

Abstract
The ability to obtain sequence-specific genetic information about rare target organisms directly from complex biological samples at the point of care would transform many areas of biotechnology. Microfluidics technology offers compelling tools for integrating multiple biochemical processes in a single device, but despite significant progress, only limited examples have shown specific, genetic analysis of clinical samples within the context of a fully integrated, portable platform. Herein we present the Magnetic Integrated Microfluidic Electrochemical Detector (MIMED) that integrates sample preparation and electrochemical sensors in a monolithic disposable device to detect RNA-based virus directly from patient samples. By combining immunomagnetic target capture, concentration and purification, reverse-transcriptase polymerase chain reaction (RT-PCR) and single-stranded DNA (ssDNA) generation in the sample preparation chamber, as well as sequence specific electrochemical DNA detection in the electrochemical cell, we demonstrate the detection of influenza H1N1 in throat swab samples at loads as low as 10 TCID50 - 4 orders of magnitude below the clinical titer for this virus. Given the availability of affinity reagents for a broad range of pathogens, our system offers a general approach for multi-target diagnostics at the point-of-care.
Keywords: Point of Care Diagnostics, Electrochemical Sensing, Pathogen Detection, Integrated Microfluidics, Integrated Sample Preparation
Introduction
There exists a general need for technologies that enable sensitive, accurate, and sequence-specific genetic detection of rare target organisms (e.g., viruses, bacteria or mammalian cells) within complex biological samples for a broad variety of biotechnology applications, including forensics,1 food safety,2 environmental monitoring3 and clinical diagnostics4–7 at the point-of-care (POC). Specifically, due to the low titers of target organisms and the complexity of clinical samples, direct detection is met with severe technical challenges.8 For example, influenza tests from untreated throat and nasopharyngeal swabs typically contain sample-degrading nucleases, PCR inhibitors, and aggregating factors.9–12 For E. coli O157:H7 stool sample tests, pathogen levels typically fall below ~105 colony-forming units per milliliter (CFU mL−1) and exist in a mixture of a background of non-pathogenic strains, PCR inhibitors and cellular debris.13–15
Thus, it is apparent that effective sample preparation, including concentration and purification of target species from complex backgrounds, holds the key for direct molecular detection at the POC. Furthermore, in order to minimize sample loss and achieve rapid detection, it is imperative to integrate sample preparation with the detection assay in a single device. Toward this end, a number of groups have explored the use of microfluidics technology as a means for integrating sample preparation, genetic amplification and molecular readout.16–18 However, this goal has proven to be technically challenging to achieve, and only a few limited examples have reported the sequence-specific genetic analysis of target species at relevant concentrations, directly from unprocessed patient samples.19–21 This rings especially true for electrochemical-based platforms, which are well suited for POC applications due to the portability, robustness, and integration with circuitry.22–25
Towards a universal solution for electrochemical sequence specific genetic detection at the point of care, we present here the Magnetic Integrated Microfluidic Electrochemical Detector (MIMED) (Fig. 1). This system integrates high-throughput immunomagnetic target capture, concentration, and purification, efficient on-chip reverse-transcriptase polymerase chain reaction (RT-PCR), single-stranded DNA (ssDNA) generation, and sequence specific electrochemical detection – all in an integrated, monolithic device. By taking advantage of the multifuncational sample preparation chamber which enables high throughput target enrichment, the prevention of non-specific enzyme adsorption, high PCR efficiency, and lossless integration with the target specifc signaling probe, this system can be configured to detect a wide range of RNA or DNA-based pathogens in unprocessed samples. As a model, we demonstrate the specific, genetic detection of H1N1 viruses directly from throat swabs within 3.5 hours and a limit of detection (LOD) of 10 TCID50 - approximately four orders of magnitude below the clinically relevant infectious dose.
Figure 1. Sample-to-answer genetic analysis of H1N1 virus.

(A) The 1×6 cm device features three fluidic ports: sample/buffer/reagent input (left), waste output (center) and E-DNA product output (right). Capture, RT-PCR and ssDNA generation are performed in the sample prep chamber; detection is performed in the electrochemical DNA detection cell. (B) A throat swab is collected and combined with influenza virus and antibody-coated magnetic beads in a tube containing RNA stabilizer. (C) The sample is pumped into the device where external magnets capture, concentrate and purify labeled viral RNP in the sample prep chamber. (D) RT-PCR mix is injected. (E) The chip is heated to denature the RNP and release the RNA. (F-G) RT-PCR is performed on-chip followed by lambda exonuclease-mediated ssDNA generation. (H) Product is pumped into the DNA detection cell, where hybridization is measured via AC voltammetry.
