

Abstract
Bacterial genomes have diverged during evolution, resulting in clearcut differences in their nucleotide composition, such as their GC content. The analysis of complete sequences of bacterial genomes also reveals the presence of nonrandom sequence variation, manifest in the frequency profile of specific short oligonucleotides. These frequency profiles constitute highly specific genomic signatures. Based on these differences in oligonucleotide frequency between bacterial genomes, we investigated the possibility of predicting the genome of origin for a specific genomic sequence. To this end, we developed a naïve Bayesian classifier and systematically analyzed 28 eubacterial and archaeal genomes. We found that sequences as short as 400 bases could be correctly classified with an accuracy of 85%. We then applied the classifier to the identification of horizontal gene transfer events in whole-genome sequences and demonstrated the validity of our approach by correctly predicting the transfer of both the superoxide dismutase (sodC) and the bioC gene from Haemophilus influenzae to Neisseria meningitis, correctly identifying both the donor and recipient species. We believe that this classification methodology could be a valuable tool in biodiversity studies.
The complete genome sequences of many organisms are now available. This permits comprehensive comparative analysis of genome structures. Recent investigations have reported differences in both the subsets of proteins the genomes encode (Rubin et al. 2000) and the frequency of occurrence of many short oligonucleotides (Karlin and Burge 1995), hereafter called “motifs”. The comparative studies have mostly focused on short motifs, such as dinucleotides (Karlin et al. 1992; Goldman 1993; Karlin and Ladunga 1994; Karlin and Burge 1995; Karlin et al. 1997; Nakashima et al. 1997, 1998), trinucleotides (Karlin et al. 1992; Goldman 1993; Karlin and Ladunga 1994; Karlin et al. 1997) and tetranucleotides (Karlin and Ladunga 1994; Karlin et al. 1997). Motifs up to eight nucleotides long were recently analyzed and compared using the chaos game representation (CGR) (Deschavanne et al. 1999). The existence of specific genomic signatures (motif frequency profiles) has been reported for all motif lengths. It has also been shown that intergenomic differences are generally higher than intragenomic differences (Karlin and Ladunga 1994; Karlin and Burge 1995; Karlin et al. 1997; Nakashima et al. 1998; Deschavanne et al. 1999). Genes from different prokaryotic and eukaryotic organisms have been classified using dinucleotide composition (Nakashima et al. 1997, 1998). In the present study, we examined the conditions for identifying the genome of origin for a specific genomic sequence, using the genomic signature concept. The naïve Bayesian classifier used here is a probabilistic technique commonly used in text classification (Robertson and Sparck-Jones 1976; Langley 1992; Lewis and Gale 1994). The methodology is illustrated in Figure 1. First, all genomes are scanned for the occurrences of all possible overlapping motifs with a length of n nucleotides (4n possible motifs). Then, a genomic sequence is chosen at random from anywhere inside a genome (coding or noncoding). From this genomic sequence, all overlapping motifs are extracted. The naïve Bayesian classifier uses the extracted motifs to predict their most probable genomic origin by comparing the frequencies of the extracted motifs with the motif frequencies of the different genomes. In the present study, we determined how the performance of the Bayesian classifier of genomic sequences depends on motif length and sequence length. We demonstrate its generalizing ability and its capacity to discriminate between closely related microorganisms using a sequence sample of only a few hundred nucleotides. We also demonstrate how these properties of the classifier can be applied to the problem of pinpointing horizontal gene transfer events (Doolittle 1999), identifying both donor and recipient (Kroll et al. 1998). As discussed by Eisen (Eisen 2000), there are very few well-documented cases of horizontal gene transfer events where both donor and recipient strains are known. The horizontal gene transfer events from H. influenzae to N. meningitis are one of the best-documented cases available and were therefore chosen as a reference system.
Figure 1.

