

Abstract
AIM: To establish whether activation of adenosine type-3 receptors (A3Rs) and inhibition of interleukin-1β-induced inflammation is beneficial in combination with antibiotic therapy to increase survival of mice challenged with anthrax spores.
METHODS: DBA/2 mice were challenged with Bacillus anthracis spores of the toxigenic Sterne strain 43F2. Survival of animals was monitored for 15 d. Ciprofloxacin treatment (50 mg/kg, once daily, intraperitoneally) was initiated at day +1 simultaneously with the administration of inhibitors, and continued for 10 d. Two doses (2.5 mg/kg and 12.5 mg/kg) of acetyl-tyrosyl-valyl-alanyl-aspartyl-chloromethylketone (YVAD) and three doses (0.05, 0.15 and 0.3 mg/kg) of 1-[2-Chloro-6-[[(3-iodophenyl) methyl]amino]-9H-purin-9-yl]-1-deoxy-N-methyl-β-D- ribofuranuronamide (Cl-IB-MECA) were tested. Animals received YVAD on days 1-4, and Cl-IB-MECA on days 1-10 once daily, subcutaneously. Human lung epithelial cells in culture were challenged with spores or edema toxin and the effects of IB-MECA on phosphorylation of AKT and generation of cAMP were tested.
RESULTS: We showed that the outcome of antibiotic treatment in a murine anthrax model could be substantially improved by co-administration of the caspase-1/4 inhibitor YVAD and the A3R agonist Cl-IB-MECA. Combination treatment with these substances and ciprofloxacin resulted in up to 90% synergistic protection. All untreated mice died, and antibiotic alone protected only 30% of animals. We conclude that both substances target the aberrant host signaling that underpins anthrax mortality.
CONCLUSION: Our findings suggest new possibilities for combination therapy of anthrax with antibiotics, A3R agonists and caspase-1 inhibitors.
Keywords: Anthrax, Mice, Antibiotics, Combination therapy, Inflammasome, Adenosine 3 receptor agonist, Caspase-1 inhibitor, AKT
INTRODUCTION
Anthrax is a life-threatening disease affecting humans and many species of animals. Major routes of infection occur through inhalation, cutaneous abrasions or ingestion of Bacillus anthracis (B. anthracis) spores. Dissemination of bacteria from the initial site of entry can cause a lethal systemic infection.
Virulence of B. anthracis represents a complex picture. It is mainly attributed to the lethal and edema toxins (LeTx and EdTx, correspondingly) encoded by the plasmid XO1, as well as the antiphagocytic capsule encoded by the plasmid XO2. LeTx is a specific protease inactivating mitogen-activated protein kinase kinase (MAPKK), while EdTx is an adenylate cyclase generating cAMP in the host cells. Numerous in vitro studies have demonstrated that anthrax toxins influence a plethora of vital cellular functions, although the molecular events leading to death of intoxicated animals are not fully understood[1].
Despite the development of effective vaccines and antibiotics, late-stage systemic anthrax is resistant to modern therapeutic interventions. The drugs currently approved for inhalation anthrax treatment are limited to fluoroquinolone and tetracycline classes of antibiotics. Although antibiotic administration is highly effective for pre- or post-exposure prophylaxis, it becomes ineffective at the later stages of infection, when complex pathological changes in the host result in a septic shock condition manifested by hypoxic organ failure and circulatory collapse[2].
Generally, antibacterial therapy is expected to benefit from the complementary treatments aimed at the correction of aberrant host responses that result from the activity of pathogenic factors during infection. This approach gains ground with regard to the development of therapies against septic shock caused by different bacteria[3,4]. In anthrax, potential host targets include MAPKK and cAMP signaling by the toxins leading to induction of apoptosis and aberrant cytokine release, accompanied by circulatory shock, and vascular and tissue lesions[1,5]. A recent report indicates that the phosphatidylinositol-3-kinase/AKT (PI3K/AKT) pathway may be an important contributor to animal survival[6]. However, the therapeutic utility of these observations remains to be tested in animal experiments.
The aim of this study was to evaluate if pharmacological correction of host signaling could increase survival of B. anthracis-challenged animals. Using a murine anthrax model, we chose to test the host pro-survival PI3K/AKT signaling and inflammatory response through interleukin (IL)-1β, taking into account the pivotal role of these pathways in the innate immunity. A principal function of AKT in the regulation of cell survival has been established in several cell types[7-9]. AKT has been implicated as anti-apoptotic in different cell death paradigms including growth factor withdrawal, chemical and physical stress, treatment with therapeutic agents, ischemic shock and infectious disease[7-9].