Experimental Section
Quantifying viral enrichment from swab samples
To compare the effects of sample prep, each experiment was replicated in triplicate in three separate one-time-use devices. Positive controls were prepared by adding 1 μL of antibody-coated beads (106) and 1 μL of viral particles (104 TCID50) to 50 μL of standard RT-PCR mixture plus 1X SYBR green. ‘Direct detection’ samples were prepared likewise, with a throat swab first added to the mix for 5 minutes. The MIMED ‘complete prep’ samples were conducted by adding a throat swab to 1 mL of 10% RNAlater solution for 5 minutes followed by addition of 1 μL of antibody-coated beads and 1 μL of viral particles. This solution was incubated for 30 min at 4 °C, then pumped through the device at 60 mL h−1 with permanent magnets applied, followed by a 1 mL wash with PBS at the same flow rate. The magnets were then removed and the beads were eluted with 50 μL of RT-PCR mixture. ‘Magnetic Particle Concentrator’ (MPC) samples were processed with 5 min of magnetic capture time in place of the above flow rates. ‘No concentration’ samples were prepared like the ‘complete prep’ samples but by stopping sample flow after the trapping chamber became full to prevent concentration. ‘No RNA stabilization’ samples were prepared with initial incubation in PBS rather than 10% RNAlater. ‘No wash’ samples were prepared with RT-PCR mix elution immediately following capture, thereby excluding the wash step. Additionally, in order to isolate the effects of eliminating washing without the PCR inhibitory effects of residual RNAlater or the degradation effects arising from absence of RNAlater, viral particles were added after completion of the ‘no wash’ MIMED sample prep. Negative controls were prepared like the MIMED samples but without adding viral particles. Quantitative RT-PCR was run with the same parameters as in the MIMED assay but with 55 cycles to enable recalcitrant samples to cross the threshold level.
Viral sample preparation
Influenza A/PR/8/34 H1N1 was propagated on MDCK (Madin- Darby Canine Kidney) cells and virus-containing supernatants were harvested when 80% of cells showed cytopathic effects. The supernatants were clarified twice by centrifugation at 4 ºC and then stored in aliquots at −80 ºC. Viral titers were determined by measuring TCID50 using MDCK cells.26 The influenza viral RNA exists in a native complex with the viral nucleoprotein, known as the ribonucleoprotein (RNP). We exploit this RNP for high-efficiency target RNA capture via the anti-influenza A nucleoprotein antibody. The lipid envelope of the harvested virus is disrupted to release the intact RNP by diluting virus-containing supernatant (108 TCID50 mL−1) 10-fold in buffer containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA and 2% NP-40 (Sigma Aldrich, St. Louis, MO).27 This step allows for safe handling and improves the access of the antibody to the target nucleoprotein, thereby increasing the efficiency of the target RNA capture. The mixture was incubated at 4 ºC for 1 h and then stored at −80 ºC in aliquots.
Viral RNP capture
We thawed the stock virus and diluted it to the desired concentration with 1X PBS. 1 μL of this dilution was added to capture buffer, followed by 5 μL (~107) of streptavidin-coated magnetic beads (diameter = 1 μm) conjugated with biotinylated anti-influenza A nucleoprotein. Samples were rotated for 30 min at 4 ºC. For initial MIMED system tests, 1 mL 1X PBS was used as capture buffer, while the simulated patient samples used 10% RNAlater (Qiagen, Valencia, CA) in 1X PBS as the capture buffer to mitigate RNA degradation. Throat swabs were collected from a healthy donor with flocked nylon swabs (VWR LabShop, Batavia, IL) and incubated in 50 μL of capture buffer for 5 minutes prior to the addition of virus and beads. After incubation, the device chamber was placed above six permanent NeFeB magnets (K&J Magnetics, Inc., Jamison, PA) while the sample solution was pumped through the chip at 60 mL h−1 via a syringe pump (Next Advance Inc., Averill Park, NY). The trapped beads were then washed by flowing 1 mL of 1X PBS through the chamber at 60 mL h−1 to remove interferents. Magnetic separation is effective for a wide range of biological targets,28,29 and this approach has proven particularly advantageous for achieving high-throughput capture with minimal loss within the context of a microfluidic channel.30–33
Magnetic trapping simulations
In the sample-prep chamber, the Reynolds number (Re) was calculated to be ~1 at a flow rate (Q) of 60 mL h−1. Thus, the magnetic beads would experience a Stokes drag force of Fd = 6πηa(vf − vp), where a is the bead diameter, and vf and vp are the velocities of the fluid and particle respectively. When a strong magnetic field (B) is applied via external neodymium iron boron (NeFeB) permanent magnets, the magnetic force exerted on the bead is taken as Fm = (4/3)πr3ρM∇B, where r is the radius, ρ is the density and M is the saturation magnetization of the bead (~23.5 Am2 kg−1).34 The governing equations of magnetostatics and incompressible flow were solved to yield the magnetic force and velocity fields. By balancing the magnetic and drag forces on a given bead, the transport equations were solved, yielding the bead concentration profile through the channel. The minimum bead residence time necessary for 100% capture was expressed as the time required for a bead to translate to the capture plane from the opposing channel. The drag on an immobilized bead was estimated as the Stokes drag force with a velocity differential equal to the flow speed one bead radius away from the channel surface.