Outline of the Bayesian classifier. (A) For a given motif length (in this figure, two base pairs), the occurrence of all overlapping motifs for each genome is recorded in the motif occurrence profile (B). The motif occurrence profile for each genome is then transformed to a motif frequency profile (C) by dividing each motif occurrence by the total number of motifs in that genome. (D) A sequence, S, of arbitrary length is taken at random from any of the genomes, consisting of a number of j overlapping motifs. (E) The probability of obtaining the motif distribution present in sequence S is separately calculated for each genome and motif. For example, the probability of obtaining motif i in E. coli, P(Mi:Ge) is estimated by the frequency of that motif in the E. coli genome, calculated in (C). The probability of obtaining the motif distribution present in sequence S is then estimated as the product of the individual probabilities of obtaining each motif (E). The classifier predicts the most probable genomic origin (F), the genome with the highest probability P(S:G).
RESULTS
Visualizing the Genomic Signature Concept
To visualize the difference in motif frequencies between and within the genomes of prokaryotic species, we performed a principal components analysis on the motif frequency profiles (Fig. 2). Different eubacterial and archaeal genomes form clusters in the three-dimensional space drawn by principal components 4, 5, and 6 (“PCA space”). Closely related microorganisms cluster together in PCA-space as shown for Helicobacter and Pyrococcus.
Figure 2.

Visualizing the genomic signature concept. Principal components analysis (PCA) was performed on the motif frequencies of 25 genomic sequences from each eubacterial and archaeal genome. The sequences are mapped into a three-dimensional PCA-space, drawn by three principal components (here components 4, 5, and 6). Each sequence was randomly chosen and had a length of 1000 bp. Closely related microorganisms cluster together in PCA-space here shown for Pyrococcus strains pabyssi and horikoshii and for Helicobacter pylori strains 26695 and J99. For clarity, arrows indicate each distinct genome cluster when similar colors were used to plot the sequences from different eubacterial and archaeal genomes. The figure was plotted using Spotfire (Spotfire Inc.).
Dependence of Classification Accuracy on Motif and Sequence Length
The performance of the classifier depends on the motif length used for establishing the genomic signature. Training was performed on 90% of the genome sequences, and the remaining 10% was used to evaluate classification accuracy. Longer motifs result in a more specific representation of the genome (Fig. 3). Classification accuracy increases with motif length (Fig. 3), and highest accuracy was achieved with eight and nine-nucleotide motifs. We classified sequences of six different lengths: 35, 60, 100, 200, 400, and 1000 nucleotides (nt), and monitored the classification accuracy. The accuracy increases with the sequence length, and sequences of 400-nt length were correctly classified in 85 of 100 cases (Fig. 3). For 100 nucleotide sequences, the mean classification accuracy is 60%, and for short sequences (35 nt) the classifier correctly predicts 36 of 100 test sequences on average. In further studies, we used nine-nucleotide motifs. Inspection of the conditional probabilities within the classifiers indicates that the classification depends not on a few species-specific motifs, but rather on the whole set of motifs (data not shown).
Figure 3.

Dependence of classification accuracy upon motif and sequence length. Motif lengths ranging from one to nine base pairs were evaluated for classification accuracy. A default (“random guess”) classifier is also shown. For each motif length, six different sequence lengths were tested. (35, 60, 100, 200, 400, and 1000 bp). The classification accuracy in percent is represented on the y-axis as the arithmetic mean over the independent genome results. One hundred sequences were randomly picked from each genome for each sequence length, and the classification accuracy was calculated as the ratio of correct predictions, divided by the total number of predictions for each genome and test runs. On the x-axis, the different sequence lengths are shown. Training was performed on 90% of the genome sequences (“training set”), and the remaining 10% (“test set”) was used to evaluate classification accuracy.