IL-1β is well known as a major cytokine in control of the host antimicrobial inflammatory response. The role of IL-1β in anthrax has long been a subject of debate[10,11]. Therefore, we expected that the results of pharmacological intervention would be instructive regarding the question of whether the IL-1β release benefits the host or the microbe.
As therapeutic means for AKT activation we chose to stimulate signaling through adenosine type-3 receptor (A3R) with selective agonists such as N6-(3-iodobenzyl) adenosine-5’-N-methyluronamide (IB-MECA) and its Cl-substituted derivative Cl-IB-MECA. These compounds previously tested in a number of animal studies and clinical trials display high-affinity receptor binding along with favorable pharmacokinetic and pharmacodynamic properties[12]. IB-MECA and Cl-IB-MECA predictably enhance phosphorylation of AKT and its downstream target, glycogen synthase kinase (GSK)-3β at Ser 9 in mice[13,14]. These A3R agonists also display other biological effects, which seem to be potentially beneficial for treatment against LeTx and EdTx intracellular signaling, through stimulation of extracellular signal-regulated kinase 1/2 phosphorylation and downregulation of cAMP level, respectively[14]. Therapeutic applications of IB-MECA and Cl-IB-MECA have demonstrated cardioprotection during hypoxia, protection from endotoxemia and colitis, as well as decreased mortality from renal and hepatic injury in sepsis[15-19].
We showed that the outcome of antibiotic treatment in a murine anthrax model can be substantially improved by co-administration of the caspase-1/4 inhibitor acetyl-tyrosyl-valyl-alanyl-aspartyl-chloromethylketone (YVAD) and the A3R agonist Cl-IB-MECA. We conclude that both substances target the aberrant host signaling that underpins anthrax mortality. We suggest that our findings open new possibilities for the development of optimized therapeutic interventions complementing anthrax antibiotic therapy.
MATERIALS AND METHODS
Treatment of human small airway lung epithelial cells with forskolin, EdTx and IB-MECA
Human small airway lung epithelial cells (HSAECs) (Cambrex, Walkersville, MD, USA) were grown according to the vendor’s protocol in Ham’s F12 medium supplemented with non-essential amino acids, pyruvate, β-mercaptoethanol and 10% fetal calf serum at 37°C in an atmosphere of 5% CO2. Cell culture reagents were from CellGro (Manassas, VA, USA). Confluent HSAECs were starved for 1-4 h in complete serum-free medium and stimulated as indicated in the figure legends. Control cells were treated with medium alone, which included DMSO used as solvent for the reagents. After incubation for the indicated period of time, the cells were lysed in 150 μL of solution made of equal volumes of T-PER Reagent (Pierce, Rockford, IL, USA) and 2 × Tris-glycine SDS sample buffer (Novex; Invitrogen, Carlsbad, CA, USA) in the presence of 2.5% β-mercaptoethanol, protease and phosphatase inhibitors. Twenty-microliter samples were analyzed by western blotting using the AKT-specific primary antibody (Cell Signaling Technology, Danvers, MA, USA) at a dilution of 1:1000, and the isotype-specific secondary antibody labeled with horseradish peroxidase. In the experiments with EdTx, the cells were starved in serum-free medium for 2 h and stimulated with 1 μg/mL Edema Factor and 5 μg/mL Protective Antigen (both from List Biological Laboratories, Campbell, CA, USA) in complete culture medium containing 250 nmol/L 3-isobutyl-1-methylxanthine (IBMX) to prevent cAMP degradation, in the presence or absence of 2.5 nM IB-MECA for 2 h. Cell lysates were analyzed using competitive cAMP ELISA (R&D Systems, Minneapolis, MN, USA). Other reagents were from Sigma-Aldrich (St Louis, MO, USA).
Animal experiments
Male 6-8-wk-old DBA/2 mice (Jackson Laboratory, Bar Harbor, ME, USA) received food and water ad libitum. Each treatment group included 10 mice. B. anthracis spores of the toxigenic Sterne strain 43F2 (Colorado Serum Company, Denver, CO, USA) were prepared as described previously[20]. All experiments with this strain were carried out at biosafety level 2. Mice were challenged with the spores intraperitoneally (107 spores/animal) on day 0. Survival of animals was monitored for 15 d. Ciprofloxacin (Sigma) treatment (50 mg/kg, once daily, intraperitoneally) was initiated at day +1 simultaneously with the administration of inhibitors, and continued for 10 d. Two doses (2.5 mg/kg and 12.5 mg/kg) of YVAD (Bachem Bioscience, King of Prussia, PA, USA) and three doses (0.05, 0.15 and 0.3 mg/kg) of Cl-IB-MECA (Sigma) were tested. Animals received YVAD on days 1-4, and Cl-IB-MECA on days 1-10 once daily, subcutaneously. Survival was monitored daily. The experimental protocols were approved by the Animal Care and Use Committees of George Mason University, Manassas, VA, and the US Department of Defense. Kaplan-Meier log-rank statistical test was applied to evaluate survival data.
