of a cysteine instead of a tyrosine at position 13626, we introduced a conservative mutation (TAC to TAT). For nitration studies, tyrosine to phenylalanine (Y39F and Y125/133/136F) and tyrosine to aspartic acid (Y39D, Y125/133/136D and Y39/125/133/136D) mutants were created. For fluorescent labeling of aS, cysteine mutations were introduced singly and in pairs at positions 9, 33, 54, 72, 92 and 115 and reacted with Alexa Fluor maleimide dyes (Invitrogen, Carlsbad, CA) essentially as described previously27. To distinguish labeling mutants from the phenylalanine and aspartic acid mutants, all cysteine mutants are referred to as ‘umodified’. Nitration of aS was achieved by adding a 10x molar excess of tetranitromethane (10% in ethanol) per tyrosine residue to 2 mg/mL fluorescently labeled aS. Labeling was done prior to nitration to avoid possible modification of free cysteine residues. Details can be found in the Supporting Information.
Fluorescence Correlation Spectroscopy (FCS) and Förster Resonance Energy Transfer (FRET)
FCS and FRET measurements were made on a lab-built instrument based on an inverted Olympus IX-71 microscope (Olympus, Tokyo, Japan) that has been described previously27.
Measurements were made in 8-well chambered coverglasses (Nunc, Rochester, NY) passivated by polylysine conjugated polyethylene glycol treatment to prevent aS adsorption to the chamber surfaces. For FCS, the lipid concentration was varied against a fixed concentration of aS. All measurements were carried out at 20°C in 20 mM Tris buffer, 130 mM NaCl at a protein concentration of 100 nM. Analysis of FCS data and calculation of the molar partition coefficients KP are described in the Supporting Information.
For FRET measurements, the aS concentration was ~90 pM in 20 mM Tris buffer, 130 mM NaCl, with 500 μM lipids for measurements of vesicle-bound conformations. Details of data analysis are in the Supporting Information.
Pulsed field gradient NMR
Hydrodynamic radii of unmodified aS, nit-Y39F, and Y125/133/135D were determined in the absence and presence of 8M urea using pulsed field gradient (PFG) NMR employing the PG-SLED sequence 28. 1H NMR spectra were collected on Varian Unity Inova 600 MHz and 500 MHz Spectrometers (Lexington, MA) in 99.9% D2O, 25 mM phosphate buffer, pH 7.4 at 25°C. The protein concentration was approximately 200 μM and 1mM TMSP was added to all samples as an internal radius standard. See Supporting Information for details on analysis.
Results
Nitration of N-terminal and C-terminal tyrosines reduces aS membrane binding equally
Nitration of aS with tetranitromethane yields a heterogeneous mixture of aS species consisting of nitrated monomers, dimers and higher oligomers formed through di-tyrosine cross-linking16. Monomeric aS was separated from oligomers and dimers by size exclusion chromatography (Figures S1 and S2) and the extent of nitration of monomer aS was determined by ESI mass spectrometry as ~15% mono-, 35% di-, 35% tri- and 15% tetra-nitrated (Table S2). We used fluorescence correlation spectroscopy (FCS) to measure the binding affinities of unmodified and nit-aS for anionic lipid vesicles. aS was labeled with Alexa Fluor 488 (AL488) and autocorrelation curves were measured as a function of lipid concentration (Figure 2A)24. At each lipid concentration, fitting the autocorrelation curves yielded the fraction of protein bound to the vesicles which was used to generate a binding curve (Figure 2B; Supporting Information for details). To quantify the strength of the binding interaction and allow for comparison between the nitrated and unmodified protein, we calculated partition coefficients, KP (Figure 2C and Supporting Information for details). We measure a decrease of ~50% in the binding affinity for nit-aS in good agreement with results obtained previously from gel filtration chromatography experiments16. It is of note that the difference in KP between unmodified and nit-aS is equivalent to ΔG≈0.5 kcal/mol (Supporting Information for details of Kp calculation). This difference is comparable to the amount of reduction of or enhancement in affinity seen for the PD-associated mutants of aS, A30P and E46K, respectively, relative to wild-type aS25. While it is not yet clear if changes the interactions between aS and cellular membranes has a role in PD, studies have shown that the A30P mutation results in altered cellular distribution of aS away from synaptic terminals29, suggesting that perturbations to membrane interactions of this order of magnitude may be meaningful in a physiological context.
Figure 2.

