

Abstract
CD31 is a transmembrane molecule endowed with T-cell regulatory functions due to the presence of 2 ITIMs. For reasons not understood, CD31 is lost by a portion of circulating T-lymphocytes, which appear prone to uncontrolled activation.
Here we show that extracellular T-cell CD31 comprising Ig-like domains 1 to 5 is cleaved and shed from the surface of human T-cells upon activation via their TCR. The shed CD31 can be specifically detected as a soluble truncated protein in human plasma. CD31 shedding results in the loss of its inhibitory function because the necessary cis-homo-oligomerization of the molecule, triggered by the trans-homophilic engagement of the distal Ig-like domain 1, cannot be established by CD31shed cells. However, we show that a juxta-membrane extracellular sequence, comprising part of the domain 6, remains expressed at the surface of CD31shed T-cells. We also show that the immunosuppressive CD31 peptide aa 551–574 is highly homophilic and possibly acts by homo-oligomerizing with the truncated CD31 remaining after its cleavage/shedding. This peptide is able to sustain phosphorylation of the CD31 ITIM686 and of SHP2 and to inhibit TCR-induced T-cell activation. Finally, systemic administration of the peptide in BALB/c mice efficiently suppresses antigen-induced T-cell-mediated immune responses in vivo.
We conclude that the loss of T-cell regulation due to CD31 shedding driven by TCR stimulation can be rescued by molecular tools able to engage the truncated juxta-membrane extracellular molecule that remains exposed at the surface of CD31shed cells.
Keywords: Amino Acid Sequence; Animals; Antigens, CD31; biosynthesis; genetics; metabolism; Cell Membrane; immunology; metabolism; Cells, Cultured; Extracellular Space; immunology; metabolism; Humans; Immunoglobulins; metabolism; Jurkat Cells; Lymphocyte Activation; immunology; Mice; Mice, Inbred BALB C; Mice, Inbred C57BL; Mice, Knockout; Molecular Sequence Data; Peptide Fragments; chemical synthesis; genetics; metabolism; Protein Structure, Tertiary; Receptors, Antigen, T-Cell; metabolism; physiology; T-Lymphocyte Subsets; immunology; metabolism
INTRODUCTION
CD31 (PECAM-1, Figure 1A) consists of a single-chain molecule comprising 6 Ig-like extracellular domains, a short transmembrane segment, and a cytoplasmic tail containing two ImmunoTyrosine-based Inhibitory Motif (ITIM) units (1). The presence of Ig-like domains and their high density at endothelial intercellular borders has previously led to the hypothesis that CD31 functions as a cell adhesion molecule (2) and experimental studies showed that CD31-specific monoclonal antibodies or recombinant soluble proteins block transendothelial migration of monocytes and neutrophils (3). In contrast, in vitro (4) and in vivo (5) studies have shown that lymphocyte CD31 expression is negatively correlated with transmigration. Indeed, it has been demonstrated experimentally that trans-homophilic CD31 engagement drives lymphocyte detachment from antigen-presenting cells (6) and inhibits T-cell receptor (TCR)-mediated signal transduction (7), pointing to an important immunoregulatory function for T-cell CD31 surface molecules. Moreover, CD31 knockout mice are prone to develop chronic inflammatory diseases (8–10).
Figure 1. Flow cytometric analysis of CD31 expression on blood leukocytes.
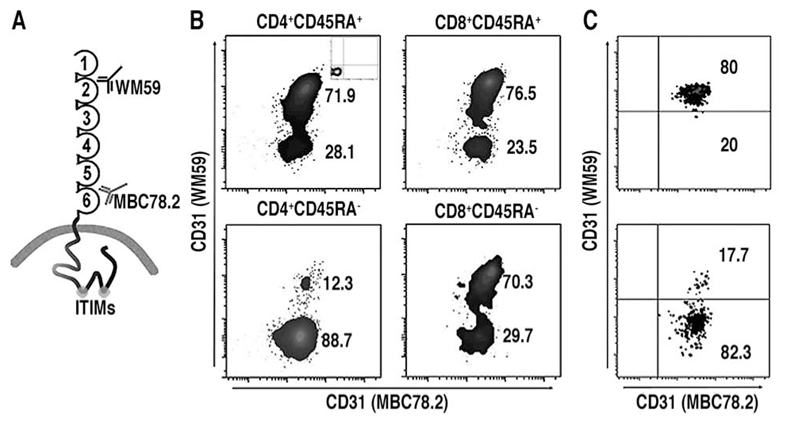
(A) Schematic representation of membrane-bound CD31 and of the monoclonal antibodies used to detect the CD31 domains 2 (WM59) and 6 (MBC 78.2) on peripheral blood leukocytes. (B) Representative example of flow cytometric analysis of human peripheral blood cells from a healthy donor. CD8+ and CD4+ subpopulations were gated within CD3+ cells and were further analyzed for the expression of CD45RA. The proportion of cells lacking domain 2 was dramatically increased in memory (CD45RA−) cells compared to naïve (CD45RA+) cells. All leukocytes were positive for CD31 domain 6. Isotype controls of WM59 and MBC 78.2 antibodies are shown in the insets. (C) Fresh peripheral blood-derived resting CD4+ T-cells (top) lost their CD31 domain 2 upon TCR-activation (bottom).
Previous studies have demonstrated that a subset of T-cells in adult human blood, mostly within the CD45RA−/CD45RO+ (memory) subpopulation (11, 12), lack CD31. Furthermore, an effective “loss” of CD31 is observed upon lymphocyte activation and differentiation (13), especially in CD4+ T cells activated via TCR stimulation (14). Nevertheless, the mechanism underlying loss of CD31 from the T cell surface is not known. Down regulation of the CD31 transcript has been documented in lymphocytes lacking surface CD31 (13). However, regulation of CD31 expression levels at the transcript level does not appear to be the mechanism responsible for down-modulation of CD31 expression in response to TCR stimulation since CD31 expression is constitutive and turn-over of the molecule takes 48 hours (15), whereas the process of CD31 disappearance upon T-cell activation takes less than 16 hours (16). These findings indicate that another mechanism is responsible for the rapid loss of CD31 that occurs upon lymphocyte activation.
We hypothesized that CD31 could undergo proteolytic cleavage and thus escape detection via the shedding of a cleaved extracellular portion of the protein (17). This process is extremely rapid (18) and is similar to the mechanism observed with other transmembrane molecules bearing Ig-like domains (19, 20). This hypothesis is supported by the fact that plasma CD31 exists in two distinct forms: a “transmembraneless” form (120 kDa) and a “truncated” form (90 kDa). The “transmembraneless” form resembles the one produced by alternative splicing of the transmembrane segment-encoding (exon 9) transcript documented in endothelial and myeloid cell lines (15). The “truncated” form (90 kDa) lacks the cytoplasmic tail (15) and could originate from proteolytic cleavage at the cell surface and shedding of the extracellular portion of CD31 into the plasma. A recent study by Eugenin et al. (21) showed that the positive staining for extracellular CD31 in inflamed brain tissues of HIV-infected patients is only minimally co-localized with the intracellular portion of CD31. The authors therefore suggested that activated inflammatory cells undergo an active shedding of extracellular CD31 which could thus contribute to the rise in soluble CD31 in the circulation of patients with inflammatory diseases (21).
In addition to the potential diagnostic value of measuring the cleaved moiety, the loss of CD31 on peripheral T-cells due to shedding, rather than its total absence, could also provide an important therapeutic potential. Herein, we show that the remaining cell surface fragment can indeed be engaged in interactions with a homotypic synthetic peptide in the aim of restoring the physiological functions of the molecule and controlling antigen-induced T-cell responses in vivo.
MATERIALS AND METHODS
Assessment of CD31+ and CD31shed peripheral blood leukocytes
Ten-color flow cytometry was performed on peripheral blood leukocytes obtained from 5 healthy individuals. The leukocytes were either maintained in basal conditions or stimulated overnight with soluble purified anti-CD3 antibody (R&D Systems) at 1 μg/ml. Cells were fixed in PBS/Formaldehyde 1%/Fetal Calf Serum 1% for 4 minutes at 37°C and subjected to erythrocyte hypotonic lysis (10 minutes at 37°C 1:10 v:v in Tris 10 mM, NH4Cl 155 mM, KHCO3 10 mM, pH 7.4), prior to processing, All experiments on human samples were approved by the Institutional Ethical Committee (www.clinicaltrials.gov; Identifier: NCT00430820). Pelleted cells were incubated for 30 minutes, at room temperature and protected from light, with a cocktail of fluorescent monoclonal antibodies directed towards CD4 (PE-Cy7), CD8 (PerCP), HLA-DR (APC-Cy7), CD45RA (Pacific Blue), CD14 (APC), CD10 (PE-Cy5) and CD31 (clone WM59, PE) from BD Biosciences and CD3 (PE-Texas Red), CD20 (AlexaFluor®700) and CD31 (clone MBC 78.2, FITC) from Invitrogen. At least 50,000 events were acquired in the lymphocyte gate using a BD LSRII® equipped with 3 lasers (405, 488 and 633 nm) and analyzed with BD FACSDiva® 6.0 software.