RESULTS
Effect of IB-MECA in cultured cells
To obtain preliminary evidence of IB-MECA activity in the PI3K pathway, the effect of this substance was tested on cultured cells. For this purpose we used the HSAECs as a cell model sensitive to B. anthracis pathogenic factors[6]. IB-MECA quickly upregulated the basal level of AKT phosphorylation in HSAECs at a wide range of concentrations (1-100 nmol/L) (Figure 1A). It also reversed the inhibition of AKT phosphorylation previously reported[6] in HSAECs infected B. anthracis spores (Figure 1B). Inhibition of ATK phosphorylation can also be induced by purified EdTx[6], therefore, we suggested that IB-MECA was able to reduce the toxin-induced elevation of the intracellular cAMP level. Data obtained in the EdTx-treated cells confirm this suggestion (Figure 1C).
Figure 1.
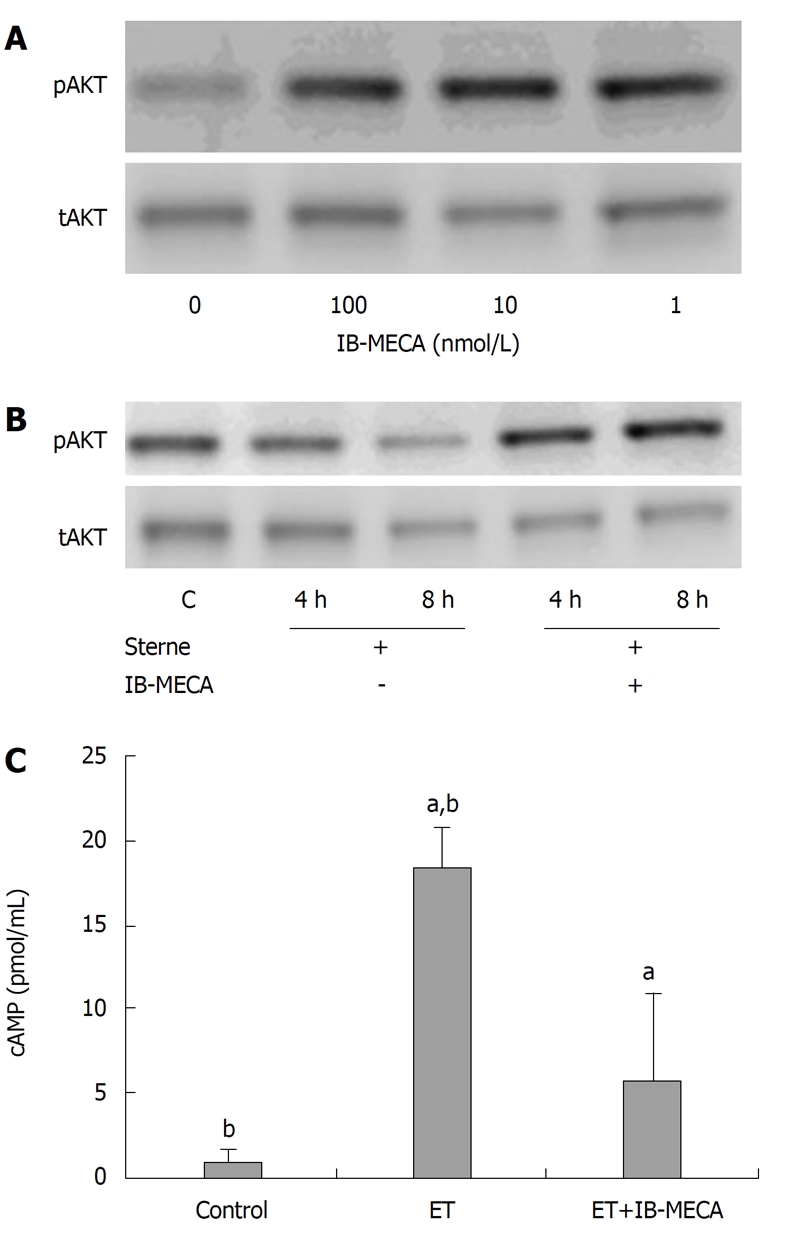
Effects of IB-MECA on phosphorylation of AKT and cAMP level in human small airway lung epithelial cells. A: IB-MECA upregulates AKT phosphorylation in cultured human small airway lung epithelial cells (HSAECs) and prevents downregulation of AKT phosphorylation caused by exposure of the cells to B. anthracis Sterne spores (MOI of 10) when added to the culture medium for 5 min at the indicated times (B); C: IB-MECA treatment (2.5 nmol/L) downregulated EdTx-mediated cAMP production in HSAECs. Cells were starved in serum-free medium for 2 h and treated with EdTx (1 μg/mL edema factor and 5 μg/mL protective antigen) in complete culture medium containing 250 nmol/L IBMX, in the presence or absence of IB-MECA for 2 h. Cell lysates were analyzed with western blotting using antibodies specific for phospho-AKT (pAKT, S473) and total AKT (tAKT) (A and B), and competitive cAMP ELISA (C). Asterisks indicate the corresponding pairs of measurements (mean ± SD, n = 3) used for statistical comparison by the two-tailed t test. aP = 0.05, bP = 0.01.
