

Abstract
The discovery of molecules that bind tightly and selectively to desired proteins continues to drive innovation at the interface of chemistry and biology. This paper describes the binding of human insulin by the synthetic receptor cucurbit[7]uril (Q7) in vitro. Isothermal titration calorimetry and fluorescence spectroscopy experiments show that Q7 binds to insulin with an equilibrium association constant of 1.5 × 106 M-1 and with 50-100-fold selectivity versus proteins that are much larger but lack an N-terminal aromatic residue, and >1000-fold selectivity versus an insulin variant lacking the N-terminal phenylalanine (Phe) residue. The crystal structure of the Q7•insulin complex shows that binding occurs at the N-terminal Phe residue and that the N-terminus unfolds to enable binding. These findings suggest that site-selective recognition is based on the properties inherent to a protein terminus, including the unique chemical epitope presented by the terminal residue and the greater freedom of the terminus to unfold, like the end of a ball of string, to accommodate binding. Insulin recognition was predicted accurately from studies on short peptides and exemplifies an approach to protein recognition by targeting the terminus.
Living systems present a highly complex array of molecular surfaces that vary in size, shape, polarity, organization and solvation, and yet each molecule finds its target destination with high fidelity. The weak intermolecular forces (e.g., coulombic, dispersive, and solvophobic) that direct these events are well understood in principle.1-3 In practice, however, the design of synthetic compounds to recognize desired protein targets presents persistent challenges en route to pharmaceuticals, medical diagnostics, and other tools for biochemistry and chemical biology. So-called “rational” approaches to this problem use detailed knowledge of the structure of a target to aid in the design of a complementary molecule.4-6 Although high-resolution structures are available for a number of protein targets, the precise structure of the surface of a protein and the surrounding solvent remains difficult to predict from the genetically encoded sequence of amino acids or from homology modeling. Therefore, general design principles for sequence-based recognition of folded proteins do not currently exist. By contrast, numerous groups have reported on the sequence-specific recognition of short peptides in aqueous solution by synthetic receptors.7-17 Short peptides are more practical than proteins for structure-activity studies of molecular recognition because they are less expensive, are easier to modify, and in many cases their structures are predictably folded/unfolded. Therefore, strategies for translating known approaches of peptide recognition into protein recognition would be exceptionally valuable. Here we show that the synthetic receptor cucurbit[7]uril (Q7, Figure 1) binds to the N-terminal phenylalanine (Phe) of human insulin, and that this recognition enables Q7 to bind more tightly to insulin than to proteins lacking an N-terminal aromatic residue. Equilibrium binding studies and X-ray crystallography reveal the molecular basis for recognition and show how the properties of a protein terminus are well suited to site-selective recognition.
Figure 1.

Amino acid sequences of the insulins and model peptides used in this study, and the chemical formula of Q7.