Subtractive measurement of soluble CD31 (Patent N° PCT/EP2009/058220)
To detect the soluble or solubilized CD31 forms containing the Ig-like domain 1 and/or 2 and/or 5 and/or 6, we have customized the cytometric bead array (CBA®, BD) technology. Functional CBA beads were coupled with purified monoclonal anti-CD31 antibodies against either the most distal Ig-like domain 1 [clone JC70A, DAKO (22)] or the most proximal Ig-like domain 6 [clone MBC 78.2 (23), also known as “PECAM-1.2”, generously provided by Dr Peter Newman, Milwaukee Blood Center-MBC-, Wisconsin]. The coupled beads (Figure 2) were incubated with the plasma of healthy subjects, culture supernatant and membrane lysates from stimulated Jurkat cells and serial dilutions of the standard (recombinant, full length extracellular CD31, aa 1–574, R&D Systems). Positive binding of circulating CD31 captured by the anti-CD31 cytometric beads was detected by a combination of three anti-CD31 monoclonal antibodies directed against Ig-like domain 2 (clone WM-59 (22), PE from BD), domain 5 (clone HC1/6 (22), FITC from Invitrogen) and domain 6 (clone MBC 78.2 (23) coupled to Pacific Blue using the kit #P30013 from Invitrogen). The median fluorescent intensity of each detecting antibody was analyzed on ≥1000 acquired beads. The three standard curves were obtained by using all detecting antibodies simultaneously on the serial dilutions of recombinant CD31 in order to overcome any bias due to differences in binding affinity of the diverse antibodies. The concentration of soluble CD31 forms comprising at least domain 1–2 (capture by JC70A, detection by WM59-PE), domain 1–5 (capture by JC70A, detection by HC1/6-FITC) domain 1–6 (capture by JC70A, detection by MBC 78.2-Pacific Blue), domain 6 (capture by MBC 78.2, detection by MBC 78.2-Pacific Blue), domain 5–6 (capture by MBC 78.2, detection by HC1/6) or domain 2–6 (capture by MBC 78.2, detection by WM59) of CD31 in samples was calculated using the linear regression formula obtained with the same capture beads and detecting antibodies on the serial standard dilutions. In plasma and culture supernatant, the concentration of CD31 domain 1–6 was subtracted from the concentration of CD31 domain 1–5 in order to obtain the amount of truncated CD31 lacking domain 6. The latter was subtracted from the concentration of CD31 domain 1–2 to calculate the value of soluble CD31 lacking both domain 5 and 6. In membrane lysates, the concentration of CD31 domain 2–6 was subtracted from the concentration of CD31 domain 5–6 to obtain the fraction lacking at least domain 2. The concentration of CD31 Δ2 was then subtracted from CD31 domain 6 to obtain the fraction lacking both domain 2 and 5.
Figure 2. Analysis of soluble and membrane-bound CD31 upon T-cell activation.
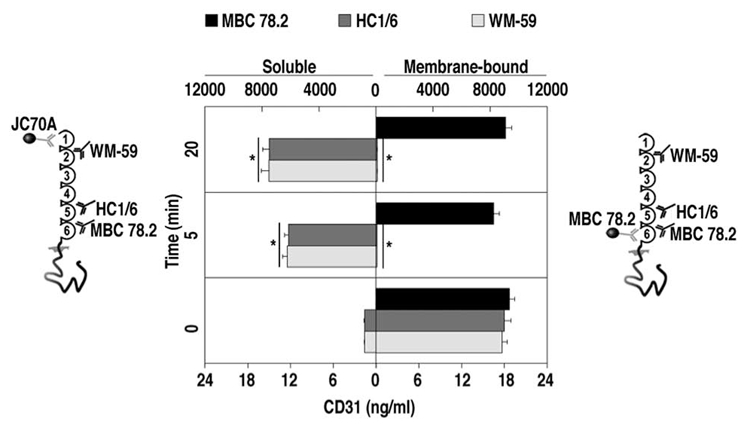
Results from 6 independent experiments are shown in the mirrored bar histograms. Soluble CD31 (left panel) and membrane-bound CD31 (right panel) were measured respectively in the supernatant and in membrane lysates of Jurkat cells stimulated via their TCR by crosslinking CD3 molecules, as previously described (7). Supernatant and membrane lysates were collected at time 0, 5 and 20 minutes after TCR stimulation. For supernatant analysis, CD31 molecules were captured by cytometric beads coupled to JC70A antibodies (domain 1). For membrane lysates, the capture was performed with cytometric beads coupled to MBC 78.2-antibodies (domain 6). CD31 molecules captured by either type of bead were revealed using a three-color CD31 antibody cocktail (WM59-domain 2 coupled to PE; HC1/6-domain 5 coupled to FITC; and MBC 78.2 couped to Pacific Blue). The same sets of capture and detecting antibodies were used to obtain the standard curves using serial dilutions of recombinant human CD31 and data are expressed as ng/ml. Before TCR stimulation (Time=0), soluble CD31 is virtually absent in the supernatant, while all three detecting antibodies are able to detect membrane-bound CD31 molecules. Upon TCR stimulation (Time =5 and 20 minutes), virtually all CD31 molecules are cleaved upstream of the epitope recognized by MBC 78.2 (domain 6) antibodies since these are the only antibodies able to reveal the membrane bound CD31 molecules at these time points. The missing fragment comprising domain 1 (capture by JC70A), domain 2 (detection by WM59) through to domain 5 (detection by HC1/6) is shed from the membrane and detected in the supernatant. Bottom X-axis applies to time=0 while the top X-axis concerns times =5 and 20 minutes.
Peptides
The human (NHASSVPRSKILTVRVILAPWKK, MW 2601.2) and the mouse (SSMRTSPRSSTLAVRVFLAPWKK, MW 2606.0) custom peptides were synthetized by GENOSPHERE (France) and by MIMOTOPES (Australia). The 5(6)-FAM human sequence was provided by MIMOTOPES. Peptides were >95% pure, as assessed by analytical reverse phase HPLC (RP-HPLC), and were checked for correct identity by aminoacid analysis and mass spectrometry (MS). Upon receipt from the fabricant, the lyophilized peptides were dissolved in sterile PBS/DMSO and the concentration was determined by UV spectroscopy at 280 nm. Endotoxin levels were consistently < 0.01 ng/μg of peptide as determined by the LAL test. The peptide stock concentration was adjusted to 10 mg/ml with sterile PBS (final concentration of DMSO ≤ 5%) and stock aliquots were stored at −20°C in a manual defrost freezer.
TIRF microscopy
Freshly purified CD4+ cells from heparinized peripheral blood obtained by positive magnetic selection (Dynabeads® FlowComp™ Human CD4 kit, Invitrogen) were stimulated by CD3 crosslinking as described below (“TCR stimulation in vitro”) in the presence of either 100 μg/ml unlabeled human CD31 peptide, 100 μg/ml 5,6 FAM human CD31 peptide or 10 μg/ml CD31 domain 6 antibody (clone MBC 78.2) directly coupled to AlexaFluor®546 (Monoclonal Antibody Labeling kit # A-20183, Invitrogen). Unstimulated cells and cells stimulated for 20 minutes were plunged immediately into 1 volume of ice-cold 2% paraformaldehyde for 15 minutes. Cells were subsequently pelleted and resuspended in 2% paraformaldehyde for an additional 15 minutes on ice. Following fixation, the cells were rinsed three times in 1x phosphate-buffered saline and incubated overnight at room temperature with 10 μg/ml AlexaFluor®546 CD31 domain 6 (clone MBC 78.2), as appropriate. After incubation, cells were washed twice, post-fixed with 1% paraformaldehyde and transferred to 35 mm glass-bottom poly-D-lysine coated dishes (#P35GC-1.5-10-C, MatTek) and left to adhere for 2 hours in phosphate-buffered saline. Three independent experiments gave similar results. Digital images were acquired using the objective PLAPO 100xO TIRFM (NA 1.45) on an Olympus IX81 inverted microscope equipped with a Total Internal Reflection Fluorescence Microscopy (TIRFM) module (488 and 561 nm) and a cooled monochromatic digital camera (Andor EMCCD DU885 iXon+) and were subsequently analyzed using ImageJ® software.