Combination treatment of mice
The mouse survival experiments included intraperitoneal challenge with B. anthracis (Sterne) spores and subcutaneous administration of selected substances. All treatments began 24 h after spore challenge to mimic the likely real-world scenario, when the exposed persons would seek medical help after a certain period of time required for the detection of exposure and administration of therapy. Treatment with Cl-IB-MECA (chosen for animal experiments over IB-MECA because of higher receptor affinity [12]) as a single daily bolus dose demonstrated a remarkable 40% protection (P = 0.02) over a narrow dose interval (Figure 2A), which perhaps reflected a need to maintain the concentration of a critical signaling mediator, such as cAMP, in a certain physiological range[19]. Combinations of Cl-IB-MECA with ciprofloxacin showed highly reliable improvements in survival relative to untreated control at all tested doses (P < 0.003), while antibiotic alone was marginally protective (P = 0.082) (Figure 2B). Relative to antibiotic alone, co-administration Cl-IB-MECA and ciprofloxacin demonstrated up to a twofold increase in survival, but this effect did not reach statistical reliability: the highest contribution of Cl-IB-MECA at 0.3 mg/kg was 88% reliable.
Figure 2.

Survival of B. anthracis Sterne-challenged DBA/2 mice after treatment. Mice challenged with B. anthracis Sterne spores (i.p.) were treated (s.c.) with indicated daily bolus doses. A: A3R agonist Cl-IB-MECA (0.05, 0.15 and 0.3 mg/kg); B: combination of Cl-IB-MECA (0.05, 0.15 and 0.3 mg/kg) with ciprofloxacin (50 mg/kg); C, D: Caspase-1 inhibitor YVAD (C: 2.5 mg/kg; D: 12.5 mg/kg) in combination with ciprofloxacin (50 mg/kg); E: Triple combination of caspase-1 inhibitor YVAD (2.5 mg/kg), A3AR agonist Cl-IB-MECA (0.15 mg/kg) and ciprofloxacin (50 mg/kg); F: Triple combination of caspase-1 inhibitor YVAD (12.5 mg/kg), A3AR agonist Cl-IB-MECA (0.3 mg/kg) and ciprofloxacin (50 mg/kg). Kaplan–Meier statistical log-rank analysis was used for survival data.
We further tested the effect of caspase-1/4 inhibitor YVAD, which was previously used in our anthrax animal studies to inhibit maturation of IL-1β precursor into a biologically-active cytokine[20]. B. anthracis-challenged mice showed that increased release of this cytokine into the circulation takes place at the pre-mortal stage, which indicates its relevance to lethal outcome[20]. However, the role of IL-1β in mortality remains controversial. Figure 2C and D show that only four doses of the inhibitor administered at days 1-4 post-infection protected up to 70% of animals (P = 0.01) in combination therapy with ciprofloxacin (days 1-10). Based upon these results, we further investigated a triple combination of YVAD, Cl-IB-MECA and ciprofloxacin (Figure 2E and F). While a combination of YVAD and Cl-IB-MECA was marginally protective, a very strong synergistic effect that resulted in 90% protection (P = 0.004) occurred with ciprofloxacin (Figure 2E). Although the increased dose of Cl-IB-MECA was not optimal for protection (Figure 2F), it demonstrated the dose-dependent nature of the therapeutic effect.