Binding of aS to lipid vesicles. A. Normalized autocorrelation curves of aS with increasing lipid concentrations showing characteristic shift to longer decay times with increasing fraction of bound protein. B. Representative hyperbolic binding curves of aS (black) and nit-aS (gray) binding to 50 nm diameter 1:1 POPC/POPS vesicles at pH 7.4. Partition coefficients of the different aS constructs: (C) Y→F, unmodified (solid) and nitrated (diagonal hatch marks); and (D) Y→D nitration mimics.
Phenylalanine is not subject to nitration, but otherwise resembles tyrosine in bulk. Thus, to determine the the origin of the decreased membrane binding affinity, we made two tyrosine to phenylalanine mutants: Y39F, where only the C-terminal tyrosines are targets for nitration and Y125/133/136F, where all the C-terminal tyrosines are replaced with phenylalanine, leaving only Y39 in the membrane binding region to be nitrated (Figure 1). While both unmodified mutants have partition coefficients comparable to unmodified wild-type aS, the binding affinity was reduced by ~50% for nit-Y125/133/136F and, interestingly, also for nit-Y39F (Figure 2C). These data indicate that selective nitration of either the N-terminal or C-terminal tyrosines influences the binding to lipid vesicles. This is particularly striking for the latter case, given that the C-terminus is generally not thought to interact directly with the lipid bilayer. Notably, the perturbation of binding we find upon nitration at the two different sites is not additive, suggesting some degree of cooperativity between the two domains.
Decreased membrane binding of N-terminally nitrated aS is mediated by electrostatics
Nitration decreases the pKa of a tyrosine residue from 10 to ~730. Therefore, at pH 7.4 nitrotyrosine is partially negatively charged, providing a possible explanation for the observed decrease in binding due to electrostatic repulsion from the negatively charged vesicles. To investigate this possibility, we created tyrosine to aspartic acid mutants (Figure 1) to mimic the negative charge aspect of tyrosine nitration, a strategy that is commonly used to mimic serine or tyrosine phosphorylation. A mutant with all tyrosines replaced by aspartic acid (Y39/125/133/136D) shows markedly reduced binding affinity - by more than an order of magnitude - relative to wild-type aS (Figure 2D). For Y39D, binding was reduced to a similar degree (Figure 2D) indicating that the increase in net negative charge in the N-terminal region of aS through nitration of Y39 reduces its affinity for anionic lipid vesicles. To further probe this effect, we measured the binding affinity of Y125/133/136F at pH 5.0, where neither tyrosine nor nitrotyrosine are charged. At pH 5.0, the partition coefficients of the unmodified and nitrated construct are similar (Figure S3). In combination, these two results provide evidence that the decreased membrane binding upon nitration is primarily due to an increase in electrostatic repulsion between the protein and the anionic lipid bilayer.
Despite the fact that it carries three additional negative charges in its C-terminus, Y125/133/136D binds vesicles with an affinity comparable to unmodified wild-type aS (Figure 2D). Moreover, at pH 5.0, where the decrease in binding affinity upon nitration is virtually eliminated for Y125/133/136F, Y39F demonstrates the same significant decrease in binding affinity upon nitration that wild-type nit-aS does (Figure S3). These results suggest that the charge of the C-terminus does not play a major role in modulating binding.
Nitration of C-terminal tyrosines alters the conformation of the C-terminus
To examine the source of the reduced binding affinity due to nitration of the C-terminus, we used single molecule Förster resonance energy transfer (smFRET) to probe the conformation of aS upon nitration. aS was labeled with a donor, AL488, and an acceptor, Alexa Fluor 594 (AL594), fluorophore at three different sets of sites: two within the membrane binding region at residues 9 and 54 (aS9–54) and at residues 54 and 92 (aS54–92), and one closer to the C-terminus at residues 72 and 115 (aS72–115). Labeling nit-aS closer to the C-terminus, for example at residue 130, resulted in artifacts in the ETeff histograms, likely due to hydrophobic interactions between the nitrotyrosines and the fluorophore. The ETeff histograms of unmodified and nitrated aS9–54 are virtually superimposable (Figures 3A and 3B), whereas nitration results in a small increase in the peak ETeff value for aS54–92 (Figures 3E and 3F) and a larger increase for aS72–115 (Figures 3I and 3J). These measurements indicate that the C-terminus and the NAC regions of nit-aS are more compact than in the unmodified protein, while the N-terminus is relatively unaffected. There is precedence for similar local rearrangements in aS structure; at low pH, the C-terminus of the protein becomes considerably more compact, while the N-terminal region is virtually unaltered31.
Figure 3.