Plasmon Surface Resonance
Homophilic peptide association and dissociation were evaluated in three independent experiments by surface plasmon resonance (BIAcore® 2000, GE). In brief, human and murine peptides were amine-coupled (2000–5000 Resonance Units, RU) to CM5 chips according to the manufacturer’s instructions. For the kinetic measurements, 6 two-fold concentrations of the same peptides (concentration range from 0.625 to 20 μg/ml) in 10 mM HEPES pH 7.4, 140 mM NaCl, 3 mM EDTA, 0.005% Tween 20 were injected over the chip surface at a flow rate of 10 μl/minutes at 25°C. The BIAevaluation® 3.0 Software (GE) was used for evaluation and data are expressed as ΔRU (Resonance Units on the peptide-coated channel minus Resonance Units on the channel coated with a control, scrambled peptide). The rate equation used for the analysis was a 1:1 binding with drifting baseline model.
TCR stimulation in vitro
For T-cell activation, human peripheral blood mononuclear cells or spleen cells from CD31+/+ and CD31−/− mice (Jackson Laboratories, C57Bl/6 background) were stimulated in vitro as previously described (24). Briefly, cells were plated in triplicate at 0.2×106 cells/well in a U bottom 96-well plate in complete medium (RPMI 1640, 1% pyruvate, 1% glutamine, 1% penicillin-streptomicyn-fungizone, 10% decomplemented fetal calf serum, all from Invitrogen) containing 1μg/ml anti-mouse CD3/CD28 or 5μg/ml anti-human CD3 antibodies (BD) as appropriate. Human or mouse CD31 peptide at 25, 50 and 100 μg/ml and scrambled peptide at 100μg/ml final concentration were deposited in the wells just before cell plating. For T-cell proliferation analysis, plated cells were cultured for 72 hours in 5% CO2 at 37°C. 3(H) thymidine (0.5 μCi/well) was added for the last 16 hours and proliferation evaluated using a Tomtec harvester and analysis on a Wallac micro beta counter. Data are from triplicates. The same conditions of culture were tested to determine the amount of apoptotic cells at 36 hours using Annexin-V and 7-AAD staining; cells were acquired using a BD LSRII® and analyzed with BD FACSDiva® 6.0 software. Data obtained from 6 independent experiments yielded similar results.
Assessment of CD31 pY686 (Patent N° PCT/EP2009/058220) and SHP2 pY542
Log-phase Jurkat cells (107 cells/condition) were stimulated to induce PECAM-1 tyrosine phosphorylation and SHP-2 binding. Cell suspensions (1 ml) were incubated with no antibody, with a human CD3ε-specific antibody (clone UCTH1, 1μg/ml, R&D Systems) alone or in presence of a human CD31-specific antibody directed to domain 6 [MBC 78.2, 50μg/ml (23)] or human CD31 peptide (100μg/ml) for 30 minutes on ice. To remove unbound antibodies, cells were washed twice with 5 ml of ice-cold 0.145 M NaCl, Bovin Serum Albumin 0.1%, 10 mM HEPES, 2.8 mM KCl, 2 mM MgCl2, 10mM D-glucose, 10mM CaCl2 and then resuspended in the same buffer to a final concentration of 2×107 cells/ml. The washing step was omitted for CD31 peptide conditions. To induce the crosslink reaction, cells were pre-warmed for 10 minutes at 37°C and then goat anti-mouse IgG (Fab′)2 fragments (Jackson Laboratories) were added to achieve a final concentration of 20–100 μg/ml. At the specified time points, cells were rapidly spun and the supernatant was replaced by an equal volume of lysis buffer (50 mM Tris, 150 mM NaCl, 1% Triton X-100 (pH 7.4), 1x protease inhibitor cocktail Sigma # P-8340, 1x phosphatase inhibitor cocktail-2 Sigma # P-5726). Lysis was carried out on ice for 30 minutes. Control conditions included cells either left unstimulated (negative control) or incubated with sodium pervanadate (Na3VO8, positive control). The last control consisted of unstimulated cells incubated with the peptide alone. Lysates were ultracentrifuged and the supernatant aliquoted and stored at −80°C until analysis. Lysates (20 μl/condition) were each incubated with Functional CBA® beads (BD) coated with a monoclonal antibody directed to the PECAM-1 C-terminal sequence (clone 235.1, kindly provided by Dr Peter Newman, MBC, Wisconsin) for 3 hours at room temperature. Beads were subsequently washed with CBA washing buffer (BD) and incubated with 2μl of undiluted rabbit anti-CD31 phospho-tyrosine 686 (pY686) sera (kindly provided by Dr Peter Newman, MBC, Wisconsin). This step was followed by two washing steps and a further incubation with AlexaFluor®488-conjugated (Fab′)2 fragments (2μg/ml in CBA washing buffer) of goat-anti-rabbit IgG (Invitrogen). For quantifying pSHP2, cells were stimulated as described above. At the same time points as above, the cells were fixed with BD Cytofix™ Buffer for 10 minutes at 37°C, permeabilized with BD™ Phosflow Perm Buffer III on ice for 30 minutes, blocked with normal mouse immunoglobulin followed by intracellular staining using 2μl of undiluted PE mouse anti-SHP2 (pY542) antibody (clone L99-921, BD Biosciences). Each condition was performed in triplicate and the experiments were repeated three times. The beads (>1000/condition) and cells (106/condition) were analyzed on an LSRII® cytometer (BD Biosciences) in the 530/30 nm (pY686) and 575/26 nm (pY542) detection channel and data are expressed as Median Fluorescence Intensity (MFI) calculated with the FACSDiva 6.0® software (BD Biosciences).
Calcium mobilization assay
Spleen cells from C57Bl/6 mice were prepared as described (24). Cells were incubated with Fluo-3AM (Invitrogen, # F1242) as per the instructions of the manufacturer. Fluorescence of calcium-bound tracer was measured in the 530/30 nm detection channel on an LSRII® cytometer (BD Biosciences). Data were acquired prior to and during 60 seconds following the addition of rat anti-mouse CD3/CD28 monoclonal antibodies (from Biolegend, 10μg/ml each) and Fab fragments of affinity purified goat anti-rat immunoglobulins (from Jackson ImmunoResearch Laboratories, 1.3 μg/ml). Measurements were taken as described above and in the presence of either rat anti-mouse CD31 antibody (clone 390, from BD Biosciences, 10 μg/ml) or mouse CD31 peptide (100μg/ml). Negative controls included rat IgG isotype control and scrambled peptide. Experiments were repeated three times and gave similar results.
Immunosuppressive effect of the peptide in vivo
Delayed Type Hypersensitivity (DTH) suppression was evaluated as described (25). Briefly, TNCB (2-chloro-1,3,5-trinitrobenzene, Fluka #79874) was dissolved in acetone/olive oil (1:1 v/v) at a concentration of 10 mg/ml. BALB/c mice (n=6/group) were primed by painting shaved regions of the abdomen with 0.2 ml of the preparation. The experiment included 3 dose groups for the CD31 peptide (25, 50, 100 μg) and 1 for the scrambled peptide (100 μg). Five days after priming, 10 μl of the TNCB-solvent mixture was painted on the right pinna, 30 minutes after subcutaneous (interscapolar) administration of the mouse CD31 peptide or the scrambled peptide diluted in 100μl of PBS. Right ear thickness was measured at 24 hours with a dial caliper (“Pocotest”, Kroeplin Längenmesstechnick). Measurement was performed 5 times and individual data are shown. The experience was repeated three times and gave similar results.
Statistical Analysis
Data are expressed as means ± SEM unless otherwise indicated in the text. Differences between paired experimental conditions were analyzed by Student t-test and were considered statistically significant when the p value was <0.05.