To test the agreement between simulations of capture efficiency and experimental results, we pumped suspensions of phycoerythrin-labeled beads in 1X PBS (107 beads mL−1) through the chip at 6.0, 60 or 600 mL h−1, followed by washing at the same flow-rate, to measure bead retention. The beads eluted at the outlet with or without magnets were measured in triplicate by flow cytometry (BD FACSAria, NJ). Capture efficiency was calculated based on the number of beads trapped as normalized against the counts from the non-magnetized control. This does not include beads that may have been lost in the interfacing common to both groups; however, such loss was measured as < 2%.
RT-PCR and ssDNA generation
We injected 50 μL RT-PCR mix (OneStep RT-PCR kit, Qiagen, Valencia, CA) containing RT-PCR buffer (10 μL, 5X initial), a phosphorylated forward primer and standard reverse primer (3 μL each, 10 μM initial), dNTP (2 μL, 10 mM initial), enzyme (2 μL, 25X initial) and deionized water (remaining volume) into the chamber over the trapped virus. The chip was mounted onto a thermoelectric cooler (TEC, Custom Thermoelectric, Bishopville, MD) linked to a PID controller (Omega Engineering, Inc., Stamford, CT), which was heated to 50 ºC for 30 minutes to denature protein-RNA complexes. The sample was then subjected to a 15 min hot start at 95 ºC followed by 38 cycles of 95, 55, and 72 ºC with 30 s dwells and average ramp rate of ~1 ºC s−1. Following amplification, ssDNA was generated. The PCR product solution was mixed 10:1 with 10X lambda exonuclease enzyme stock (New England Biolabs, Ipswich, MA) and incubated in the reaction chamber for 20 min at 37 ºC directly with no purification steps. The ssDNA generation efficiency was measured by fluorescence (Gel Logic EDAS 200, Kodak, Rochester, NY). Fluid transport was conducted either manually via syringes or via automated syringe pump (PhD 2000, Harvard Apparatus, Holliston, MA). Mixing was facilitated by introducing reagents into the device in pre-loaded syringes and pumping back and forth. Additional fluid volume exceeding the device chamber capacity was simply retained in the syringe. Exploiting the syringes as mixing chambers exhibited efficiency indistinguishable from traditional pipette-aided mixing in a tube, and negated the need for on-chip mixers, thereby significantly decreasing the complexity and cost of the disposable chip, increasing the value at the POC.
On-Chip E-DNA measurements
The working electrodes of the electrochemical detection cell feature DNA oligonucleotide probes complementary to the 20-base-pair region in Segment 7 of Influenza A/PR/8/34/H1N1, which have been immobilized via gold-thiol chemistry. All voltammetric scans were conducted in the E-DNA cell in the presence of 1X high-salt incubation E-DNA buffer (HSIEB, 1.5 M NaCl, 100 mM phosphate, 1 mM Mg2+) to maintain consistent salt concentration and pH. To establish baseline signals, the DNA detection cell was flushed with 1X HSIEB and Alternating Current Voltammetry (ACV) scans were taken prior to sample injection. Subsequently, the PCR product was drawn into a syringe containing an equal volume of 2X HSIEB, mixed, and injected into the DNA detection cell for hybridization with probes for 30 minutes, at room temperature after which ACV signals were measured. Finally, the E-DNA probes were regenerated by pumping 1 mL of 50 mM NaOH followed by 5 mL of deionized water through the cell, and the sensor was scanned again in the presence of 1X HSIEB. ACV was performed between −0.7 V and −0.2 V, at a frequency of 100 Hz, an amplitude of 10 mV and sensitivity of 500 nA.
Results and Discussion
MIMED system overview
The entire influenza H1N1 detection was performed within a single MIMED device from throat swab samples. The MIMED device is microfabricated with PDMS and glass materials and contains two physical modules: the sample-prep chamber (35 μL) and electrochemical DNA detection cell (7 μL) (Fig. 1A). Briefly, the target capture, concentration and purification, RT-PCR amplification and ssDNA generation are performed within the sample-prep chamber. This chamber is designed for (1) high throughput magnetic capture enabled by reduced drag and large magnetic field gradients, (2) the prevention of non-specific enzyme adsorption by the incorporation of low PDMS surface area35,36 without ferromagnetic structures37,33 and (3) high PCR efficiency enabled by uniform heating across the low-aspect ratio channel. The amplicon detection is achieved by the E-DNA probes in the DNA detection cell. The E-DNA probes undergo specific target binding-induced conformation change,37,38 and the detection cell is designed for seamless integration with the sample preparation chamber without the need for intermediate purification steps. The cell contains platinum counter (CE) and reference (RE) electrodes, and two gold working (WE) for duplicate measurement of E-DNA probe signal. The details of the device and probe fabrication are provided in the supporting information (Fig. S1).