DISCUSSION
A therapeutic paradigm of synergistic treatment combining antibiotic and substances restoring the homeostasis of the infected host represents an exciting approach to combat pathogenic organisms[3,4,21,22], including the weaponized versions purposefully engineered to resist conventional antimicrobial therapies[23]. In the current study, we showed for the first time that combination therapy with substances stimulating A3R and inhibiting IL-1β maturation is able to improve strongly survival of B. anthracis-challenged mice in the presence of low doses of antibiotic, which cannot control infection on its own. The activity of A3R agonist IB-MECA in vitro leads to upregulation of AKT phosphorylation and allows us to explain the positive effect of Cl-IB-MECA on animal survival, in agreement with our previous data showing that downregulation of PI3K during infection contributes to mortality[6]. We also demonstrate that A3R stimulation by IB-MECA results in the anti-EdTx effect, because it reduces the increased level of intracellular cAMP generated by EdTx. Previously AKT has been shown to be negatively regulated by cAMP in T lymphocytes or by LeTx and EdTx in the lung epithelial cells[6,24]. It is therefore likely that the activity of Cl-IB-MECA might be relevant to the physiological effects of cAMP on the organs of infected animals.
It is important to emphasize that the optimal therapeutic effect of a given physiological modulator that influences multifunctional cellular mediators such as cAMP or IL-1β might be displayed in a narrow dosage range reflecting particular homeostatic requirements and its interaction with different host systems. For example, a cardioprotective effect of IB-MECA and Cl-IB-MECA can be reversed to apoptosis in the concentration range between 1 and 10 μmol/L[19]. Mathematical modeling based on the analyses of biological circuits and pharmacological parameters allows us to explain increased therapeutic efficacy of combination treatments[21,22]. We expect that the results of Figure 2 that demonstrate the proof of therapeutic concept could be further optimized in future experiments that are aimed at better understanding of the signaling mechanisms that underlie the observed synergism.
Our results allow an intriguing explanation of some previously unexplained experimental observations. The epinephrine-like activity of culture filtrate from B. anthracis[22] has long implicated the central nervous system (CNS) effect of anthrax toxins, and indicated the existence of unknown bacterial AR agonists[25,26]. cAMP is a secondary messenger of the vascular β-adrenergic receptor (AR) stimulation with the agonist epinephrine. The latter is known to cause initial vasoconstriction of the terminal arterioles and venules, followed by their gradual hyporeactivity and dilation resulting in terminal shock[25,27]. We hypothesize that the epinephrine-like effect could be explained by activity of cAMP produced by EdTx. Chronic exposure to cAMP can be cytotoxic and can mimic the well-known effect of catecholamines, including norepinephrine, through β1-AR[27]. LeTx is expected to aggravate the adrenergic effect of EdTx, because LeTx as the inhibitor of p38 signaling would abrogate the anti-apoptotic effect of β2-AR stimulation coupled to PI3K and AKT[28,29]. Our hypothesis that cAMP-mediated signaling through AKT is one of the key axes in anthrax pathogenesis can explain the protective effect of β-AR agonist isoproterenol, which can prevent the life-threatening decrease in blood pressure caused by intensive vasodilation in monkeys administered lethal doses of crude anthrax toxin[26].
We chose IL-1β as an additional therapeutic target due to its ultimate role in innate immunity. As a component of the inflammatory cascade, IL-1β is essential for antimicrobial defenses. IL-1β has long been in the focus of anthrax researchers, and recent data have shown that the host response to LeTx helps protect against infection[30]. In agreement with this, knockout animals deleted of caspase-1 and thus unable to respond with IL-1β release are more susceptible to B. anthracis infection than the wild-type counterparts[30]. However, our results argue in favor of an overall negative impact of IL-1β on survival. This discrepancy can be explained by taking into account that the initial inflammatory response positively contributes to survival, but this benefit can be overwhelmed by the pathogenic impact of excessive inflammation during disease progression. The knockout animals are devoid of this late response and therefore cannot be used to assess fully the contribution of IL-1β-related inflammation to their death, in contrast to normal animals.