Effect of nitration on conformation of aS in solution and bound to lipid vesicles. ETeff histograms of unmodified (rows 1 and 3, red fits) or nitrated (rows 2 and 4, blue fits) aS were measured in solution (upper two rows, dark gray) and in the presence of 100% POPS vesicles (lower two rows, light gray). In order to probes specific regions of the protein, aS was labeled at various positions: A–D, residues 9 and 54; E–H, residues 54 and 92; I–L, residues 72 and 115; M&N, residues 33 and 115. The peak at ETeff≈1 seen in some panels (for example, K and L) is the result of a trace contaminant present in some of the vesicle solutions. All histograms are normalized so that the area under the Gaussian fit curve (red or blue) equals 1.
The effect of nitration on the conformation of the C-terminus is even more apparent in comparing the vesicle-bound conformations of the proteins. Upon binding to anionic vesicles, the ETeff of unmodified aS72–115 shows a large decrease in the peak position relative to the unbound protein (Figures 3I and 3K). This increase in distance is consistent with a coil-helix conversion of residues 72–95 upon binding, which is expected to increase the mean distance between the fluorophore pair. In contrast, the peak ETeff value for vesicle-bound nit-aS72–115 is surprisingly high (ETeff≈0.72; Figure 3L) and is very similar to nit-aS72–115 in solution (ETeff≈0.74; Figure 3J). The conformational changes observed for nit-aS72–115 are not due to a change in the electrostatic properties of the C-terminus, as smFRET measurements of the Y125/133/136D construct (which mimics the charge effect of nitration) show more extended conformational ensembles both in buffer and on vesicles, similar to those of the unmodified wild-type protein (Figure S5).
We considered several potential explanations for this observation. The first possibility is that the protein was not bound to the vesicles. This, however, is highly unlikely; based on the partition coefficients measured and the concentration of lipids used we expect >90–95% of the protein to be bound. The second possibility is that the conformation of the α-helical region of aS (residues 1–95) is different for the unmodified and nitrated protein. However, measurements of the aS9–54 and aS54–92 constructs show large peak ETeff shifts upon vesicle binding for both the unmodified and nitrated proteins, but only minor differences between the membrane bound nitrated and unmodified proteins (compare: Figures 3A and 3C with Figures 3B and 3D; Figures 3E and 3G with Figures 3F and 3H). These results indicate that the structure of the membrane binding region of the protein is largely unaffected by nitration and that the major conformation of the nitrated protein is the same extended α-helix as the unmodified protein, which we have observed previously27. Thus, the most likely explanation for the high peak ETeff value (ETeff≈0.72) measured for nit-aS72–115 is a further compaction of the C-terminus (residues 96–140) upon membrane binding that compensates for the increase in distance between residues 72 and 95 due to α-helix formation.
It is also possible that nitration results in an altered conformation of some fraction of the membrane bound protein. In Figure 3H, there are clearly two populations of nit-aS54–92 molecules. The major population with mean ETeff≈0.34 matches well to the peak measured for membrane bound unmodified aS54–92 (Figure 3G) and is consistent with an extended α-helical conformation aS. Although our measurement conditions favor almost fully bound protein, the minor population with mean ETeff≈0.75 could be attributed to a population of unbound protein. However, there is a small shift in its peak value compared to the solution measurement (ETeff≈0.72; Figure 3F), which means it could also represent a bound population of protein with an alternative conformation. Recent work indicates that the membrane binding region of aS may consist of two α-helices with differential affinities for the membrane32,33: the first spanning residues ~1–25 and the second spanning residues ~30–95. Nitration could preferentially destabilize the binding of this second region, resulting in a membrane bound structure with an ETeff corresponding to that of the smaller peak (ETeff≈0.75). Similarly, the broad peak centered at ETeff≈0.72 for membrane bound nit-aS72–115 could be overlapping histograms from two different membrane bound conformations with similar ETeff that cannot be distinguished by our approach. This scenario is particularly plausible given recent work which demonstrates that under some conditions, aS populates both extended and ‘bent’ δ-helical conformations on lipid vesicles34. Regardless, independent of the detailed description of the bound states, the significance of our data is that they demonstrate that modifications to the C-terminus which affect its structure are also capable of modulating functional interactions (in this case, binding a lipid vesicle) of the N-terminus. This is particularly striking given that the C-terminus lacks any significant stable structure, even upon binding to vesicles35–37.