MIMED-based H1N1 virus genetic analysis
To mimic a clinical sample of known viral load, each swab was obtained from a healthy donor and combined with the desired viral titer in vial of RNA stabilization medium with antibody-coated magnetic beads. Influenza RNA exists in a stable complex with the nucleoprotein, known as the ribonucleoprotein (RNP). Thus, to capture the target RNA, we capture the RNP complex via anti-nucleoprotein antibody (Fig. 1B). After the beads/sample incubation, the sample is injected into the MIMED sample preparation chamber where magnetic forces capture and concentrate the magnetically-labeled target. During trapping, we remove interferents from the swab sample by continuous washing in the microchamber (Fig. 1C). Next we inject RT-PCR mix containing a phosphorylated primer into the chamber (Fig. 1D). The RNP complex is thermally denatured to enable reverse transcription of the captured RNA target (Fig. 1E). The newly obtained complementary DNA (cDNA) target is then PCR amplified to yield up to a ~300 nM amplicon concentration within the microchamber (Fig. 1F). We selectively digest the phosphorylated strands40 from the dsDNA amplicons with lambda exonuclease41 to obtain ssDNA necessary for subsequent sequence-specific detection (Fig. 1G). Finally, this ssDNA product is mixed with high-salt buffer and delivered to the DNA detection cell to hybridize with a redox-labeled, electrode-bound E-DNA signaling probe (Fig. 1H). Target hybridization induces a conformational change in the E-DNA probe, forcing the redox label away from the WE, decreasing faradic current. The relative current change due to the amplified target DNA corresponds to the initial viral quantity in the samples. To verify that the signal was the result of target hybridization, we flushed dehybridization buffer through the cell, removing the target and regenerating the sensor.
Viral RNA capture, concentration and purification from swab samples
Clinical samples usually contain a low concentration of target among a high concentration of background including target-degrading components and interferents, which inhibit downstream enzymatic processes.9–12 Thus rare target detection from unprocessed patient samples at the POC requires efficient sample preparation, which we achieved through a combination of viral RNP concentration, RNA stabilization, and removal of interferents. In the clinical setting, swab samples are typically eluted directly into a vial of transport medium. In our assay we replaced the transport medium with a solution containing 1) non-ionic detergent to dissolve the viral envelope and release intact RNPs containing target RNA,27 2) RNA stabilizer to protect this target RNA from degradation, and 3) antibody-coated magnetic beads to capture the released and protected RNP. The sample is injected into the device and high-gradient magnetic capture was used to concentrate the viral RNA on-chip, obviating the need for time-consuming benchtop procedures such as phenol-chloroform extraction.42 PCR inhibitors present in the swab sample were then removed via a continuous washing with buffer within the device.
The MIMED system shows remarkable RNA enrichment from swab samples (Fig. 2); for example, the MIMED sample-prep performance nearly matches the ideal lossless positive control (an equal quantity of purified viral particles doped directly into PCR mix), with ΔCT = 0.7. We use ΔCT (defined as the difference in threshold cycle value between a given sample and the positive control) to quantify the signals as it directly relates the difference in nucleic acid template copy number between samples.43 PCR efficiency was determined from the standard curve to be ε = 1.94, indicating efficient amplification compared to the theoretical maximum of ε = 2, and corresponding to 10-fold difference in template copy number for each ΔCT = 3.48. As a comparison, the same sample preparation procedure performed in a magnetic particle concentrator (MPC, Invitrogen) enriched less effectively (ΔCT = 1.8) possibly due to inferior washing or bead loss. Dramatic signal loss was evident upon omission of any of the three MIMED sample preparation steps. For example, the absence of RNA concentration or stabilization resulted in ΔCT penalties of 3.3 and 7.1, respectively, the later indicating that RNA targets were significantly degraded by nucleases and aggregating factors present in the swab samples.10,44–46 Excluding the washing step resulted in ΔCT = 15.8, rendering the samples to be indistinguishable from the zero-virus negative control (ΔCT = 14.7) potentially due to degrading factors and PCR inhibitors. The omission of all three steps—directly spiking virus-treated swab samples into PCR mix — yielded a ΔCT of 14.9, indicating the absence of a positive signal from target RNA due to background interferents. These results clearly illustrate that each element in MIMED sample preparation is necessary.
Figure 2. Enrichment of nucleic acids from swab samples as measured by the change in PCR threshold cycles.

ΔCT values were determined with respect to the positive control, which consisted of viral particles spiked directly into PCR mix without swab-based interferents. MIMED sample enrichment, consisting of concentration, RNA stabilization, and continuous washing, approached the positive control ( ΔCT = 0.7), indicating efficient capture and purification of nucleic acids. Performing these steps in a magnetic particle concentrator (MPC) resulted in moderate sample loss ( ΔCT = 1.8). Excluding any one MIMED preparation step incurred significant enrichment penalties. Forgoing all three steps yields result equivalent to the zero-virus negative control.