Overall, we presented experimental evidence of a highly effective post-exposure treatment strategy that targets the host response mediated through specific pro-survival pathways. The inhibitors that we tested demonstrate a synergistic effect on survival in combination with a low dose of the current state-of-care antibiotic, ciprofloxacin. Instead of blocking the enzymatic activity of anthrax toxins (as the predominant strategy in the field during recent decades), both the caspase inhibitor and the A3R agonist modulate the host cell inflammatory and apoptotic signaling mediators[16]. We therefore conclude that the anti-toxin component of anthrax therapy does not have to be limited to the development of specific inhibitors of LeTx proteolytic activity. In fact, there has been a paucity of reports on the efficacy of the LeTx inhibitors in spore-challenged animals[31].
Although the specific mechanisms of animal protection and the predictive power of our in vitro model are far from being completely understood and warrant further analyses, we conclude that interference with host cell response represents an important part of anthrax pathogenic strategy. Anthrax toxins and other pathogenic factors of B. anthracis cause vascular collapse, edema, liver failure, and immune suppression due to their effects on a broad range of cell types, including the endothelial, epithelial, hepatic and immune cells[5]. In follow-up studies, we plan to carry out immunohistochemical analyses to characterize changes in the activity of AKT, caspase-1 and IL-β to define the biologically relevant cell targets. Extending our results to other animal models is necessary to confirm the relevance of this conclusion to human exposure.
ACKNOWLEDGMENTS
Authors thank Drs. Svetlana Hopkins and Li Dong for help with experiments.
COMMENTS
Background
Anthrax is a lethal disease of humans and many animal species. Its pathophysiology is characterized by intense damage to many host organs and tissues due to activity of several pathogenic factors, including the lethal and edema toxins.
Research frontiers
Anthrax pharmacological treatment is mainly limited to administration of antibiotics, which become ineffective at the late stages of disease due to intoxication of the infected organism. One way of mitigating the pathogenic consequences of intoxication is to restore the homeostatic balance that is upset by infection in the patient’s body. However, this possibility to treat anthrax has not been previously explored.
Innovations and breakthroughs
The authors present experimental evidence of a highly effective post-exposure treatment of murine anthrax that targets the host response mediated through specific pro-survival pathways. They tested model substances that demonstrated a synergistic effect on survival in combination with a low dose of the current state-of-care antibiotic, ciprofloxacin. These drugs do not block the enzymatic activity of anthrax toxins, but inhibit their proinflammatory signaling through interleukin (IL)-1β and cAMP pathways.
Applications
Although the specific mechanisms of animal protection and the predictive power of the tested in vitro model require further analyses, the authors can conclude that interference with host cell response holds promise as a new approach to improve outcome of anthrax treatment. The results identify the inflammatory and cell survival mediators as candidate targets of future therapeutic interventions.
Terminology
Adenosine type-3 receptors (A3Rs) transmit several cell signals to improve survival and eliminate pathogens. AKT responds to A3R signals, and AKT inhibition in anthrax can be mitigated by substances such as N6-(3-iodobenzyl) adenosine-5’-N-methyluronamide, which activate A3Rs. Caspase-1 activates IL-1β, and treatment of animals with caspase-1 inhibitor helps reduce inflammation when it reaches a host-damaging level. Treatment with several drugs against multiple targets is highly effective because it results in a synergistic effect.
Peer review
In this interesting pharmacological study, Dr. Popov and colleagues showed that inhibition of caspase and IL-1β signaling significantly improved survival of mice inoculated with anthrax and treated with antibiotic. This finding opens new possibilities for development of optimized therapeutic interventions complementing anthrax antibiotic therapy.