Q7 is the seven-membered macrocycle in the cucurbit[n]uril (Qn) family of water-soluble, synthetic receptors, which have gained importance for their capacity to bind small guests very tightly and selectively in aqueous solution.18-20 The nonpolar interior and carbonyl-lined portals of Qn receptors work in concert to bind organic cations by including the hydrophobic portion of the guest within the apolar cavity and stabilizing the cationic group(s) with the portal oxygens. This cooperation between cavity and portal has proved ideal for binding to short peptides with an N-terminal phenylalanine (Phe), tryptophan (Trp), or tyrosine (Tyr), in a sequence-specific fashion.14-17,21 In focused structure-activity studies with Q7 and Q8,15,17 all peptides had a single aromatic group and an N-terminal ammonium group, but only those with an N-terminal aromatic residue, which fixes both groups in close proximity, were bound selectively by Q7 and Q8. Therefore, the terminus provided a binding site that is structurally unique at the level of a single-residue and thus known from the sequence of amino acids. On the basis of these studies we predicted that a Qn receptor should bind selectively to an N-terminal aromatic residue versus all other surface-exposed aromatic residues on a folded protein and thus provide a mechanism for site-specific protein recognition. In the present study, human insulin was identified as a desirable model on which to test this hypothesis because it has a rare Phe residue at the N-terminus of its B-chain (i.e., PheB1), it has been thoroughly characterized, and it is stably folded under ambient conditions.22
Isothermal titration calorimetry (ITC) experiments revealed that Q7 binds to recombinant human insulin 1 in a 1:1 stoichiometric ratio with an equilibrium association constant (Ka) value of 1.5 × 106 M-1 (Figure 2). Although we anticipated binding at the PheB1 position, insulin also has on its surface two Phe and four Tyr residues, all of which are possible binding sites. The series of compounds 2-5 (Figure 1) was designed to probe the location of binding. Protein 2 is a variant of human insulin with glutamic acid (Glu) at positions B1 and B27 (i.e., GluB1, GluB27).23,24 Peptides 3-5 are models of the N-terminal Phe (3), non-terminal Phe (4), and non-terminal Tyr (5) residues on the surface of insulin.
Figure 2.
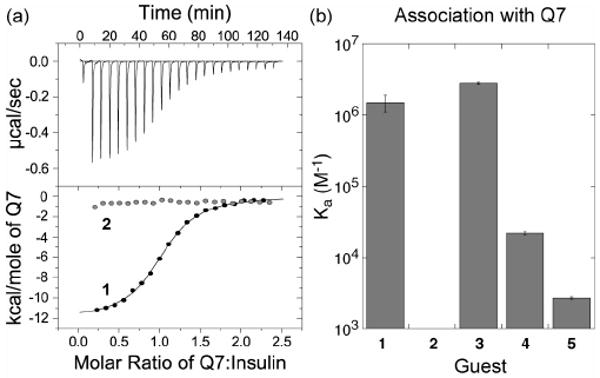
(a) Representative ITC data for the titration of Q7 to native human insulin 1. The top plot of power versus time was integrated to yield the bottom plot of molar enthalpy versus the ratio of Q7:insulin. The gray data points in the bottom plot are for the analogous experiment on variant insulin 2, showing no measurable binding. (b) Bar plot of the equilibrium association constants of analytes 1 – 5 in complex with Q7. Error bars are standard deviations from at least three measurements. The ITC technique has a practical lower limit of 103 M-1.
ITC measurements (Figure 2 and Supporting Information) showed no measurable binding of Q7 to variant insulin 2, which has no N-terminal Phe, and thus a decrease in association constant by more than three orders of magnitude compared to native insulin 1. This result makes a strong case for PheB1 as the site of binding. Positive control peptide 3 (Ka = 2.8 × 106 M-1), which has an N-terminal Phe, binds to Q7 with similar affinity as native insulin 1. Peptides 4 (2.2 × 104 M-1) and 5 (2.7 × 103 M-1) were bound less tightly than peptide 3 by two and three orders of magnitude, respectively, showing that non-terminal Phe and Tyr residues are possible, albeit weak, sites of binding for Q7. Therefore, we were surprised that Q7 showed no measurable affinity for variant insulin 2, which suggests that the two non-terminal Phe and four non-terminal Tyr residues on the surface of 2 are significantly less accessible to binding than the analogous residues in peptides 4 and 5.