Nitration of C-terminal tyrosines results in an increase in hydrodynamic radius of aS
The smFRET experiments described above were designed to probe local structure and conformational changes. Using a fourth construct, spanning residues 33 and 115 (aS33–115), we found a small decrease in the peak ETeff value upon nitration (Figure 3M and Figure 3N), which indicates that nitration results in a structure that is globally ~10% more extended in solution than the unmodified protein (see Supporting Information for details of calculation). In order to better characterize and to determine the cause of this expansion, the diffusion coefficients of unmodified aS, nit-Y39F, and Y125/133/136D were determined using PFG-NMR (Table 1, Figure S6) and used to calculate the hydrodynamic radii (RH) of the proteins. For unmodified aS we determined RH=27.4±0.4 Å, in good agreement with published results38. Nitration of the C-terminal tyrosines (nit-Y39F) resulted in an increase, RH=29.0±0.3 Å, consistent with the expansion suggested by smFRET data of aS33–115. The NMR data is supported by FCS data which measures a slower diffusion time for nit-aS and nit-Y39F but not for nit-Y125/133/136F, as compared to their unmodified counterparts (Table S3). Furthermore, measurements on Y125/133/136D show that this construct resembles the wild-type protein, indicating that the difference in RH seen in the nitrated protein is not due to electrostatic effects (Table 1). In the presence of 8M urea, aS expands to RH=34.7±0.3 Å whereas nit-Y39F expands only to 32.9±0.6 Å. The Y125/133/126D mutant expands to a similar extent as the nitrated protein, indicating that this more compact conformation with high denaturant concentrations, in contrast to the expansion seen in buffer, is most likely mediated by charge effects. These data show that nitration results in substantial, specific changes in the conformation of the protein that are not associated with canonical secondary or tertiary structure (Figure S4).
Table 1.
Hydrodynamic radii (Å) of aS, nit-Y39F and Y125/133/136D at 25°C with and without 8M urea determined by PFG-NMR (Fits to the raw data in Figure S6).
| Tris buffer | + 8M urea | |
|---|---|---|
| aS | 27.4±0.4 | 34.7±0.3 |
| nit-Y39F | 29.0±0.3 | 32.9±0.6 |
| Y125/133/136D | 27.8±0.2 | 32.6±0.1 |
Discussion
Increasing evidence suggests that oxidative stress plays an important role in the pathogenesis of PD and that aS may be a specific target for oxidative modification. A number of mechanisms for the role of nitrated aS in PD have been proposed, including an increase of aS toxicity due to the enhanced seeding ability of nit-aS, less efficient clearance by the proteasome, and an increase in the cytosolic concentration due to its decreased affinity for membranes16. Because of the central importance of binding to cellular membranes to aS function, in this work we have focused on understanding the mechanism by which membrane binding is perturbed upon nitration of tyrosine residues.
Previous work found that the decreased membrane affinity of nitrated aS was mediated by Y39, the sole tyrosine located in the membrane binding region (Figure 1), and was proposed to be caused by electrostatic and/or steric effects16. Our results show that mimicking the charge properties of nitration by replacing Y39 with aspartic acid (Y39D) had the same effect as nitration of Y39, allowing us to identify electrostatic effects as the primary cause of reduced affinity. However, the additional small reduction in affinity observed for nit-Y125/133/136F at pH 5.0 relative to unmodified Y125/133/136F (Figure S3) suggests that other properties of the nitro-group at Y39 may also have a role. While a helical wheel model of membrane bound aS derived from EPR39, places Y39 on the solvent exposed face of the helix, where effects of bulkiness of the nitrotyrosine group on interactions with the lipid membrane should be minimized, other studies point towards a buried Y39. An intrinsic fluorescence study finds that binding of aS to SDS micelles results in a more hydrophobic environment for Y3940, and that access of the enzyme tyrosinase, which may have a direct role in oxidative reactions relevant to PD, to Y39 is restricted when aS is bound to lipid vesicles41. Both of these studies indicate that Y39 may be at least partially buried in the lipid bilayer where the increased size and decreased hydrophobicity of the nitrated phenol group are expected to be unfavorable.
More intriguing is our finding that nitration of the C-terminal tyrosines, which are located ~30–40 residues away from the end of the membrane binding region (~residue 95) (Figure 1), results in a comparable decrease in binding affinity as the N-terminal nitration (Figure 2C). In one recent NMR study, the C-terminus was proposed to interact directly with lipid membranes at lipid/aS molar ratios ≥1532. Although we cannot exclude this scenario, we would expect that an increase in the net negative charge of the C-terminus (by nitration or aspartic acids) would result in a decrease in binding affinity for anionic lipid membranes. Our experiments clearly show that this decrease cannot be assigned to electrostatic effects (Figure 2D and Figure S3).