Footnotes
Peer reviewers: Jose M Andreu, Professor, Centro de Investigaciones Biológicas, CSIC, Ramiro de Maeztu 9, 28006 Madrid, Spain; Maria Luisa Mangoni, PhD, Assistant Professor, Department of Biochemical Sciences “A. Rossi Fanelli”, Sapienza University of Rome, Piazzale Aldo Moro, 5-00185-Rome, Italy
Supported by The US Department of Defense grant DAMD17-03-C-0122
S- Editor Cheng JX L- Editor Kerr C E- Editor Ma WH
References
- 1.Moayeri M, Leppla SH. The roles of anthrax toxin in pathogenesis. Curr Opin Microbiol. 2004;7:19–24. doi: 10.1016/j.mib.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 2.Cui X, Moayeri M, Li Y, Li X, Haley M, Fitz Y, Correa-Araujo R, Banks SM, Leppla SH, Eichacker PQ. Lethality during continuous anthrax lethal toxin infusion is associated with circulatory shock but not inflammatory cytokine or nitric oxide release in rats. Am J Physiol Regul Integr Comp Physiol. 2004;286:R699–R709. doi: 10.1152/ajpregu.00593.2003. [DOI] [PubMed] [Google Scholar]
- 3.Wittebole X, Collienne C, Castanares-Zapatero D, Laterre PF. Adjunctive therapies for severe sepsis. Int J Antimicrob Agents. 2008;32 Suppl 1:S34–S38. doi: 10.1016/j.ijantimicag.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 4.Rice TW. Treatment of severe sepsis: where next? Current and future treatment approaches after the introduction of drotrecogin alfa. Vasc Health Risk Manag. 2006;2:3–18. doi: 10.2147/vhrm.2006.2.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moayeri M, Leppla SH. Cellular and systemic effects of anthrax lethal toxin and edema toxin. Mol Aspects Med. 2009;30:439–455. doi: 10.1016/j.mam.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Popova T, Espina V, Bailey C, Liotta L, Petricoin E, Popov S. Anthrax infection inhibits the AKT signaling involved in the E-cadherin-mediated adhesion of lung epithelial cells. FEMS Immunol Med Microbiol. 2009;56:129–142. doi: 10.1111/j.1574-695X.2009.00558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C. PI3K/Akt and apoptosis: size matters. Oncogene. 2003;22:8983–8998. doi: 10.1038/sj.onc.1207115. [DOI] [PubMed] [Google Scholar]
- 8.Brazil DP, Park J, Hemmings BA. PKB binding proteins. Getting in on the Akt. Cell. 2002;111:293–303. doi: 10.1016/s0092-8674(02)01083-8. [DOI] [PubMed] [Google Scholar]
- 9.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 10.Erwin JL, DaSilva LM, Bavari S, Little SF, Friedlander AM, Chanh TC. Macrophage-derived cell lines do not express proinflammatory cytokines after exposure to Bacillus anthracis lethal toxin. Infect Immun. 2001;69:1175–1177. doi: 10.1128/IAI.69.2.1175-1177.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kang TJ, Basu S, Zhang L, Thomas KE, Vogel SN, Baillie L, Cross AS. Bacillus anthracis spores and lethal toxin induce IL-1beta via functionally distinct signaling pathways. Eur J Immunol. 2008;38:1574–1584. doi: 10.1002/eji.200838141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim HO, Ji XD, Siddiqi SM, Olah ME, Stiles GL, Jacobson KA. 2-Substitution of N6-benzyladenosine-5'-uronamides enhances selectivity for A3 adenosine receptors. Cardiovasc Drug J Med Chem. 1994;37:3614–3621. doi: 10.1021/jm00047a018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park SS, Zhao H, Jang Y, Mueller RA, Xu Z. N6-(3-iodobenzyl)-adenosine-5'-N-methylcarboxamide confers cardioprotection at reperfusion by inhibiting mitochondrial permeability transition pore opening via glycogen synthase kinase 3 beta. J Pharmacol Exp Ther. 2006;318:124–131. doi: 10.1124/jpet.106.101477. [DOI] [PubMed] [Google Scholar]
- 14.Matot I, Weiniger CF, Zeira E, Galun E, Joshi BV, Jacobson KA. A3 adenosine receptors and mitogen-activated protein kinases in lung injury following in vivo reperfusion. Crit Care. 2006;10:R65. doi: 10.1186/cc4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Panjehpour M, Karami-Tehrani F. An adenosine analog (IB-MECA) inhibits anchorage-dependent cell growth of various human breast cancer cell lines. Int J Biochem Cell Biol. 2004;36:1502–1529. doi: 10.1016/j.biocel.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 16.Hasko G, Nemeth ZH, Vizi ES, Salzman AL and Szabo C. An agonist of adenosine A3 receptors decreases interleukin-12 and interferon-gamma production and prevents lethality in endotoxemic mice. Eur J Pharmacol. 1998;358:261–268. doi: 10.1016/s0014-2999(98)00619-0. [DOI] [PubMed] [Google Scholar]