RESULTS
A portion of the membrane proximal extracellular CD31 is always present at the surface of blood lymphocytes
In order to establish whether the loss of CD31 was only restricted to a part or extended to the totality of its 6 extracellular Ig-like domains, we have performed multicolor flow cytometry analysis of peripheral blood leukocytes from 5 healthy donors. We used two different CD31 antibodies specifically recognizing either the membrane-distal Ig-like domain 2 [clone WM59, defined on p364 of Leukocyte Typing VI and in ref (22)] or the membrane-proximal Ig-like domain 6 [clone MBC 78.2/PECAM-1.2 (23)] (Figure 1A). We have simultaneously used a panel of lineage markers as well as the expression of CD45RA and HLA-DR. Most CD14+ (monocytes) and SSChighCD10+ (granulocytes) cells were positively detected by both CD31 monoclonal antibodies (data not shown). However, a variable portion of CD3+ (T) and CD20+ (B) lymphocytes were WM59− and therefore lacked at least the membrane-distal Ig-like domains 1 and 2 of the CD31 molecule (data not shown). CD4+CD3+ T-cells displayed the highest fraction of WM59− lymphocytes (≥ 30%) which increased dramatically from naïve (CD45RA+) to memory (CD45RA−) cells (Figure 1B). Recently activated (CD45RA− HLA-DR+) lymphocytes were virtually all WM59− (data not shown). All of these cells however expressed at least the portion of the membrane-proximal Ig-like domain 6 of CD31, irrespective of their state of maturation/activation (Figure 1B).
Loss of the domain 2 of CD31 could be experimentally induced by antibody-mediated TCR engagement. This led to a shift in phenotype of >80% of the peripheral blood resting T-cells from CD31 WM59+/MBC 78.2+ to WM59−/MBC 78.2+ phenotype (Figure 1C).
A truncated CD31 lacking the membrane-proximal extracellular domain 6 accumulates in human plasma and in the supernatant of activated T-cells
We set up an assay to enable the discrimination between soluble CD31 molecules containing or lacking domain 6, by subtraction. Indeed, domain 6 which is normally present on all peripheral blood leukocytes, should be missing from the plasma CD31 formed after cleavage and shedding from cells (Figure 1B and 1C). Alternatively, domain 6 should still be present in the soluble CD31 form after splicing (15). We used cytometric beads and a combination of fluorescent specific monoclonal antibodies that map to the different CD31 domains: domain 1 [clone JC70A (22)], domain 2 [clone WM59 (22)], domain 5 [clone HC1/6 (22)] and domain 6 [clone PECAM-1.2/MBC 78.2 (23)]. We found that the plasma of healthy donors (n=12) contained 12.87±0.89 ng/ml of soluble CD31 molecules comprising at least Ig-like domains 1 and 2. A fraction of plasma CD31 (6.69±2.10 ng/ml) contained all the 6 extracellular Ig-like domains. This fraction, detected by both JC70A (domain 1) and MBC 78.2 (domain 6) monoclonal antibodies, likely corresponds to the spliced “transmembraneless” plasma CD31 (15). A smaller fraction (6.18±2.36 ng/ml) of plasma CD31 failed detection by MBC 78.2 monoclonal antibody (domain 6) but was detected to a large extent (≥76%) by HC1/6 monoclonal antibody (domain 5). Only a minimal fraction (1.43±0.91 ng/ml) of plasma CD31 was detected by the WM59 antibody (domain 2), while undetectable by both MBC 78.2 (domain 6) and HC1/6 (domain 5) antibodies.
In order to assess whether CD31 extracellular cleavage and shedding was linked to the activation of T-cells, we have stimulated Jurkat cells by CD3 crosslinking for 0, 5 and 20 minutes. We then analyzed the integrity of CD31 molecules both in the supernatant and in the cell lysate (membrane proteins). For this purpose the immuno-subtractive analysis was based on the capture of CD31 membrane-distal (supernatant) or membrane-proximal (membrane lysate) Ig-like domains by cytometric beads and simultaneous detection of the other Ig-like domains by a cocktail of three fluorescent monoclonal antibodies (Figure 2). The soluble CD31 molecule concentration in the supernatant (comprising at least Ig-like domains 1 and 2) increased dramatically 5 minutes after T-cell stimulation (from 1.61 ± 0.05 to 6234 ± 323 ng/ml) and kept increasing up to 20 minutes (7507 ± 538 ng/ml) after stimulation. Most (86–88%) of the soluble protein comprised also the Ig-like domain 5 but lacked domain 6 (Figure 2). In parallel, >99% of the T-cell-bound CD31 molecules captured via the detection of domain 6 lacked both Ig-like domains 5 and 2 (Figure 2).
A peptide homotypic of the residual extracellular CD31 fragment on CD31shed T-cells rescues the CD31 ITIM/SHP2 inhibitory pathway
A CD31 domain-6-derived synthetic peptide, corresponding to the 23 juxta-membrane positioned aminoacids of the extracellular human CD31 sequence (NHASSVPRSKILTVRVILAPWKK) (26), showed a dose-dependent suppressor effect on proliferation of human peripheral blood T-cell stimulated in vitro by using anti-CD3ε antibodies (Figure 3A). The anti-proliferative effect of the peptide did not correlate with its pro-apoptotic effect, which was minimal (data not shown). Surface plasmon resonance (BIAcore®) analysis demonstrated that this peptide is homophilic (Figure 3B). The BIAcore curves followed concentration-dependent association and dissociation models of the peptide over its homotypic sequence. At low concentrations (up to 1.25 μg/ml), the curves showed steady values of ΔRU over time (Figure 3B) following a 1:1 binding (homo-dimerization) model. Above 2.5 μg/ml, the curves showed increasing ΔRU values during the association phase and did not drop to baseline upon peptide wash-out indicating a >1:1 binding (homo-oligomerization) model. For the analysis, different binding models (different rate equations) were tested in the global curve fitting procedure. The model best describing the experimental data was a 1:1 binding with a drifting baseline model. However, this model could only be applied to low concentrations of the analyte (≤ 1.25 μg/ml). At concentrations >2.5μg/ml, the BIAcore curves suggest peptide polymerization, implying that peptide monomers are in equilibrium with peptide dimers, trimers, tetramers, etc. As a consequence, it was not possible to evaluate the association and dissociation constants of the peptides at the biologically active concentrations (≥10μg/ml) that have been used in our experiments.
Figure 3. A homophilic peptide derived from the residual extracellular fragment on CD31shed inhibits T-cell activation and rescues CD31 signalling.
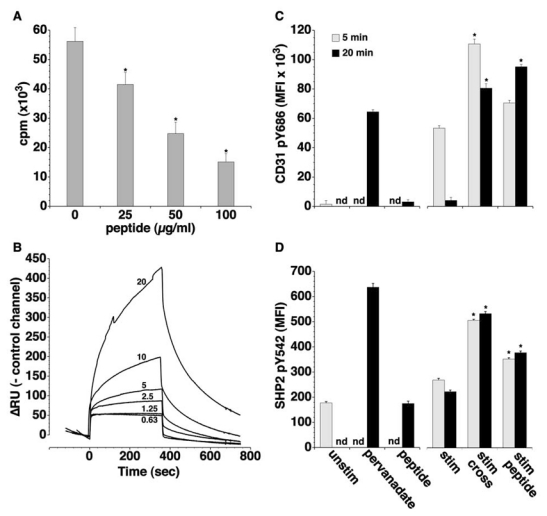
(A) Proliferative response to TCR engagement of human peripheral blood mononuclear cells in the presence of increasing doses of human CD31 peptide. *p<0.05 vs dose “0”. CD31 peptide inhibits cell proliferation in a dose-dependent manner. (B) Real-time (BIAcore®) analysis of the human CD31 peptide homophilic interaction at two-fold stepwise dilutions of the peptide (0.63, 1.25, 2.5, 5, 10 and 20 μg/ml). Data are normalized with respect to the control channel (scrambled peptide) and expressed as ΔRU (Resonance Units). (C,D) Flow cytometric assessment of human CD31 pY686 (C) and of SHP2 pY542 (D) on cultured Jurkat cells. Unstim= membrane lysate from untreated Jurkat cells, pervanadate= incubation with sodium pervanadate (Na3VO8) at 100μM for 20 minutes, peptide= incubation with the peptide alone (100μg/ml) for 20 minutes, stim= crosslinking of CD3 molecules by a monoclonal mouse anti-human CD3ε + goat-anti-mouse IgG F(ab′)2 fragments, stim/cross= crosslinking of CD3 with CD31 molecules via their domain 6, stim/peptide= crosslinking of CD3 in the presence of 100μg/ml human CD31 peptide. Quantification of CD31 pY686 was performed on solubilized membrane-bound CD31; SHP2 pY542 was analyzed by intracellular staining. Data are expressed as Median Fluorescent Intensity (MFI). nd= not determined. The crosslinking of the TCR alone and with CD31 molecules induces a rapid and transient phosphorylation of the CD31 inhibitory motifs (ITIM’s) while the peptide treatment is able to sustain phosphorylation for at least 20 minutes after the stimulation (*p<0.001 between “stim peptide” and “stim” at 20 minutes). In parallel, the peptide increases the SHP2 Y542 phosphorylation to an extent comparable to that observed with CD31 antibody-mediated crosslinking. *p<0.001 between “stim peptide” and “stim”.