These results raise the question – how does nitrative modification of the C-terminal tyrosines decrease membrane binding of aS? For this discussion, we will consider that aS consists of two domains: membrane binding (residues 1–95) and C-terminal (residues 96–140). Our data show that nitration of the C-terminal tyrosines alters the conformational ensemble of the disordered C-terminal domain; moreover, nitration of these residues results in a decrease in binding affinity mediated by the membrane binding domain of the protein. This leads us to suggest that the mechanism of perturbation is through long-range allosteric communication between the two domains of aS.
An allosteric system is one in which the binding of a ligand (or covalent modification) to one site alters structure, function and/or flexibility at a distal site. It requires communication between the two sites and, historically, has been thought to be mediated by a change in structure of the protein in response to the ligand. More recently, allosteric regulation has been proposed to occur simply via shifting the population of protein conformational states in the dynamic ensemble42–46. Although the model of dynamic allostery was proposed for structured proteins it could just as well describe the alteration of an ensemble of disordered structures. In fact, recent work suggests that allosteric coupling may actually be most efficient in proteins where disorder is present in the domains containing the binding/modification sites, as is the case for nit-aS, and that it is mediated by changes in the relative populations of states, independent of physically linked interactions47. In this theoretical work, the authors propose a model for allosteric coupling in disordered proteins whereby binding to or modification of domain A stabilizes conformational states of A favored by the binding/modification. The population of states that bind to ligand B may be stabilized, destabilized, or unaffected. A direct analogy drawn from our experimental work is that nitration of the C-terminus (modification of domain A) shifts the dynamic ensemble of C-terminal states towards more compact conformations and destabilizes or reduces the population of states in the membrane-binding domain (domain B) that favor binding to the vesicles.
We can consider this model in more detail and provide several potential mechanistic explanations of how the population of states in the membrane binding domain are affected by modification of the C-terminal domain in the context of what is known about the solution structure of aS. Introduction of a nitro-group increases the bulkiness of tyrosine (the phenolic ring has a surface area of ~30 32 and the nitrated ring ~50 32)48 which may affect its flexibility and rotational mobility49. Thus, it is possible that dynamics of the protein chain relevant to membrane binding are altered by nitration. Moreover, PFG-NMR and smFRET provide evidence for both global (Table 1) and local (Figure 3) conformational changes in nit-aS, which suggests that nitration may modify or disrupt long-range interactions within the protein. The importance of dynamics and long-range contacts has long been recognized as important to aS aggregation50–53 and it is likely that they will play a role in functional interactions of aS, such as membrane-binding, as well. Indeed, there are several examples of aS modifications where there appears to be a correlation between modulation of long-range interactions and membrane binding affinity. To illustrate, the PD mutant E46K shows increased C- to N-terminal contacts54 as well as increased membrane binding13, while A30P has reduced contacts and a reduced membrane binding affinity13,55. Phosphorylation of S87 was also shown to reduce long-range interactions with a concomitant expansion of the protein as well as to perturb membrane binding14. Interestingly, this effect could not be reproduced by mimicking phosphorylation by replacing serine with a glutamic acid. Our current study, along with these others, provides compelling evidence that long-range interactions and dynamics may be generally relevant for modulating binding of aS to membranes.
Lastly, the N-terminal region of aS contains nascent or transiently populated α-helical structure35 which could also be affected by C-terminal nitration. This nascent α-helical structure was perturbed by the disease mutant A30P, but not A53T, correlating with their ability to bind lipid membranes56. Although we do not observe a stable conformational change in the N-terminal region (Figure 3A and 3B), it is plausible that there is a change in the conformational ensemble of this region – either the specific residues that sample α-helical conformations or the dynamics of conformational interchange – that smFRET does not detect. More directly, there could be a shift in the distribution of conformational states populated by the N-terminus when bound to the vesicle, such that lower affinity conformations are favored.
Overall, our study provides evidence for a novel mechanism for regulation of aS binding to lipid membranes through allosteric communication between the disordered C-terminus and the membrane binding domain. The important role we suggest for the C-terminus is supported by observations that it can interact with a variety of ligands including copper, iron57, and calcium ions58, polyamines59, and with various proteins, notably microtubule-associated proteins60,61 and synaptobrevin-262. Based on our results, we propose that allosteric regulation by the modification of specific residues or binding of ligands to the C-terminus may be a general mechanism for rapid, reversible control of membrane binding by aS. In this study, the effect observed upon nitrative modification is inhibitory, but alternate ligands/modifications could act as activators. The association of aS with cellular membranes, and changes in those interactions in the mutant or modified forms, is emerging as of central importance to the normal function of aS, as well as to its pathogenic role in PD63. Thus, the mechanism of modulation of membrane binding through allostery that we describe here is likely to have general implications for understanding the underlying molecular mechanisms of aS function as well as dysfunction.