Characterization of integrated magnetic capture
The high-throughput, low-loss, immunomagnetic sample purification within the MIMED device is achieved by ensuring that the magnetophoretic force (Fm) is sufficient to (1) efficiently attract immunomagnetically-labeled viruses to the trapping surface from any point in the flow stream (2) exceed the fluidic drag (Fd) for sustained retention during washing.30,47 Using finite element simulation (COMSOL Multiphysics, Stockholm, Sweden) we calculated that the magnetic field gradient (∇B) in the vertical direction is >300 T m−1 across the chamber, exerting a force of ~10 pN on the magnetic beads (Supporting Information, Fig. S2A).34 By balancing magnetophoretic and fluidic drag forces in the vertical direction, we determined that a ~1 s residence time is necessary to attract all magnetic beads in solution to the trapping surface. Due to the wide-channel geometry, our device is capable of operating at a volume throughput up to 600 mL h−1 (Fig. S2B). Next, we investigated the retention of beads during the washing procedure by calculating the sum of the forces in the x-direction (Fdx + Fmx) experienced by the magnetically labeled virus captured on the trapping surface, as shown in Fig. 3B. In order for trapping to occur, the magnetic force must exceed the maximum drag force (Fmx > Fdx). Fdx on a captured bead is directly proportional to the fluid velocity and is expected to exceed Fmx at high flow rates, resulting in bead loses. We experimentally confirmed that capture occurs at the left of the trapping regions, where the Fmx field converges and equilibrates with Fdx (Fig. 3C). Consistent with our simulation, approximately 100 ± 0.3 % of the beads were captured at flow rates of 6.0 and 60 mL h−1 (Fig. 3D). At 600 mL h−1, the capture efficiency decreased to 42 ± 16 %. Thus, we selected 60 mL h−1 as the nominal flow rate for target capture and washing steps, enabling a short processing time of 1 minute for 1 mL samples.
Figure 3. Simulation of magnetic capture.

(A) Section view of the MIMED trapping chamber illustrates the magnetic gradient across the channel wherein beads experience a pull-down force of ~10 -pN. (B) Sum of the magnetic (Fmx) and drag forces (Fdx) exerted on a stationary bead at 6, 60, 600 mL h −1. At 6 and 60 mL h−1, Fm exceeds Fd throughout all three trapping regions, enabling efficient bead capture (shaded regions). However, at 600 mL h−1, this only occurs by a narrow margin in the last trapping region (< 1 pN), suggesting potential for sample loss. (C) Experimental verification of simulation predictions of bead capture at the three stable equilibria established by the permanent magnets. (D) Efficiency of bead capture is measured vs. flow rate. Triplicate trials indicated ~100±0.3% bead capture at 6.0 and 60 mL h−1. At 600 mL h-1, capture efficiency decreases to ~42±16%.
On-chip RNP denaturation, RT-PCR and ssDNA generation
Genetic detection from extremely low concentrations of captured viral RNA targets requires nucleic acid amplification (Fig. 1D-G). In order to perform the amplification, we used a thermostable reverse transcriptase to thermally denature the RNP, releasing target RNA for concurrent reverse transcription to produce a cDNA template for subsequent PCR. In contrast to linear asymmetric PCR amplification for generation of ssDNA,22,25,48 MIMED achieves higher ssDNA concentration via exponential PCR amplification and subsequent enzymatic dsDNA digestion. The dsDNA PCR amplicons were converted to ssDNA on chip by selective digestion of 5’-phosphorylated strands with lambda exonuclease49 at high purity and efficiency > 90%. On-chip RT-PCR and ssDNA generation reactions proved reproducible, with efficiencies comparable to benchtop controls, and required no additional reagents or intermediate purification steps (Fig. S3A). Furthermore, we observed only less than 5% reduction in sample volume during thermocycling in the device. In order to achieve the high efficiency, we designed the internal surface area of MIMED device to consist mostly of glass. Only ~6% of the internal surface area is PDMS, which is known to cause enzyme adsorption and significant sample loss at elevated temperatures. 36 Furthermore, we avoided the use of exposed ferromagnetic structures in the chamber as they have been reported to non-specifcally adsorb proteins.39
E-DNA sensor characterization
E-DNA signaling is highly specific due to target binding-induced changes in the dynamics of the probe DNA, and the relative scarcity of electroactive contaminants in the interrogation potential range.37,38 Therefore, it offers direct detection of amplified ssDNA target in the PCR mixture without any intermediate separation or purification steps. For samples containing H1N1 virus ranging from 10–1000 TCID50, typical amplified ssDNA concentrations range from 10 to 300 nM. To determine the time necessary to resolve this concentration range via E-DNA detection, we challenged the sensor with synthetic 62-base ssDNA target identical to the H1N1 amplicon. We incubated the DNA targets at different concentrations in the electrochemical cell in high-salt buffer and collected square wave voltammograms (SWV) at 30 s intervals (Fig. S3B). As expected, E-DNA responses were logarithmic with regard to concentration and approximately linear over time (for t < 10 min). Importantly, 10 nM ssDNA could be resolved within 30 min, indicating potential to detect 10 TCID50 viral samples.
MIMED performance in throat swab samples
Complete MIMED assays were conducted directly with throat swab samples containing a range of H1N1 viral concentrations (Fig. 4). As a negative control, we used samples without spiked virus. This produced < 1% change in the E-DNA signal (Fig. 4A), indicating the absence of specific amplification product and the complete lack of viral particles in the sample. Conversely, we obtained sensor signals of 28, 21, and 4.2 % from samples respectively spiked with H1N1 virus at 1000, 100 or 10 TCID50, indicating the presence of specific product, corresponding to the range of initial viral content (Fig. 4B-C). This result confirms the capacity of MIMED to achieve unambiguous detection at concentrations as low as 10 TCID50. This performance is reproducible; triplicate independent measurements performed with separate samples on separate devices yielded average signals of 31 ± 5.2 %, 16 ± 4.2 %, 3.0 ± 1.5 % and 0.91 ± 0.47 % for samples containing H1N1 viruses at 1000, 100, 10 and 0 TCID50, respectively. Importantly, the MIMED system directly offers a detection limit significantly below clinical titers of ~105 TCID50 for throat swab samples9 and a sensitivity improvement greater than two orders of magnitude over recently published values for rapid antigen tests against swine-origin influenza virus.50
Figure 4. Limit of detection of MIMED is ~10 TCID50.