- 17.Mabley J, Soriano F, Pacher P, Hasko G, Marton A, Wallace R, Salzman A and Szabo C. The adenosine A3 receptor agonist, N6-(3-iodobenzyl)-adenosine-5'-N-methyluronamide, is protective in two murine models of colitis. Eur J Pharmacol. 2003;466:323–329. doi: 10.1016/s0014-2999(03)01570-x. [DOI] [PubMed] [Google Scholar]
- 18.Lee HT, Kim M, Joo JD, Gallos G, Chen JF, Emala CW. A3 adenosine receptor activation decreases mortality and renal and hepatic injury in murine septic peritonitis. Am J Physiol Regul Integr Comp Physiol. 2006;291:R959–R969. doi: 10.1152/ajpregu.00034.2006. [DOI] [PubMed] [Google Scholar]
- 19.Xu Z, Jang Y, Mueller RA, Norfleet EA. IB-MECA and cardioprotection. Cardiovasc Drug Rev. 2006;24:227–238. doi: 10.1111/j.1527-3466.2006.00227.x. [DOI] [PubMed] [Google Scholar]
- 20.Popov SG, Popova TG, Grene E, Klotz F, Cardwell J, Bradburne C, Jama Y, Maland M, Wells J, Nalca A, et al. Systemic cytokine response in murine anthrax. Cell Microbiol. 2004;6:225–233. doi: 10.1046/j.1462-5822.2003.00358.x. [DOI] [PubMed] [Google Scholar]
- 21.Jia J, Zhu F, Ma X, Cao Z, Li Y, Chen YZ. Mechanisms of drug combinations: interaction and network perspectives. Nat Rev Drug Discov. 2009;8:111–128. doi: 10.1038/nrd2683. [DOI] [PubMed] [Google Scholar]
- 22.Fitzgerald JB, Schoeberl B, Nielsen UB, Sorger PK. Systems biology and combination therapy in the quest for clinical efficacy. Nat Chem Biol. 2006;2:458–466. doi: 10.1038/nchembio817. [DOI] [PubMed] [Google Scholar]
- 23.Inglesby TV, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Friedlander AM, Hauer J, McDade J, Osterholm MT, O'Toole T, et al. Anthrax as a biological weapon: medical and public health management. Working Group on Civilian Biodefense. JAMA. 1999;281:1735–1745. doi: 10.1001/jama.281.18.1735. [DOI] [PubMed] [Google Scholar]
- 24.Grader-Beck T, van Puijenbroek AA, Nadler LM, Boussiotis VA. cAMP inhibits both Ras and Rap1 activation in primary human T lymphocytes, but only Ras inhibition correlates with blockade of cell cycle progression. Blood. 2003;101:998–1006. doi: 10.1182/blood-2002-06-1665. [DOI] [PubMed] [Google Scholar]
- 25.Williams RP, Hill HR, Hawkins D, Chao KC, Neuenschwander J, Lipscomb HS. Epinephrinelike activity of culture filtrate from Bacillus anthracis. Fed Proc. 1967;26:1545–1548. [PubMed] [Google Scholar]
- 26.Remmele NS, Klein F, Vick JA, Walker JS, Mahlandt BG, Lincoln RE. Anthrax toxin: primary site of action. J Infect Dis. 1968;118:104–113. doi: 10.1093/infdis/118.1.104. [DOI] [PubMed] [Google Scholar]
- 27.Singh K, Xiao L, Remondino A, Sawyer DB, Colucci WS. Adrenergic regulation of cardiac myocyte apoptosis. J Cell Physiol. 2001;189:257–265. doi: 10.1002/jcp.10024. [DOI] [PubMed] [Google Scholar]
- 28.Communal C, Colucci WS, Singh K. p38 mitogen-activated protein kinase pathway protects adult rat ventricular myocytes against beta -adrenergic receptor-stimulated apoptosis. Evidence for Gi-dependent activation. J Biol Chem. 2000;275:19395–19400. doi: 10.1074/jbc.M910471199. [DOI] [PubMed] [Google Scholar]
- 29.Zhu WZ, Zheng M, Koch WJ, Lefkowitz RJ, Kobilka BK, Xiao RP. Dual modulation of cell survival and cell death by beta(2)-adrenergic signaling in adult mouse cardiac myocytes. Proc Natl Acad Sci U S A. 2001;98:1607–1612. doi: 10.1073/pnas.98.4.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moayeri M, Crown D, Newman ZL, Okugawa S, Eckhaus M, Cataisson C, Liu S, Sastalla I, Leppla SH. Inflammasome sensor Nlrp1b-dependent resistance to anthrax is mediated by caspase-1, IL-1 signaling and neutrophil recruitment. PLoS Pathog. 2010;6:e1001222. doi: 10.1371/journal.ppat.1001222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shoop WL, Xiong Y, Wiltsie J, Woods A, Guo J, Pivnichny JV, Felcetto T, Michael BF, Bansal A, Cummings RT, et al. Anthrax lethal factor inhibition. Proc Natl Acad Sci U S A. 2005;102:7958–7963. doi: 10.1073/pnas.0502159102. [DOI] [PMC free article] [PubMed] [Google Scholar]