Supplementary Material
Figure 4.

Cartoon illustrating changes in aS conformation upon nitration and the effects upon membrane binding. The four constructs described in the Results section are marked with gray arrows; upon nitration, the arrows are color coded to indicate a measured change in distance: gray = no change; blue = increase; red= decrease.
Acknowledgments
We thank Eric Paulson and Sean Whittier for technical assistance with the PFG-NMR experiments and Abhinav Nath for help with acquiring the mass spectrometry data and critical reading of the manuscript. This work was supported by a postdoctoral fellowship from the Max Kade Foundation (to E.S.), institutional training grant NIH GM007223 (to A.J.T. and J.M.D.), and funding from the Ellison Medical Foundation (to E.R).
Footnotes
Supporting Information Available: Experimental details of nitration and vesicle preparation. Details of smFRET and FCS measurements and analysis. Size exclusion chromatogram and SDS-PAGE of nit-aS species. Partition coefficients of nitrated aS at pH 5.0. CD spectra of nit-aS. Anisotropy and lifetime of singly labeled constructs. Mass spectrometry data of nit-aS species. SmFRET measurements of Y125/133/136D construct. Fits to PFG-NMR data. Diffusion times of nit-aS constructs determined by FCS. Complete References 3, 4, 5, and 14. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Zarranz JJ, Alegre J, Gomez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atares B, Llorens V, Gomez Tortosa E, del Ser T, Munoz DG, de Yebenes JG. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- 2.Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 3.Polymeropoulos MH, et al. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 4.Singleton AB, et al. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 5.Cooper AA, et al. Science. 2006;313:324–328. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Outeiro TF, Lindquist S. Science. 2003;302:1772–1775. doi: 10.1126/science.1090439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murphy DD, Rueter SM, Trojanowski JQ, Lee VM. J Neurosci. 2000;20:3214–3220. doi: 10.1523/JNEUROSCI.20-09-03214.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kahle PJ, Neumann M, Ozmen L, Haass C. Ann N Y Acad Sci. 2000;920:33–41. doi: 10.1111/j.1749-6632.2000.tb06902.x. [DOI] [PubMed] [Google Scholar]
- 9.Davidson WS, Jonas A, Clayton DF, George JM. J Biol Chem. 1998;273:9443–9449. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- 10.Perrin RJ, Woods WS, Clayton DF, George JM. J Biol Chem. 2000;275:34393–34398. doi: 10.1074/jbc.M004851200. [DOI] [PubMed] [Google Scholar]
- 11.Fortin DL, Troyer MD, Nakamura K, Kubo S, Anthony MD, Edwards RH. J Neurosci. 2004;24:6715–6723. doi: 10.1523/JNEUROSCI.1594-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jensen PH, Nielsen MS, Jakes R, Dotti CG, Goedert M. J Biol Chem. 1998;273:26292–26294. doi: 10.1074/jbc.273.41.26292. [DOI] [PubMed] [Google Scholar]
- 13.Bodner CR, Maltsev AS, Dobson CM, Bax A. Biochemistry. 2010;49:862–871. doi: 10.1021/bi901723p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paleologou KE, et al. J Neurosci. 2010;30:3184–3198. doi: 10.1523/JNEUROSCI.5922-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pronin AN, Morris AJ, Surguchov A, Benovic JL. J Biol Chem. 2000;275:26515–26522. doi: 10.1074/jbc.M003542200. [DOI] [PubMed] [Google Scholar]
- 16.Hodara R, Norris EH, Giasson BI, Mishizen-Eberz AJ, Lynch DR, Lee VM, Ischiropoulos H. J Biol Chem. 2004;279:47746–47753. doi: 10.1074/jbc.M408906200. [DOI] [PubMed] [Google Scholar]
- 17.Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, Lee VM. Science. 2000;290:985–989. doi: 10.1126/science.290.5493.985. [DOI] [PubMed] [Google Scholar]