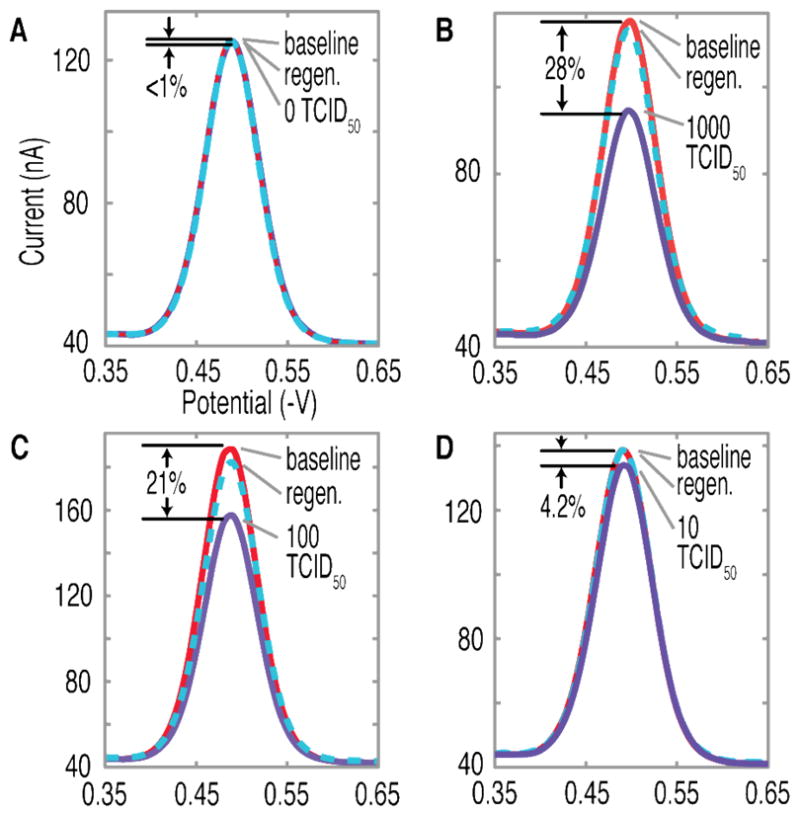
Swab samples containing 1000, 100 and 10 TCID50 returned peak faradic current changes of 28, 21 and 4.2% respectively, relative to 0.5% for negative control. All sensors could be regenerated to baseline levels, verifying that signal was the result of target hybridization.
Conclusion
We demonstrate an integrated microfluidic system, which enables sequence-specific viral RNA-based pathogen detection with high sensitivity and specificity from unprocessed throat swab samples. Using H1N1 influenza virus as a model, we have obtained a LOD of ~10 TCID50 from throat swab samples directly, which is four orders of magnitude below clinically relevant viral titers, and more than two orders below rapid tests for swine-origin influenza virus. This performance was achieved by integrating immunomagnetic target capture, concentration and purification, RT-PCR amplification, and sequence-specific electrochemical detection in a single monolithic disposable device. The MIMED device is designed as an inexpensive disposable unit, which interfaces with an instrument containing supporting peripherals such as pumps and heaters. The sample preparation uses a simple microchamber without chemical or physical modifications enabling high-throughput sample capture, minimal enzyme adsorption, favorable downstream enzymatic reactions, and high PCR efficiency. Total assay time is ~3.5 hour, and the RT-PCR represents a rate-limiting step (~150 minutes). We believe further assay optimization and rapid thermal cycling strategies51 may significantly reduce the assay time. Currently, the MIMED system is tuned for highest sensitivity,52 however, for applications where large dynamic range is required, our system can be readily operated in parallel, for example, with undiluted sample for detecting lower titers and diluted sample for higher titers. Importantly, given the availability of affinity reagents for broad range of pathogenic targets,53 we believe our MIMED system represents a universal strategy towards multiplexed genetic detection of biological sample at the point of care.54
Supplementary Material
Acknowledgments
We are grateful for financial support from the Institute of Collaborative Biotechnologies through the Army Research Office, and the National Institutes of Health. We thank Jonathan Adams, Adriana Patterson, and Ryan White for valuable discussions, the Plaxco Lab for assistance in electrode preparation, and the UCSB Nanofabrication Facility.