To assess whether the homophilic peptide could engage the signaling of CD31 in spite of its shedding, we evaluated the level of phosphorylation of the CD31 ITIM tyrosine at position 686 and of the SHP2 tyrosine at position 542 in stimulated Jurkat cells (Figure 3C,D). Crosslinking of CD3ε alone induced a discrete increase in CD31 pY686, relative to the unstimulated control. However the presence of CD31 antibody directed against domain 6 or of the CD31 peptide (Figure 3C) during the first 5 minutes of stimulation induced a higher level of CD31 pY686 phosphorylation. After 20 minutes of stimulation phosphorylation was further increased in the presence of the CD31 peptide. In parallel, the peptide treatment led to a significant and durable increase in intracellular SHP2 Y542 phosphorylation induced by CD3ε cross-linking (Figure 3D), which was comparable to that obtained by antibody-mediated CD31 domain 6 crosslinking. The peptide alone did not change either tyrosine-phosphorylation levels as compared to unstimulated cells (Figure 3C,D).
The mouse CD31 peptide 551–574 binds to the homotypic sequence and inhibits lymphocyte activation in vitro and in vivo
To further test our hypothesis it was necessary to perform in vivo experiments and therefore we synthesized the murine equivalent of CD31 (551–574) peptide (NH2 - SSMRTSPRSSTLAVRVFLAPWKK - COOH) and evaluated its properties in vitro and in vivo. As the human equivalent, the murine CD31 peptide showed consistent homophilic interaction as analyzed by surface plasmon resonance (Figure 4A). In response to the co-engagement of CD3 and of the costimulatory molecule CD28 in spleen lymphocytes, the peptide was able to inhibit calcium mobilization to the same extent as CD31 cross-linking by specific monoclonal antibodies (Figure 4B). Furthermore, the peptide inhibited in a dose-dependent manner the proliferation of spleen cells in response to stimulation of the T-cell-receptor in vitro. At low doses, the effect of the peptide was exclusively due to its homophilic binding with CD31 molecules since no effect was observed on spleen cells from CD31−/− mice. However, at high concentrations the peptide can also bind to other (low affinity) heterophilic ligands as suggested by its effect at 100 μg/ml dose on cells lacking CD31. The scrambled peptide (NH2 - SMPAVRSRFSATSLVTLKSRWPK – COOH), used at the highest dose, was ineffective on both CD31+/+ and CD31−/− cells (Figure 4C). The immunosuppressive effect of the peptide was also documented in vivo, using a model of delayed type hypersensitivity. In this model the antigenic challenge is first exerted over the abdominal skin and the response is easily quantified after a second challenge, one week later, by measuring the thickness of the pinna of the ear (Figure 4D). The effect of the treatment was calculated as % suppression = (1−ΔTE/ΔTS) × 100 where ΔT= (ear thickness 24 hr after elicitation) – (baseline ear thickness), E=sensitized animals, S=treated animals (25). A minor reduction of the ear thickness (33±4%) was observed in the mice treated with 100μg of the scrambled peptide. The injection of 25, 50 and 100μg of peptide prior to the elicitation inhibited the DTH response by 48±5 (p<0.05 vs scrambled), 80±4 (p<0.001 vs scrambled) and 90±3% (p<0.001 vs scrambled), respectively. n=6 mice/group.
Figure 4. The mouse equivalent of the CD31 homotypic peptide inhibits T-cell responses in vitro and in vivo.
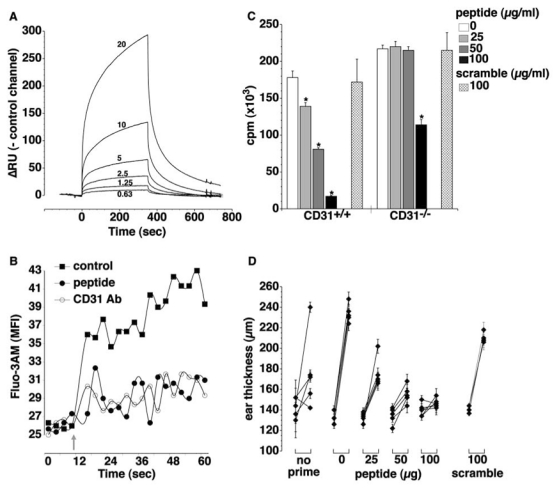
(A) BIAcore® analysis of the mouse CD31 peptide homophilic interaction at different concentrations (0.63, 1.25, 2.5, 5, 10 and 20 μg/ml). Real time binding is evaluated as for the equivalent human CD31 peptide detailed in Figure 3. Data are normalized against the control channels and expressed as ΔRU (Resonance Units). (B) Intracellular calcium mobilization induced by crosslinking of mouse CD3ε and CD28, determined by flow cytometry in Fluo-3AM®-loaded spleen cells. Data are expressed as Median Fluorescence Intensity (MFI) detected in the FITC channel (BP 530/30 nm). Grey arrow = addition of anti-CD3/CD28 antibodies and crosslinker alone (■=control) or together with either mouse CD31 peptide at 100μg/ml (●) or CD31 antibody, clone Mab390 (○). The peptide treatment inhibits, upon TCR stimulation, the intracellular calcium influx to the same extent as CD31 crosslinking. (C) Inhibitory effect of the mouse peptide on the proliferative response of CD31+/+ and CD31−/− spleen cells. Crisscross column= 100μg/ml of the scrambled peptide. *p<0.05 vs previous peptide dose. (D) Immunosuppressive effect of the mouse peptide in vivo on a model of delayed type hypersensitivity. Mice were treated with different doses of the peptide (0, 25, 50, 100 μg) or with a scramble peptide (100μg). The graphic shows individual right ear thickness (average of 5 measurements±SEM) of each mouse, before and 24 hours after elicitation. The control group of mice did not receive the priming (no prime).
Peptide treatment affects surface CD31 redistribution and clustering upon TCR stimulation
In order to evaluate whether the peptide acts by homo-oligomerization with the truncated CD31, we observed the CD31 redistribution on the membrane of TCR-stimulated cells by immunostaining the domain 6 of the molecule and TIRF microscopy. This technique allows high resolution visualization of processes that occur in the cell membrane. CD31 molecules, that were homogeneously distributed on the surface of unstimulated cells (Figure 5A), showed a modest redistribution and cluster formation upon TCR stimulation (Figure 5B). The crosslinking of TCR with the CD31 slightly increased cluster formation as compared to TCR stimulation alone (Figure 5C) while the peptide treatment led to larger clusters (Figure 5D). In order to visualize the localization of the peptide, we have treated the cells with a fluorescent peptide, in parallel conditions. Analysis of the fluorescence intensity profiles on lines drawn across the cell membrane showed that the peptide oligomerizes and localizes within CD31 clusters as suggested by the shift of the two fluorescence profiles (Figure 6E–F).
Figure 5. Peptide treatment affects surface CD31 redistribution and clustering upon TCR stimulation.