Lack-of-Knowledge Experiments Support Global Motif Patterning in Bacterial Genomes
Because the classifier does not depend on alignments, but instead uses motif frequencies, it can be trained using only a subset of the sequence. This subset is subsequently excluded when performing the classification task. This approach assumes that the intergenomic differences in motif frequency between genomes are greater than the intragenomic differences, as previous studies indicate (Karlin and Ladunga 1994; Karlin and Burge 1995; Nakashima et al. 1998; Deschavanne et al. 1999). We thus excluded regions from the different genomes when training the classifier (i.e., when recording motif frequencies) and then randomly picked genomic sequences from the excluded regions for assessing the classification accuracy (Fig. 4). We systematically increased the percentage of the genome excluded when training the classifier to find its limits (Fig. 4). Even when 90% of a genome is excluded during the training, the classifier still produces reliable results. The decrease in classification accuracy could, to some extent, be compensated for by increasing the sequence sample length (Fig. 4).
Figure 4.

Lack-of-knowledge experiments. The genomic percentage of the genome excluded from the training phase was systematically increased and the classification accuracy was monitored. The percentage of genome excluded when training the classifier ranged from 5% to 90%. The classification accuracy in percent is represented as the arithmetic mean over all genomes and sampled sequences and is plotted on the y-axis. For each genome, we sampled 100 random sequences for each sequence length, resulting in 2500 predictions for each plotted value. Different sequence lengths (35, 60, 100, 200, 400, and 100 bp) are plotted on the x-axis. Classification was based on nine-nucleotide motifs.
Classification of Closely Related Microorganisms
We investigated whether the classifier was able to correctly discriminate different strains of the same species, exemplified by H. pylori strains 26695 and J99, N. meningitis (serotype B strain MC58 and serotype A strain Z2491), Pyrococcus (abyssi and horikoshi OT3), and Chlamydia trachomatis (strain Nigg and Serovar D [D/UW-3/Cx]). Each new classifier trained had to correctly discriminate between two different strains of the same species with highly similar motif frequency profiles. The results are presented in Figure 5. For both N. meningitis and H. Pylori, roughly 200 nucleotides were needed for accuracy around 90% (Fig. 5), but for Pyrococcus and Chlamydia, 60 nucleotides sufficed for discrimination with 90% accuracy. However, prediction accuracy was also enhanced because only two classes were to be discriminated, compared to the previous experiments with 25 classes. These results suggest a “hierarchical classification” procedure that first classifies a sequence to correct species and then, using a species-specific classifier, correctly identifies the strain.
Figure 5.

Classification of closely related microorganisms. Classification accuracy between different strains of the same species. The classification accuracy in percent is represented on the y-axis as the mean of the ratio of correct predictions, divided by the total number of predictions for each genome and test runs. The x-axis represents the different sequence lengths (35, 60, 100, 200, 400, and 1000) in base pairs. We sampled 100 genomic sequences for each genome and sequence length.
Identifying Donor and Recipient Strains in Horizontal Gene Transfer Events
The possibility of using the classifier for identifying regions of horizontally transferred genes was examined. Many different methods have been proposed for finding putative cases of horizontal gene transfer (Mrazek and Karlin 1999; Eisen 2000; Garcia-Vallve et al. 2000). However, most methods are designed to search genomes for putative horizontally transferred genes without identifying the donor (Eisen 2000). A general problem in analyzing horizontal gene transfer events is the validation of the results (Eisen 2000). However, one case where strong evidence for horizontal gene transfer exists is from H. influenzae to N. meningitis (Kroll et al. 1998; Eisen 2000). The SodC gene and the Bio gene cluster show strong homology to H. influenza genes, and the 29-nt long Haemophilus Uptake Sequence (HmUS) was found downstream of both of the genes (Kroll et al. 1998). The horizontal gene transfer events between H. influenza and N. meningitis were used to evaluate our classifier for identifying both the donor and the recipient in a horizontal gene transfer event.