Footnotes
SUPPORTING INFORMATION AVAILABLE. Characterization data, experimental and fabrication details. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Horsman KM, Bienvenue JM, Blasier KR, Landers JP. Journal of forensic sciences. 2007;52:784. doi: 10.1111/j.1556-4029.2007.00468.x. [DOI] [PubMed] [Google Scholar]
- 2.Palchetti I, Mascini M. Analytical and Bioanalytical Chemistry. 2008;391:455. doi: 10.1007/s00216-008-1876-4. [DOI] [PubMed] [Google Scholar]
- 3.Gardeniers JGE, van den Berg A. Anal Bioanal Chem. 2004;378:1700. doi: 10.1007/s00216-003-2435-7. [DOI] [PubMed] [Google Scholar]
- 4.Holland Ca, Kiechle FL. Current opinion in microbiology. 2005;8:504. doi: 10.1016/j.mib.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 5.Yang S, Rothman R. The Lancet Infectious Diseases. 2004;4:337. [Google Scholar]
- 6.Kling J. Nature biotechnology. 2006;24:891. doi: 10.1038/nbt0806-891. [DOI] [PubMed] [Google Scholar]
- 7.Bissonnette L, Bergeron MG. Clin Microbiol Infect. 16:1044. doi: 10.1111/j.1469-0691.2010.03282.x. [DOI] [PubMed] [Google Scholar]
- 8.Wilson IG. Applied and Environmental Microbiology. 1997;63:3741. doi: 10.1128/aem.63.10.3741-3751.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fouchier RAM, Bestebroer TM, Herfst S, Van der Kemp L, Rimmelzwaan GF, Osterhaus ADME. Journal of Clinical Microbiology. 2000;38:4096. doi: 10.1128/jcm.38.11.4096-4101.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spackman E, Suarez DL. Methods Mol Biol. 2008;436:13. doi: 10.1007/978-1-59745-279-3_3. [DOI] [PubMed] [Google Scholar]
- 11.Andreoletti L, Hober D, Belaich S, Lobert PE, Dewilde A, Wattre P. Journal of Virological Methods. 1996;62:1. doi: 10.1016/0166-0934(96)02080-0. [DOI] [PubMed] [Google Scholar]
- 12.Hartshorn KL, Ligtenberg A, White MR, van Eijk M, Hartshorn M, Pemberton L, Holmskov U, Crouch E. Biochemical Journal. 2006;393:545. doi: 10.1042/BJ20050695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karch H, JanetzkiMittmann C, Aleksic S, Datz M. Journal of Clinical Microbiology. 1996;34:516. doi: 10.1128/jcm.34.3.516-519.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holland JL, Louie L, Simor AE, Louie M. J Clin Microbiol. 2000;38:4108. doi: 10.1128/jcm.38.11.4108-4113.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lou Q, Chong SK, Fitzgerald JF, Siders JA, Allen SD, Lee CH. J Clin Microbiol. 1997;35:281. doi: 10.1128/jcm.35.1.281-283.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yager P, Edwards T, Fu E, Helton K, Nelson K, Tam MR, Weigl BH. Nature. 2006;442:412. doi: 10.1038/nature05064. [DOI] [PubMed] [Google Scholar]
- 17.Myers FB, Lee LP. Lab on a chip. 2008;8:2015. doi: 10.1039/b812343h. [DOI] [PubMed] [Google Scholar]
- 18.Chen L, Manz A, Day PJR. Lab on a chip. 2007;7:1413. doi: 10.1039/b708362a. [DOI] [PubMed] [Google Scholar]
- 19.Easley CJ, Karlinsey JM, Bienvenue JM, Legendre La, Roper MG, Feldman SH, Hughes Ma, Hewlett EL, Merkel TJ, Ferrance JP, Landers JP. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:19272. doi: 10.1073/pnas.0604663103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang CS, Xing D. Nucleic Acids Research. 2007;35:4223. doi: 10.1093/nar/gkm389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaigala GV, Huskins RJ, Preiksaitis J, Pang XL, Pilarski LM, Backhouse CJ. Electrophoresis. 2006;27:3753. doi: 10.1002/elps.200600061. [DOI] [PubMed] [Google Scholar]
- 22.Yeung SW, Lee TMH, Cai H, Hsing IM. Nucleic Acids Research. 2006:34. doi: 10.1093/nar/gkl702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang J. Biosensors & Bioelectronics. 2006;21:1887. doi: 10.1016/j.bios.2005.10.027. [DOI] [PubMed] [Google Scholar]
- 24.Drummond TG, Hill MG, Barton JK. Nature biotechnology. 2003;21:1192. doi: 10.1038/nbt873. [DOI] [PubMed] [Google Scholar]
- 25.Liu RH, Yang JN, Lenigk R, Bonanno J, Grodzinski P. Analytical Chemistry. 2004;76:1824. doi: 10.1021/ac0353029. [DOI] [PubMed] [Google Scholar]
- 26.Flint SJ, Racaniello VR, Enquist L, Skalka WAM. Principles of Virology: Molecular Biology, Pathogenesis, and Control of Animal Viruses. 2. ASM Press; 2003. [Google Scholar]