- 18.Souza JM, Choi I, Chen Q, Weisse M, Daikhin E, Yudkoff M, Obin M, Ara J, Horwitz J, Ischiropoulos H. Arch Biochem Biophys. 2000;380:360–366. doi: 10.1006/abbi.2000.1940. [DOI] [PubMed] [Google Scholar]
- 19.Danielson SR, Held JM, Schilling B, Oo M, Gibson BW, Andersen JK. Anal Chem. 2009;81:7823–7828. doi: 10.1021/ac901176t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shavali S, Combs CK, Ebadi M. Neurochem Res. 2006;31:85–94. doi: 10.1007/s11064-005-9233-x. [DOI] [PubMed] [Google Scholar]
- 21.McCormack AL, Mak SK, Shenasa M, Langston WJ, Forno LS, Di Monte DA. J Neuropathol Exp Neurol. 2008;67:793–802. doi: 10.1097/NEN.0b013e318180f0bd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu Z, Xu X, Xiang Z, Zhou J, Zhang Z, Hu C, He C. PLoS One. 2010;5:e9956. doi: 10.1371/journal.pone.0009956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Souza JM, Giasson BI, Chen Q, Lee VM, Ischiropoulos H. J Biol Chem. 2000;275:18344–18349. doi: 10.1074/jbc.M000206200. [DOI] [PubMed] [Google Scholar]
- 24.Rhoades E, Ramlall TF, Webb WW, Eliezer D. Biophys J. 2006;90:4692–4700. doi: 10.1529/biophysj.105.079251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Middleton ER, Rhoades E. Biophys J. 2010;99:2279–2288. doi: 10.1016/j.bpj.2010.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Masuda M, Dohmae N, Nonaka T, Oikawa T, Hisanaga S, Goedert M, Hasegawa M. FEBS Lett. 2006;580:1775–1779. doi: 10.1016/j.febslet.2006.02.032. [DOI] [PubMed] [Google Scholar]
- 27.Trexler AJ, Rhoades E. Biochemistry. 2009;48:2304–2306. doi: 10.1021/bi900114z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones JA, Wilkins DK, Smith LJ, Dobson CM. J Biomol NMR. 1997;10:199–203. [Google Scholar]
- 29.Fortin DL, Troyer MD, Nakamura K, Kubo S, Anthony MD, Edwards RH. J Neurosci. 2004;24:6715–6723. doi: 10.1523/JNEUROSCI.1594-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sokolovsky M, Riordan JF, Vallee BL. Biochemistry. 1966;5:3582–3589. doi: 10.1021/bi00875a029. [DOI] [PubMed] [Google Scholar]
- 31.Trexler AJ, Rhoades E. Biophys J. 2010;99:3048–3055. doi: 10.1016/j.bpj.2010.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bodner CR, Dobson CM, Bax A. J Mol Biol. 2009;390:775–790. doi: 10.1016/j.jmb.2009.05.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Drescher M, Godschalk F, Veldhuis G, van Rooijen BD, Subramaniam V, Huber M. ChemBioChem. 2008;9:2411–2416. doi: 10.1002/cbic.200800238. [DOI] [PubMed] [Google Scholar]
- 34.Robotta M, Braun P, van Rooijen B, Subramaniam V, Huber M, Drescher M. ChemPhysChem. 2011;12:267–269. doi: 10.1002/cphc.201000815. [DOI] [PubMed] [Google Scholar]
- 35.Eliezer D, Kutluay E, Bussell R, Jr, Browne G. J Mol Biol. 2001;307:1061–1073. doi: 10.1006/jmbi.2001.4538. [DOI] [PubMed] [Google Scholar]
- 36.Chandra S, Chen X, Rizo J, Jahn R, Sudhof TC. J Biol Chem. 2003;278:15313–15318. doi: 10.1074/jbc.M213128200. [DOI] [PubMed] [Google Scholar]
- 37.Bussell R, Jr, Eliezer D. J Mol Biol. 2003;329:763–778. doi: 10.1016/s0022-2836(03)00520-5. [DOI] [PubMed] [Google Scholar]
- 38.Paleologou KE, Schmid AW, Rospigliosi CC, Kim HY, Lamberto GR, Fredenburg RA, Lansbury PT, Jr, Fernandez CO, Eliezer D, Zweckstetter M, Lashuel HA. J Biol Chem. 2008;283:16895–16905. doi: 10.1074/jbc.M800747200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jao CC, Der-Sarkissian A, Chen J, Langen R. Proc Natl Acad Sci U S A. 2004;101:8331–8336. doi: 10.1073/pnas.0400553101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bisaglia M, Tessari I, Pinato L, Bellanda M, Giraudo S, Fasano M, Bergantino E, Bubacco L, Mammi S. Biochemistry. 2005;44:329–339. doi: 10.1021/bi048448q. [DOI] [PubMed] [Google Scholar]