The availability of complete sequences for both N. meningitis serotypes A and B further gave us the possibility to constrain the method by conducting the in silico experiment on two highly similar genomes. Two percent of the N. meningitis genomes (serotype A 1.8% and serotype B 2.3%) was classified as being of H. influenzae origin. Scanning the whole genomes of N. meningitis serotypes A and B for the 29-bp HmUS gave us three perfect hits in each genome and a few HmUS containing only a few mismatches. All HmUS hits (perfect matches and with one mismatch) were located in regions of the N. meningitis genome that our tool classified as being of H. influenzae origin. Both the gene regions described by Kroll et al. (1998) were correctly classified as being of H. influenzae origin in the genomes of both serotypes A and B, demonstrating that the classifier can correctly identify both the recipient and the donor in a horizontal gene transfer event (Fig 6). Interestingly, three additional regions were classified as being of H. influenzae origin in both N. meningitis genomes (Fig. 6, Table 1). In all three cases, one or more HmUS were also found within the genomic region. The identified regions contained a putative virulence-associated protein (NMA1725), putative restriction enzyme (NMA1591), putative methyltransferase (NMA1590), and a conserved hypothetical protein (NB1979). For the latter three genes, BLASTX searches identified striking homologs in Haemophilus proteins (Table 1). Those genes are likely to represent previously undetected instances of horizontal gene transfer from H. influenzae to N. meningitis.
Figure 6.

Identification of putative horizontal gene transfer events. Identification of horizontal gene transfer events in the Neisseria meningitis genome exemplified by (A) SodC (putative horizontal transferred gene [Kroll et al. 1998]) and (B) a conserved hypothetical protein. Nucleotide coordinates indicate the positions of the genes in the genome. Results are of “sliding window” classification using 500-bp windows with 250-bp overlap. Black, unfilled windows were classified as “being of N. meningitis origin,” and solid black windows as “being of H. influenzae origin.” Haemophilus Uptake Sequences (HmUS) positions are indicated by small arrows. The numbers of perfect matches to the 29-nucleotide consensus are shown in parentheses. BLASTX results show similar positions in H. influenzae.
Table 1.
List of Putative Horizontally Transferred Genes
| Gene | ORF | Classification (kb) | BLAST | BLASTX | HmUS |
|---|---|---|---|---|---|
| SodC1 | NMB1398/NMA1617 | 1/1 | M84012 (E = 0.0; 87%) | M84012 (E = 4e-95) | 28/29 | 28/29 |
| bio gene cluster -bioC1 | NMB0474/NMA2011 | 0.75/1.5 | U32830 (E = 0.0; 88%) | P45248 (E = 1e-134) | 29/29 | 29/29 |
| Conserved hypothetical protein | NMB1979/NMA0465 | 1.25/1.25 | U32837 (E = 0.0; 92%) | HI1643 (E = 3e-158) | 28/29 | 27/29 |
| Type III restriction enzyme | CA09003/NMA1591 | 2.75/3.5 | U32786 (E = 0.0; 92%) | HI1055 (E = 0.0) | 28/29 | 27/29 |
| Type III methyltransferase | —/NMA1590 | 0.5/0.5 | U32786 (E = 0.0; 92%) | HI1056 (E = 0.0) | 28/29 | 27/29 |
| Virulence associated protein | U23782/NMA1725 | 1.75/1.25 | U32728 (E = 2e-45; 95%) | — | 2 ✻ 29/29 | 2 ✻ 29/29 |
Genes identified as horizontally transferred from Haemophilus influenzae to Neisseria meningitis. ORF and Bayesian classification data are provided for both N. meningitis serotype A and, in parentheses for serotype B. Classification column indicates block length identified as being of H. influenzae origin. The genomic regions identified in N. meningitis were compared for homology using BLAST toward H. influenzae and the homologous sequences reported with accession number, e-value and percent identical nucleotides. The best hit after searching genomic regions using BLASTX with accession number and e-value. HmUS indicates the number of perfect matching nucleotides between the N. meningitis region and the consensus. The two N. meningitis genomes gave almost identical results (data not shown).
Putative horizontal gene transfer events identified by another study (Kroll et al. 1998).