- 27.Kawakami K, Ishihama A. J Biochem-Tokyo. 1983;93:989. doi: 10.1093/oxfordjournals.jbchem.a134254. [DOI] [PubMed] [Google Scholar]
- 28.Miltenyi S, Muller W, Weichel W, Radbruch A. Cytometry. 1990;11:231. doi: 10.1002/cyto.990110203. [DOI] [PubMed] [Google Scholar]
- 29.Palecek E, Fojta M. Talanta. 2007;74:276. doi: 10.1016/j.talanta.2007.08.020. [DOI] [PubMed] [Google Scholar]
- 30.Adams JD, Kim U, Soh HT. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:18165. doi: 10.1073/pnas.0809795105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Csordas A, Gerdon AE, Adams JD, Qian JR, Oh SS, Xiao Y, Soh HT. Angewandte Chemie-International Edition. 2010;49:355. doi: 10.1002/anie.200904846. [DOI] [PubMed] [Google Scholar]
- 32.Qian JR, Lou XH, Zhang YT, Xiao Y, Soh HT. Analytical Chemistry. 2009;81:5490. doi: 10.1021/ac900759k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pamme N. Lab on a chip. 2006;6:24. doi: 10.1039/b513005k. [DOI] [PubMed] [Google Scholar]
- 34.Fonnum G, Johansson C, Molteberg A, Morup S, Aksnes E. Journal of Magnetism and Magnetic Materials. 2005;293:41. [Google Scholar]
- 35.Kim JA, Lee JY, Seong S, Cha SH, Lee SH, Kim JJ, Park TH. Biochem Eng J. 2006;29:91. [Google Scholar]
- 36.Shin YS, Cho K, Lim SH, Chung S, Park SJ, Chung C, Han DC, Chang JK. J Micromech Microeng. 2003;13:768. [Google Scholar]
- 37.Fan CH, Plaxco KW, Heeger AJ. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:9134. doi: 10.1073/pnas.1633515100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xiao Y, Lubin AA, Heeger AJ, Plaxco KW. Angewandte Chemie-International Edition. 2005;44:5456. doi: 10.1002/anie.200500989. [DOI] [PubMed] [Google Scholar]
- 39.Williams DF, Askill IN, Smith R. J Biomed Mater Res. 1985;19:313. doi: 10.1002/jbm.820190312. [DOI] [PubMed] [Google Scholar]
- 40.Reske T, Mix M, Bahl H, Flechsig GU. Talanta. 2007;74:393. doi: 10.1016/j.talanta.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 41.Little JW. Journal of Biological Chemistry. 1967;242:679. [PubMed] [Google Scholar]
- 42.Chomczynski P, Sacchi N. Anal Biochem. 1987;162:156. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 43.Bernard PS, Wittwer CT. Clin Chem. 2002;48:1178. [PubMed] [Google Scholar]
- 44.Krafft AE, Russell KL, Hawksworth AW, McCall S, Irvine M, Daum LT, Connoly JL, Reid AH, Gaydos JC, Taubenberger JK. Journal of Clinical Microbiology. 2005;43:1768. doi: 10.1128/JCM.43.4.1768-1775.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kumar SV, Hurteau GJ, Spivack SD. Clinical Cancer Research. 2006;12:5033. doi: 10.1158/1078-0432.CCR-06-0501. [DOI] [PubMed] [Google Scholar]
- 46.Spackman E, Senne DA, Myers TJ, Bulaga LL, Garber LP, Perdue ML, Lohman K, Daum LT, Suarez DL. Journal of Clinical Microbiology. 2002;40:3256. doi: 10.1128/JCM.40.9.3256-3260.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu YL, Adams JD, Turner K, Cochran FV, Gambhir SS, Soh HT. Lab on a chip. 2009;9:3604. doi: 10.1039/b820985e. [DOI] [PubMed] [Google Scholar]
- 48.Lee TMH, Carles MC, Hsing IM. Lab on a chip. 2003;3:100. doi: 10.1039/b300799e. [DOI] [PubMed] [Google Scholar]
- 49.Ferguson BS, Buchsbaum SF, Swensen JS, Hsieh K, Lou XH, Soh HT. Analytical Chemistry. 2009;81:6503. doi: 10.1021/ac900923e. [DOI] [PubMed] [Google Scholar]
- 50.Chan KH, Lai ST, Poon LLM, Guan Y, Yuen KY, Peiris JSM. J Clin Virol. 2009;45:205. doi: 10.1016/j.jcv.2009.05.034. [DOI] [PubMed] [Google Scholar]
- 51.Zhang CS, Xing D. Nucleic Acids Research. 2007;35:4223. doi: 10.1093/nar/gkm389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Petric M, Comanor L, Petti CA. J Infect Dis. 2006;194:S98. doi: 10.1086/507554. [DOI] [PubMed] [Google Scholar]
- 53.Olsvik O, Popovic T, Skjerve E, Cudjoe KS, Hornes E, Ugelstad J, Uhlen M. Clin Microbiol Rev. 1994;7:43. doi: 10.1128/cmr.7.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pavlovic E, Lai RY, Wu TT, Ferguson BS, Sun R, Plaxco KW, Soh HT. Langmuir. 2008;24:1102. doi: 10.1021/la702681c. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.