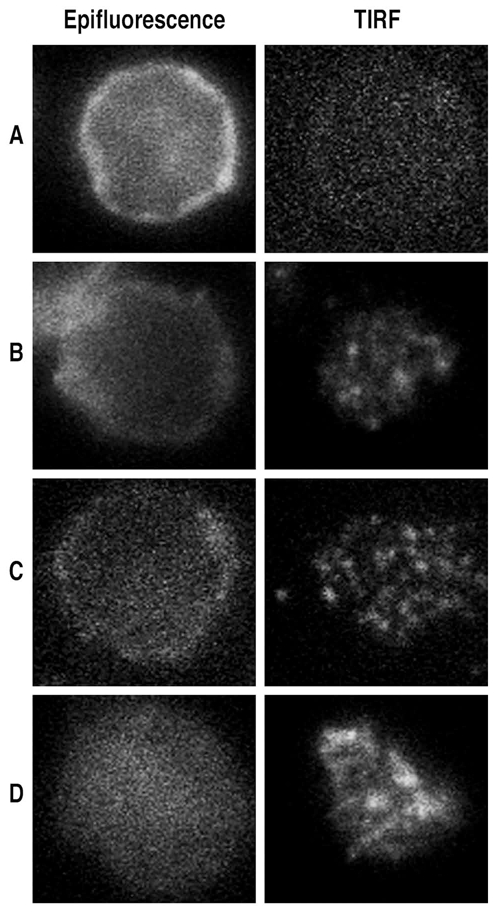
Epifluorescence and TIRF analysis of CD31 domain 6 membrane distribution on CD4+ Tcells isolated from peripheral blood. Cells were either were left in basal conditions (A) or stimulated by crosslinking the CD3 (B) in the presence of 10 μg/ml of anti-CD31 domain 6 antibody (clone MBC 78.2) directly coupled to AlexaFluor®546 (C) or 100μg/ml of human CD31 peptide (D). After 20 minutes all cells were fixed, rinsed and, for the conditions A, B and D, cells were stained with anti-CD31 domain 6 antibody (clone MBC 78.2) AlexaFluor®546-conjugated. Cells were then transferred to poly-D-lysine coated glass-bottom dishes and CD31 membrane clustering was visualized by TIRF. Antibody-mediated crosslinking of the CD31 domain 6 (C) did not change the appearance of the clustering spots as compared to T-cell stimulation alone (B). TIRF analysis of the cells stimulated in the presence of the peptide (D) showed larger clustering spots.
Figure 6. The CD31 peptide clusters on the plasma membrane and it accumulates between CD31 clusters upon TCR stimulation.
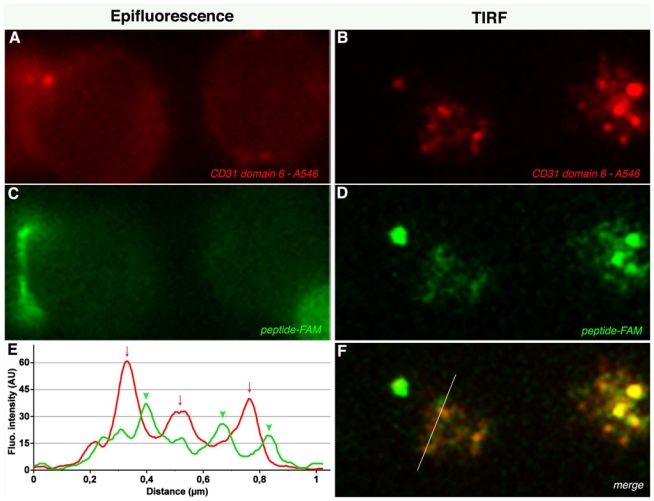
Epifluorescence and TIRF analysis of CD31 domain 6 (A, B) and of CD31 fluorescent peptide (5,6 FAM) (C, D) distribution on CD4+ T-cell membrane. Cells were isolated from peripheral blood and stimulated by crosslinking the CD3 molecules in presence of 100 μg/ml 5,6 FAM human CD31 peptide. Twenty minutes after stimulation cells were fixed, rinsed and stained with anti-CD31 domain 6 antibody (clone MBC 78.2) AlexaFluor®546-conjugated. (F) Merge of B and D. (E) Exemple of the analysis of fluorescence intensity profiles obtained by line-scans across the cell in the red and green channels. The x-axis is the scanning coordinate in μm, and the y-axis plots the fluorescence intensities in arbitrary units. Upon TCR stimulation, the CD31 domain 6 and the CD31 human peptide form clusters on the plasma membrane (panels B and D) and, as shown in panel E, CD31 domain 6 clusters (arrows, red line) localize next to and in alternation with peptide clusters (arrow heads, green line).
DISCUSSION
Experimental studies had previously shown that the CD31 inhibitory pathway mediated by the ITIMs is engaged during T-cell responses elicited by TCR stimulation (7). We have recently shown that the lack of T-cell CD31-signaling favors auto-immune responses in vitro and in vivo (24, 27) supporting a T-cell regulatory role for this transmembrane ITIM-bearing receptor (1). In this study we show that TCR stimulation drives the cleavage and shedding of the extracellular T-cell CD31 comprising Ig-like domains 1 to 5. We also show that the immunosuppressive CD31 peptide aa 551–574 (26, 28) is highly homophilic and probably acts by homo-oligomerizing with the truncated CD31 remaining after its cleavage/shedding.
Although CD31 is known to interact with heterophilic ligands on blood and vascular cells (29–32), the original feature of CD31 signaling resides in its trans- and cis-homophilic nature. According to the model proposed by Newton et al. (33), a low-affinity trans-homophilic engagement of the distal domains is necessary to drive the cis-homodimerization of the membrane-proximal portion of the molecule. This, in turn, would be the critical step for triggering the phosphorylation of the ITIMs, and the recruitment and activation of SH2-containing phosphatases. This hypothesis is supported by the fact that cross-linking of the CD31 molecule is necessary to induce the phosphorylation of ITIMs (34, 35) and inhibition of antigen-driven T-cell activation (7).
CD31 expression is constitutive but peripheral blood T-cells of the memory/activated phenotype lack this molecule at their surface (11, 13). We demonstrate here that the assumption that CD31 molecules are absent on activated T-lymphocytes is based on incomplete information. Indeed, the observed loss is due to a cleavage and shedding of the extracellular CD31 comprising domains 1–5. Since the membrane-proximal 6th domain remains anchored to all peripheral blood T-lymphocytes, we propose to use the term CD31shed rather that CD31 “negative” cells for the designation of those lymphocytes that are not detected by monoclonal antibodies directed towards the distal CD31 Ig-like domains. CD31 shedding occurs rapidly upon T-cell activation and leads to an accumulation of the truncated molecule in the supernatant. The same truncated form of CD31 is also specifically detected in human plasma and could serve as a biomarker of pathologic T-cell activation.
We propose that the broad overlap of values between patients and controls in the previous studies concerning the predictive value of soluble CD31 levels (36–40) is due to the fact that i) circulating CD31 represents a mixture of the transmembraneless and the truncated form and ii) it was not possible, until now, to discriminate between the two soluble forms of CD31.
In a therapeutic perspective, it is therefore necessary to find alternative strategies to rescue CD31-mediated T-cell regulation in situations in which pathogenic T-cells have already undergone activation and hence CD31 shedding.
Previously, it had been documented that the presence of antibodies targeting the 23 juxta-membrane amino acid sequence of CD31 (LYP21) inhibited the mixed lymphocyte reaction (MLR) in a specific and dose-dependent manner (26). Intriguingly, a synthetic 23-mer peptide corresponding to the epitope of the LYP21 antibody (aa 551–574) equally inhibited mixed lymphocyte reactions, which showed dispersed small aggregates of cells, rather than the single large aggregate observed in control mixed lymphocyte reactions (26). Thus it appears that the CD31 peptide is able to reproduce the physiologic active detachment signal which is mutually driven in live leukocytes by CD31-CD31 interactions (6). However, stimulated T-cells typically lose CD31 at their surface (13) and Zehnder et al. demonstrated that the peptide was effective also on CD31 negative enriched T cells (26). As a consequence, the mechanism involved in the immunosuppressive effect exerted by the CD31 peptide 551–574 was left unresolved.
The present study demonstrates that the extracellular portion of CD31 recognized by MBC 78.2/PECAM-1.2 antibody [domain 6 (23)], which is located N-terminal of the aa 551–574 (26), remains expressed on CD31shed T cells. We have also documented that the CD31 551–574 immunosuppressive peptide is highly homophilic. We therefore hypothesized that it can homo-oligomerize with the truncated CD31 on CD31shed cells and thus acts by mimicking the second step of the CD31 homophilic engagement described by Newton et al (33): the cis-homo-dimerization of the molecule. Indeed, the analysis of the CD31 domain 6 distribution showed that the peptide clusters localize in between CD31 domain 6 clusters. We thus postulate that the peptide rescues the CD31-ITIM inhibitory signaling in CD31shed T-cells by “bridging” the distant truncated CD31 molecules over the cell surface. This hypothesis is supported by the fact that either antibody-mediated CD31 domain 6 crosslinking or treatment with the CD31 551–574 peptide durably enhanced the phosphorylation of CD31 ITIMs and of SHP2 of T-cells and inhibited their TCR-induced activation and proliferation in vitro. Our data cannot rule out that the peptide clusters may exert other and as yet unknown effects in T-cells. Of note, we also show that the CD31 peptide inhibits antigen-induced T-cell responses in vivo (in the delayed type hypersensitivity model) pointing to a potential immunosuppressive therapeutic effect for this peptide in chronic inflammatory diseases.