DISCUSSION
We investigated the possibility of classifying genomic sequences based on motif frequency distributions. The classifier presented needs a sample of only 400 nucleotides to correctly classify its origin from 25 totally sequenced bacteria with >85% accuracy. This demonstrates that genome characteristics are captured in the frequencies of overlapping motifs in very short sequences. The lack-of-knowledge experiments demonstrate the feasibility of using the classifier on partial genome sequences. The classifier produced the best results when representing the genomes with eight- or nine-nucleotide motifs, although the optimum motif length is likely to depend on the amount of genomic data available. Longer genomic sequences permit a more specific motif representation, particularly if the motif length is increased. The classifier is able to generalize the genomic motif distribution from a sampled region of the genome to other regions, a functional consequence of the observation that overall variation in motif frequency within a genome is lower than the variation between genomes of different species.
Motif frequency classification does not depend on alignment methods (BLAST, Smith-Waterman) because it is position-independent (“scrambled”). It is therefore computationally inexpensive. Because the bacterial genome sequences are stored in the form of a motif frequency table, the original sequence entry is not required for comparison to the target sequence, in contrast to optimal alignment methods. The genome representation does not grow with more genomic sequences, only with new species identified (i.e., new classes). The genomes are represented as motif frequency vectors with a set dimensionality, which enables further preprocessing (vector transformations) to find better genome representation and possible improvements of classification accuracy.
In the present configuration, the classifier was used for identifying horizontal gene transfer events in whole genome sequences. The classifier was able to correctly identify both the donor and recipient strains in known horizontal gene transfer events from H. influenzae to N. meningitis, in contrast to most methods that only detect genes with abnormal sequence composition without predicting a likely donor. Using the classifier, we found three new potential examples of horizontal gene transfer from H. influenzae into N. meningitis. Finding both HmUS in the proposed regions as well as highly homologous genes in the H. influenzae genome supported the classifier results.
Surprisingly, short sequences with only 60 base pairs were correctly classified in 46 of 100 cases. Because of the remarkable resolution of the classifier, it is intriguing to speculate whether it could be applied to diagnostics of microbial diseases. New techniques for high-throughput sequencing of short genomic sequences have been developed (Ronaghi et al. 1996). The classifier could possibly be used to complement existing diagnostic tools in conjunction with these new sequencing techniques.
It is also interesting to speculate whether this method could be applied to analyze bacterial species composition in complex mixtures such as water, soil, and feces, where traditional culturing techniques allow only the identification of a restricted subset of the prokaryotes present. For this purpose it is of importance that the classifier can discriminate between prokaryotic and potentially contaminating eukaryotic DNA, as our preliminary experiments demonstrated (data not shown). Preliminary experiments indicate that principal component analysis is a useful strategy to further analyze and visualize the differences in motif frequency distributions between bacterial genomes.
Finally, it should be stressed that although further improvements may be necessary for different applications, the methodology is general and could easily be applied to other classification tasks on biological sequences.
METHODS
Data
The complete genomic sequence of 28 archae and eubacterium organisms, with genome sizes ranging from 580 kb for Mycoplasma genitalia to 4.639 kb for Escherichia coli, were obtained from GenBank and TIGR at 05/00. The genomes were “scanned” for overlapping motif occurrence using motif lengths, m, of one to nine nucleotides, and frequency tables for each motif, Mj, in each genome were calculated for each motif length. For example, when using a motif length of nine nucleotides, 49 (262.144) possible unique motifs (“words”) exist. Species with multiple strains sequenced (N. meningitis, Pyrococcus, and H. pylori) were considered as one class and classification correct if any of the two strains were predicted (resulting in 25 different classes). We then designed new classifiers that only discriminate between different strains of the same species.