- 41.Tessari I, Bisaglia M, Valle F, Samori B, Bergantino E, Mammi S, Bubacco L. J Biol Chem. 2008;283:16808–16817. doi: 10.1074/jbc.M709014200. [DOI] [PubMed] [Google Scholar]
- 42.Lockless SW, Ranganathan R. Science. 1999;286:295–299. doi: 10.1126/science.286.5438.295. [DOI] [PubMed] [Google Scholar]
- 43.Reichheld SE, Yu Z, Davidson AR. Proc Natl Acad Sci U S A. 2009;106:22263–22268. doi: 10.1073/pnas.0911566106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Volkman BF, Lipson D, Wemmer DE, Kern D. Science. 2001;291:2429–2433. doi: 10.1126/science.291.5512.2429. [DOI] [PubMed] [Google Scholar]
- 45.Malmendal A, Evenas J, Forsen S, Akke M. J Mol Biol. 1999;293:883–899. doi: 10.1006/jmbi.1999.3188. [DOI] [PubMed] [Google Scholar]
- 46.Sinha N, Nussinov R. Proc Natl Acad Sci U S A. 2001;98:3139–3144. doi: 10.1073/pnas.051399098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hilser VJ, Thompson EB. Proc Natl Acad Sci U S A. 2007;104:8311–8315. doi: 10.1073/pnas.0700329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Savvides SN, Scheiwein M, Bohme CC, Arteel GE, Karplus PA, Becker K, Schirmer RH. J Biol Chem. 2002;277:2779–2784. doi: 10.1074/jbc.M108190200. [DOI] [PubMed] [Google Scholar]
- 49.Cho MK, Kim HY, Bernado P, Fernandez CO, Blackledge M, Zweckstetter M. J Am Chem Soc. 2007;129:3032–3033. doi: 10.1021/ja067482k. [DOI] [PubMed] [Google Scholar]
- 50.Grupi A, Haas E. J Mol Biol. 2011;405:1267–1283. doi: 10.1016/j.jmb.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 51.Bernado P, Bertoncini CW, Griesinger C, Zweckstetter M, Blackledge M. J Am Chem Soc. 2005;127:17968–17969. doi: 10.1021/ja055538p. [DOI] [PubMed] [Google Scholar]
- 52.Dedmon MM, Lindorff-Larsen K, Christodoulou J, Vendruscolo M, Dobson CM. J Am Chem Soc. 2005;127:476–477. doi: 10.1021/ja044834j. [DOI] [PubMed] [Google Scholar]
- 53.McClendon S, Rospigliosi CC, Eliezer D. Protein Sci. 2009;18:1531–1540. doi: 10.1002/pro.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rospigliosi CC, McClendon S, Schmid AW, Ramlall TF, Barre P, Lashuel HA, Eliezer D. J Mol Biol. 2009;388:1022–1032. doi: 10.1016/j.jmb.2009.03.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bertoncini CW, Fernandez CO, Griesinger C, Jovin TM, Zweckstetter M. J Biol Chem. 2005;280:30649–30652. doi: 10.1074/jbc.C500288200. [DOI] [PubMed] [Google Scholar]
- 56.Bussell R, Jr, Eliezer D. J Biol Chem. 2001;276:45996–46003. doi: 10.1074/jbc.M106777200. [DOI] [PubMed] [Google Scholar]
- 57.Binolfi A, Rasia RM, Bertoncini CW, Ceolin M, Zweckstetter M, Griesinger C, Jovin TM, Fernandez CO. J Am Chem Soc. 2006;128:9893–9901. doi: 10.1021/ja0618649. [DOI] [PubMed] [Google Scholar]
- 58.Nielsen MS, Vorum H, Lindersson E, Jensen PH. J Biol Chem. 2001;276:22680–22684. doi: 10.1074/jbc.M101181200. [DOI] [PubMed] [Google Scholar]
- 59.Bertoncini CW, Jung YS, Fernandez CO, Hoyer W, Griesinger C, Jovin TM, Zweckstetter M. Proc Natl Acad Sci U S A. 2005;102:1430–1435. doi: 10.1073/pnas.0407146102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jensen PH, Hager H, Nielsen MS, Hojrup P, Gliemann J, Jakes R. J Biol Chem. 1999;274:25481–25489. doi: 10.1074/jbc.274.36.25481. [DOI] [PubMed] [Google Scholar]
- 61.Jensen PH, Islam K, Kenney J, Nielsen MS, Power J, Gai WP. J Biol Chem. 2000;275:21500–21507. doi: 10.1074/jbc.M000099200. [DOI] [PubMed] [Google Scholar]
- 62.Burre J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Sudhof TC. Science. 2010;329:1663–1667. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Auluck PK, Caraveo G, Lindquist S. Ann Rev Cell Dev Biol. 2010;26:211–233. doi: 10.1146/annurev.cellbio.042308.113313. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.