In conclusion, we demonstrate that, upon cell activation, the loss of T-cell CD31 is due to its cleavage and shedding of the truncated portion of its ectodomain into biological fluids. CD31 shedding results in the loss of its inhibitory function, as the necessary trans-homophilic engagement of the molecule cannot be established by CD31shed cells because they lack the distal Ig-like domain 1. However, cis-homo-oligomerization, which is induced by the trans-homophilic engagement of the molecule, is the key event in CD31 inhibitory signal transduction. It appears therefore possible to use a homophilic peptide or an appropriate peptidomimetic compound to mimic the cis-homo-oligomerization step and rescue the lost physiological immunoregulatory function of CD31 on activated T-cells. Such a therapeutic approach may be useful for treating debilitating chronic immuno-inflammatory diseases in which CD31-signaling is suspected to play an important protective role, such as in rheumatoid arthritis (9), multiple sclerosis (8), inflammatory liver disease (41) and atherothrombosis (24, 27, 42).
Acknowledgments
This work was supported in part by grants from the “Fondation de France” (Engt 2006-005656 and 2008-002724), the “Fondation pour la Recherche Médicale” (DCV20070409268) and the “Agence Nationale de la Recherche” (project “RELATE” and project “BROSCI”). G.F. is the recipient of a training grant from the “Ministère affaires étrangères” (Egide N°636511F) and of the “Groupe de Reflexion sur la Recherche Cardio-vasculaire et la Féderation Française de Cardiologie”. E.G. was the recipient of a research grant from the “Fondation pour la Recherche Médicale” (FDT20071211595).
We thank Drs Mary Osborne-Pellegrin (Inserm U698) and Frans Nauwelaers (BD Biosciences Europe, Scientific Affairs) for their help in editing the manuscript, Sébastien Lacroix-Desmazes (Inserm U872) for his help with the BIAcore® analysis, Pascal Roux (Plate forme d’imagerie dynamique-Imagopole, Institut Pasteur) for the TIRF acquisition.
Nonstandard abbreviations
- DTH
Delayed Type Hypersensitivity
- CBA
cytometric bead array
- TNCB
2-chloro-1,3,5-trinitrobenzene
References
- 1.Newman PJ. Switched at birth: a new family for PECAM-1. J Clin Invest. 1999;103:5–9. doi: 10.1172/JCI5928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Newman PJ, Berndt MC, Gorski J, White GC, 2nd, Lyman S, Paddock C, Muller WA. PECAM-1 (CD31) cloning and relation to adhesion molecules of the immunoglobulin gene superfamily. Science. 1990;247:1219–1222. doi: 10.1126/science.1690453. [DOI] [PubMed] [Google Scholar]
- 3.Muller WA, Weigl SA, Deng X, Phillips DM. PECAM-1 is required for transendothelial migration of leukocytes. J Exp Med. 1993;178:449–460. doi: 10.1084/jem.178.2.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bird IN, Spragg JH, Ager A, Matthews N. Studies of lymphocyte transendothelial migration: analysis of migrated cell phenotypes with regard to CD31 (PECAM-1), CD45RA and CD45RO. Immunology. 1993;80:553–560. [PMC free article] [PubMed] [Google Scholar]
- 5.Tada Y, Koarada S, Morito F, Ushiyama O, Haruta Y, Kanegae F, Ohta A, Ho A, Mak TW, Nagasawa K. Acceleration of the onset of collagen-induced arthritis by a deficiency of platelet endothelial cell adhesion molecule 1. Arthritis Rheum. 2003;48:3280–3290. doi: 10.1002/art.11268. [DOI] [PubMed] [Google Scholar]
- 6.Brown S, Heinisch I, Ross E, Shaw K, Buckley CD, Savill J. Apoptosis disables CD31-mediated cell detachment from phagocytes promoting binding and engulfment. Nature. 2002;418:200–203. doi: 10.1038/nature00811. [DOI] [PubMed] [Google Scholar]
- 7.Newton-Nash DK, Newman PJ. A new role for platelet-endothelial cell adhesion molecule-1 (CD31): inhibition of TCR-mediated signal transduction. J Immunol. 1999;163:682–688. [PubMed] [Google Scholar]
- 8.Graesser D, Solowiej A, Bruckner M, Osterweil E, Juedes A, Davis S, Ruddle NH, Engelhardt B, Madri JA. Altered vascular permeability and early onset of experimental autoimmune encephalomyelitis in PECAM-1-deficient mice. J Clin Invest. 2002;109:383–392. doi: 10.1172/JCI13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wong MX, Hayball JD, Hogarth PM, Jackson DE. The inhibitory co-receptor, PECAM-1 provides a protective effect in suppression of collagen-induced arthritis. J Clin Immunol. 2005;25:19–28. doi: 10.1007/s10875-005-0354-7. [DOI] [PubMed] [Google Scholar]
- 10.Goel R, Schrank BR, Arora S, Boylan B, Fleming B, Miura H, Newman PJ, Molthen RC, Newman DK. Site-specific effects of PECAM-1 on atherosclerosis in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2008;28:1996–2002. doi: 10.1161/ATVBAHA.108.172270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stockinger H, Schreiber W, Majdic O, Holter W, Maurer D, Knapp W. Phenotype of human T cells expressing CD31, a molecule of the immunoglobulin supergene family. Immunology. 1992;75:53–58. [PMC free article] [PubMed] [Google Scholar]
- 12.Thiel A, Schmitz J, Miltenyi S, Radbruch A. CD45RA-expressing memory/effector Th cells committed to production of interferon-gamma lack expression of CD31. Immunol Lett. 1997;57:189–192. doi: 10.1016/s0165-2478(97)00056-4. [DOI] [PubMed] [Google Scholar]
- 13.Demeure CE, Byun DG, Yang LP, Vezzio N, Delespesse G. CD31 (PECAM-1) is a differentiation antigen lost during human CD4 T-cell maturation into Th1 or Th2 effector cells. Immunology. 1996;88:110–115. doi: 10.1046/j.1365-2567.1996.d01-652.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kohler S, Thiel A. Life after the thymus: CD31+ and CD31- human naive CD4+ T-cell subsets. Blood. 2009;113:769–774. doi: 10.1182/blood-2008-02-139154. [DOI] [PubMed] [Google Scholar]
- 15.Goldberger A, Middleton KA, Oliver JA, Paddock C, Yan HC, DeLisser HM, Albelda SM, Newman PJ. Biosynthesis and processing of the cell adhesion molecule PECAM-1 includes production of a soluble form. J Biol Chem. 1994;269:17183–17191. [PubMed] [Google Scholar]
- 16.Zehnder JL, Hirai K, Shatsky M, McGregor JL, Levitt LJ, Leung LL. The cell adhesion molecule CD31 is phosphorylated after cell activation. Down-regulation of CD31 in activated T lymphocytes. J Biol Chem. 1992;267:5243–5249. [PubMed] [Google Scholar]
- 17.Ilan N, Mohsenin A, Cheung L, Madri JA. PECAM-1 shedding during apoptosis generates a membrane-anchored truncated molecule with unique signaling characteristics. Faseb J. 2001;15:362–372. doi: 10.1096/fj.00-0372com. [DOI] [PubMed] [Google Scholar]
- 18.Yun PL, Decarlo AA, Chapple CC, Hunter N. Functional implication of the hydrolysis of platelet endothelial cell adhesion molecule 1 (CD31) by gingipains of Porphyromonas gingivalis for the pathology of periodontal disease. Infect Immun. 2005;73:1386–1398. doi: 10.1128/IAI.73.3.1386-1398.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garton KJ, Gough PJ, Philalay J, Wille PT, Blobel CP, Whitehead RH, Dempsey PJ, Raines EW. Stimulated shedding of vascular cell adhesion molecule 1 (VCAM-1) is mediated by tumor necrosis factor-alpha-converting enzyme (ADAM 17) J Biol Chem. 2003;278:37459–37464. doi: 10.1074/jbc.M305877200. [DOI] [PubMed] [Google Scholar]