Naïve Bayesian Classifier
The ordered set of nucleotides in each bacterial genome analyzed is referred to as a “class”. We use the term “classifier” for each statistical tool, trained using a specific genomic sequence dataset to discriminate between the “classes”. Bayesian statistics handle conditional probabilities, that is, given that event A occurred, how likely is event B to occur, P(B∖A). Using this framework, the probability of finding a sequence, S, in a genome, Gi, can be used to calculate the probability of a sequence to belong to a certain genome, P(Gi∖S), by using Bayes' rule (Equation 1).
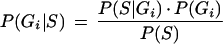 |
1 |
The aim of the naïve Bayesian classifier is, given a sequence, S, to predict its most probable genomic origin (see also Fig. 1). A linear sequence of N nucleotides consists of N-(m-1) overlapping motifs of length m, and the probability of finding sequence S in genome Gi can be expressed as the product of the N-(m-1) probabilities of finding each motif Mj in genome Gi (eq. 2). The naïve Bayesian classifier assumes each motif to be independent of the other motifs, which is clearly false. In fact, in most real-world tasks the independence assumptions is violated, but the method has still proven successful (Domingos and Pazzani 1997).
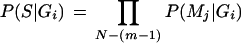 |
2 |
The classifier assigns genomic origin to sequences by taking the maximum P(Gi∖S) value, calculated for all available genomes. The probability of finding sequence S, P(S), is constant (independent of the class) and could therefore be excluded. If excluded, the methodology is equivalent to the maximum a posteriori estimate (Durbin et al. 1998). When applying the classification method to biological samples, the a priori probability of finding the different classes should reflect the hypothesis of the relative abundance of different microorganisms. When the a priori probabilities of finding the different classes are equal, the procedure is equivalent to the maximum likelihood estimate (Durbin et al. 1998). Our implementation of the Bayesian classifiers trained on available bacterial genomes will be accessible at http://www.mtc.ki.se/groups/ernberg/GenomeClass.html.
Horizontal Gene Transfer
A sliding window of 500 bp (with 250 bp overlap) was used to scan the N. meningitis serotype A and B genomes for regions with possible horizontal gene transfer events. The criteria used to detect horizontal gene transfer events were at least two consecutive windows classified as of H. influenzae origin. The 29 nt Haemophilus Uptake Sequences (HmUS) AAGTGC GGTnRWWWWWnnnnnnRWWWWW (Kroll et al. 1998) are highly overrepresented in the genome of H. influenzae and serve as a ligand for surface DNA receptors (Deich and Smith 1980). The occurrences of HmUS have therefore been used as a genomic marker for H. influenzae (Kroll et al. 1998). We scanned the genomes of N. meningitis A and B and H. influenzae for HmUS occurrence, allowing two mismatches from the consensus sequence. We also scanned the genomes of E. coli and Rickettsia as a control. For all identified regions of possible horizontal gene transfer, we searched the databases for homologs using BLASTX.
Acknowledgments
We thank Per Liden and Mikael Huss at Virtual Genetics Laboratory for helpful comments on the manuscript. This work was supported in part by the Swedish Cancer Society and the Swedish Technical Research Council (TFR).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL rickard.sandberg@vglab.com; FAX 46-8-30-55-80.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.186401.
REFERENCES
- Deich RA, Smith HO. Mechanism of homospecific DNA uptake in Haemophilus influenzae transformation. Mol Gen Genet. 1980;177:369–374. doi: 10.1007/BF00271475. [DOI] [PubMed] [Google Scholar]
- Deschavanne PJ, Giron A, Vilain J, Fagot G, Fertil B. Genomic signature: Characterization and classification of species assessed by chaos game representation of sequences. Mol Biol Evol. 1999;16:1391–1399. doi: 10.1093/oxfordjournals.molbev.a026048. [DOI] [PubMed] [Google Scholar]
- Domingos P, Pazzani M. On the optimality of the simple Bayesian classifier under zero-one loss. Machine Learn. 1997;29:103–130. [Google Scholar]
- Doolittle WF. Phylogenetic classification and the universal tree [see comments] Science. 1999;284:2124–2129. doi: 10.1126/science.284.5423.2124. [DOI] [PubMed] [Google Scholar]
- Durbin R, Eddy S, Krogh A, Mitchison G. Biological sequence analysis. Cambridge: Cambridge University Press; 1998. [Google Scholar]
- Eisen JA. Horizontal gene transfer among microbial genomes: New insights from complete genome analysis. Curr Opin Genet Dev. 2000;10:606–611. doi: 10.1016/s0959-437x(00)00143-x. [DOI] [PubMed] [Google Scholar]
- Garcia-Vallve S, Romeu A, Palau J. Horizontal gene transfer in bacterial and archaeal complete genomes. Genome Res. 2000;10:1719–1725. doi: 10.1101/gr.130000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman N. Nucleotide, dinucleotide and trinucleotide frequencies explain patterns observed in chaos game representations of DNA sequences. Nucleic Acids Res. 1993;21:2487–2491. doi: 10.1093/nar/21.10.2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin S, Burge C. Dinucleotide relative abundance extremes: A genomic signature. Trends Genet. 1995;11:283–290. doi: 10.1016/s0168-9525(00)89076-9. [DOI] [PubMed] [Google Scholar]
- Karlin S, Burge C, Campbell AM. Statistical analyses of counts and distributions of restriction sites in DNA sequences. Nucleic Acids Res. 1992;20:1363–1370. doi: 10.1093/nar/20.6.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin S, Ladunga I. Comparisons of eukaryotic genomic sequences. Proc Natl Acad Sci. 1994;91:12832–12836. doi: 10.1073/pnas.91.26.12832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin S, Mrazek J, Campbell AM. Compositional biases of bacterial genomes and evolutionary implications. J Bacteriol. 1997;179:3899–3913. doi: 10.1128/jb.179.12.3899-3913.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroll JS, Wilks KE, Farrant JL, Langford PR. Natural genetic exchange between Haemophilus and Neisseria: Intergeneric transfer of chromosomal genes between major human pathogens. Proc Natl Acad Sci. 1998;95:12381–12385. doi: 10.1073/pnas.95.21.12381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley, P. 1992. An analysis of Bayesian classifiers. AAAI-92.
- Lewis, D. and Gale, W. 1994. A sequential algorithm for training text classifiers. SIGIR-94.
- Mrazek J, Karlin S. Detecting alien genes in bacterial genomes. Ann N Y Acad Sci. 1999;870:314–329. doi: 10.1111/j.1749-6632.1999.tb08893.x. [DOI] [PubMed] [Google Scholar]
- Nakashima H, Nishikawa K, Ooi T. Differences in dinucleotide frequencies of human, yeast, and Escherichia coli genes. DNA Res. 1997;4:185–192. doi: 10.1093/dnares/4.3.185. [DOI] [PubMed] [Google Scholar]
- Nakashima H, Ota M, Nishikawa K, Ooi T. Genes from nine genomes are separated into their organisms in the dinucleotide composition space. DNA Res. 1998;5:251–259. doi: 10.1093/dnares/5.5.251. [DOI] [PubMed] [Google Scholar]
- Robertson SE, Sparck-Jones K. Relevance weighting of search terms. J Am Soc Inf Sci. 1976;27:129–146. [Google Scholar]
- Ronaghi M, Karamohamed S, Pettersson B, Uhlen M, Nyren P. Real-time DNA sequencing using detection of pyrophosphate release. Anal Biochem. 1996;242:84–89. doi: 10.1006/abio.1996.0432. [DOI] [PubMed] [Google Scholar]
- Rubin GM, Yandell MD, Wortman JR, Gabor Miklos GL, Nelson CR, Hariharan IK, Fortini ME, Li PW, Apweiler R, Fleischmann W, et al. Comparative genomics of the eukaryotes. Science. 2000;287:2204–2215. doi: 10.1126/science.287.5461.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]