- 20.Mendez MP, Morris SB, Wilcoxen S, Greeson E, Moore B, Paine R., 3rd Shedding of soluble ICAM-1 into the alveolar space in murine models of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2006;290:L962–970. doi: 10.1152/ajplung.00352.2005. [DOI] [PubMed] [Google Scholar]
- 21.Eugenin EA, Gamss R, Buckner C, Buono D, Klein RS, Schoenbaum EE, Calderon TM, Berman JW. Shedding of PECAM-1 during HIV infection: a potential role for soluble PECAM-1 in the pathogenesis of NeuroAIDS. J Leukoc Biol. 2006;79:444–452. doi: 10.1189/jlb.0405215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fawcett J, Buckley C, Holness CL, Bird IN, Spragg JH, Saunders J, Harris A, Simmons DL. Mapping the homotypic binding sites in CD31 and the role of CD31 adhesion in the formation of interendothelial cell contacts. J Cell Biol. 1995;128:1229–1241. doi: 10.1083/jcb.128.6.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yan HC, Pilewski JM, Zhang Q, DeLisser HM, Romer L, Albelda SM. Localization of multiple functional domains on human PECAM-1 (CD31) by monoclonal antibody epitope mapping. Cell Adhes Commun. 1995;3:45–66. doi: 10.3109/15419069509081277. [DOI] [PubMed] [Google Scholar]
- 24.Caligiuri G, Groyer E, Khallou-Laschet J, Al Haj Zen A, Sainz J, Urbain D, Gaston AT, Lemitre M, Nicoletti A, Lafont A. Reduced immunoregulatory CD31+ T cells in the blood of atherosclerotic mice with plaque thrombosis. Arterioscler Thromb Vasc Biol. 2005;25:1659–1664. doi: 10.1161/01.ATV.0000172660.24580.b4. [DOI] [PubMed] [Google Scholar]
- 25.Shevach EM, Coligan JE, Bierer B, Margulies DH. Vivo Assays for Lymphocyte Function 2001;Chapter 4 [Google Scholar]
- 26.Zehnder JL, Shatsky M, Leung LL, Butcher EC, McGregor JL, Levitt LJ. Involvement of CD31 in lymphocyte-mediated immune responses: importance of the membrane-proximal immunoglobulin domain and identification of an inhibiting CD31 peptide. Blood. 1995;85:1282–1288. [PubMed] [Google Scholar]
- 27.Caligiuri G, Rossignol P, Julia P, Groyer E, Mouradian D, Urbain D, Misra N, Ollivier V, Sapoval M, Boutouyrie P, Kaveri SV, Nicoletti A, Lafont A. Reduced immunoregulatory CD31+ T cells in patients with atherosclerotic abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. 2006;26:618–623. doi: 10.1161/01.ATV.0000200380.73876.d9. [DOI] [PubMed] [Google Scholar]
- 28.Chen Y, Schlegel PG, Tran N, Thompson D, Zehnder JL, Chao NJ. Administration of a CD31-derived peptide delays the onset and significantly increases survival from lethal graft-versus-host disease. Blood. 1997;89:1452–1459. [PubMed] [Google Scholar]
- 29.Muller WA, Berman ME, Newman PJ, DeLisser HM, Albelda SM. A heterophilic adhesion mechanism for platelet/endothelial cell adhesion molecule 1 (CD31) J Exp Med. 1992;175:1401–1404. doi: 10.1084/jem.175.5.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vernon-Wilson EF, Aurade F, Brown SB. CD31 promotes beta1 integrin-dependent engulfment of apoptotic Jurkat T lymphocytes opsonized for phagocytosis by fibronectin. J Leukoc Biol. 2006;79:1260–1267. doi: 10.1189/jlb.1005571. [DOI] [PubMed] [Google Scholar]
- 31.Wong CW, Wiedle G, Ballestrem C, Wehrle-Haller B, Etteldorf S, Bruckner M, Engelhardt B, Gisler RH, Imhof BA. PECAM-1/CD31 trans-homophilic binding at the intercellular junctions is independent of its cytoplasmic domain; evidence for heterophilic interaction with integrin alphavbeta3 in Cis. Mol Biol Cell. 2000;11:3109–3121. doi: 10.1091/mbc.11.9.3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sachs UJ, Andrei-Selmer CL, Maniar A, Weiss T, Paddock C, Orlova VV, Choi EY, Newman PJ, Preissner KT, Chavakis T, Santoso S. The neutrophil-specific antigen CD177 is a counter-receptor for platelet endothelial cell adhesion molecule-1 (CD31) J Biol Chem. 2007;282:23603–23612. doi: 10.1074/jbc.M701120200. [DOI] [PubMed] [Google Scholar]
- 33.Newton JP, Buckley CD, Jones EY, Simmons DL. Residues on both faces of the first immunoglobulin fold contribute to homophilic binding sites of PECAM-1/CD31. J Biol Chem. 1997;272:20555–20563. doi: 10.1074/jbc.272.33.20555. [DOI] [PubMed] [Google Scholar]
- 34.Couty JP, Rampon C, Leveque M, Laran-Chich MP, Bourdoulous S, Greenwood J, Couraud PO. PECAM-1 engagement counteracts ICAM-1-induced signaling in brain vascular endothelial cells. J Neurochem. 2007;103:793–801. doi: 10.1111/j.1471-4159.2007.04782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Newman PJ, Newman DK. Signal transduction pathways mediated by PECAM-1: new roles for an old molecule in platelet and vascular cell biology. Arterioscler Thromb Vasc Biol. 2003;23:953–964. doi: 10.1161/01.ATV.0000071347.69358.D9. [DOI] [PubMed] [Google Scholar]
- 36.Khare A, Shetty S, Ghosh K, Mohanty D, Chatterjee S. Evaluation of markers of endothelial damage in cases of young myocardial infarction. Atherosclerosis. 2005;180:375–380. doi: 10.1016/j.atherosclerosis.2004.12.040. [DOI] [PubMed] [Google Scholar]
- 37.Wei H, Fang L, Chowdhury SH, Gong N, Xiong Z, Song J, Mak KH, Wu S, Koay E, Sethi S, Lim YL, Chatterjee S. Platelet-endothelial cell adhesion molecule-1 gene polymorphism and its soluble level are associated with severe coronary artery stenosis in Chinese Singaporean. Clin Biochem. 2004;37:1091–1097. doi: 10.1016/j.clinbiochem.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 38.Khare A, Shetty S, Ghosh K, Mohanty D, Chatterjee S. Evaluation of markers of endothelial damage in cases of young myocardial infarction. Atherosclerosis. 2005;180:375–380. doi: 10.1016/j.atherosclerosis.2004.12.040. [DOI] [PubMed] [Google Scholar]
- 39.Kuenz B, Lutterotti A, Khalil M, Ehling R, Gneiss C, Deisenhammer F, Reindl M, Berger T. Plasma levels of soluble adhesion molecules sPECAM-1, sP-selectin and sE-selectin are associated with relapsing-remitting disease course of multiple sclerosis. J Neuroimmunol. 2005;167:143–149. doi: 10.1016/j.jneuroim.2005.06.019. [DOI] [PubMed] [Google Scholar]
- 40.Figarella-Branger D, Schleinitz N, Boutiere-Albanese B, Camoin L, Bardin N, Guis S, Pouget J, Cognet C, Pellissier JF, Dignat-George F. Platelet-endothelial cell adhesion molecule-1 and CD146: soluble levels and in situ expression of cellular adhesion molecules implicated in the cohesion of endothelial cells in idiopathic inflammatory myopathies. J Rheumatol. 2006;33:1623–1630. [PubMed] [Google Scholar]
- 41.Goel R, Boylan B, Gruman L, Newman PJ, North PE, Newman DK. The proinflammatory phenotype of PECAM-1-deficient mice results in atherogenic diet induced- steatohepatitis. Am J Physiol Gastrointest Liver Physiol. 2007;293:G1205–1214. doi: 10.1152/ajpgi.00157.2007. [DOI] [PubMed] [Google Scholar]
- 42.Groyer E, Nicoletti A, Ait-Oufella H, Khallou-Laschet J, Varthaman A, Gaston AT, Thaunat O, Kaveri SV, Blatny R, Stockinger H, Mallat Z, Caligiuri G. Atheroprotective effect of CD31 receptor globulin through enrichment of circulating regulatory T-cells. J Am Coll Cardiol. 2007;50:344–350. doi: 10.1016/j.jacc.2007.04.040. [DOI] [PubMed] [Google Scholar]