We sought structural evidence to support the ITC data and to explore a molecular basis for recognition. Toward this end, we determined the crystal structure of the Q7•1 complex (Figure 3). The asymmetric unit of the crystal (Figure 3a) contains two insulin molecules (1a and 1b) and one molecule of Q7, which is associated with insulin 1a at the N-terminus of the B chain. Akin to previously reported structures of Q8 in complex with Phe-Gly-Gly and Trp-Gly-Gly,15 the Q7•insulin structure shows the aromatic sidechain of PheB1 included within the cavity of Q7. In addition, ValB2 and AsnB3 contribute weak stabilizing interactions by contacting atoms of the Q7 exterior. Overall, the binding of Q7 buries approx. 200 Å2 of insulin's solvent-accessible surface area.25 The presence of Q7-bound insulin 1a and unbound insulin 1b in the asymmetric unit of the crystal allows us to study perturbations to the structure of insulin upon binding to Q7. Overall, the structures of 1a and 1b are strikingly similar, with a root mean square deviation (RSMD) of 0.37 Å over 35 alpha carbons. The largest deviations are observed at the B1-B4 residues of 1a, which unfold from the surface of the protein to accommodate Q7 (Figure 3b).
Figure 3.
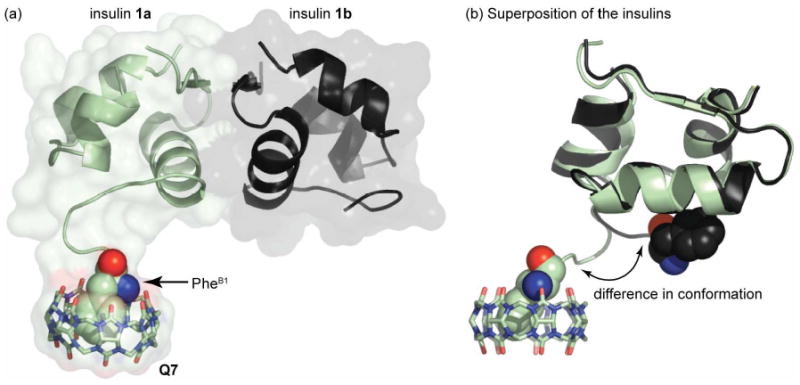
Crystal structure of the Q7• 1 complex. (a) Asymmetric unit of the crystal, with the two molecules of insulin, 1a (green) and 1b (black), rendered as ribbons and the associated Q7 as sticks. The solvent-accessible surface is shown as semi-transparent to reveal the overall shape of the complex. The PheB1 position of 1a, which is bound inside Q7, is rendered as space-filling, with carbon in green, oxygen in red, and nitrogen in blue; the N-terminal nitrogen is the blue sphere. (b) Superposition of the two insulins illustrates the strong homology between 1a and 1b with clear divergence at the N-terminus of the B-chain. In this image, the PheB1 position of both insulins is rendered as space-filling for comparison.
These results corroborate the calorimetric data and provide a structural basis for recognition at the N-terminus of the B chain of insulin. As observed with small peptides,15 stable binding requires the simultaneous inclusion of the hydrophobic sidechain of PheB1 within the cavity of Q7 and interaction of the N-terminal ammonium group with the portal oxygens of Q7. The PheB1 residue therefore presents a unique binding epitope comprising the aromatic sidechain and the N-terminal ammonium group, which cannot exist anywhere else on the protein. Moreover, the ability of the insulin N-terminus to partially unfold may be critical to binding in the sterically demanding context of a folded protein. The singular connection of the terminal residue to the protein, like the end of a ball of string, facilitates the adoption of a fully solvent-exposed, random-coil structure, akin to short peptides. This result is likely not unique to insulin because protein termini are typically solvent-exposed, more so than charge alone would predict.26
Based on the mechanism of binding established here, we expected that Q7 would not exhibit high affinity binding to proteins lacking an N-terminal aromatic residue. A fluorescence indicator displacement assay27 was used to test this hypothesis. We chose acridine orange because it has relatively little nonspecific association with proteins, it is effective at neutral pH, and its Ka value for Q7 (2.0 × 105 M-1) is similar yet lower than that of insulin.28 The emission of acridine orange increases when bound to Q7. Therefore, upon introducing a competitive analyte to a solution containing the Q7•acridine orange complex, the displacement of acridine orange from Q7 yields a decrease in fluorescence intensity that indicates the binding of analyte (Figure 4a).
Figure 4.
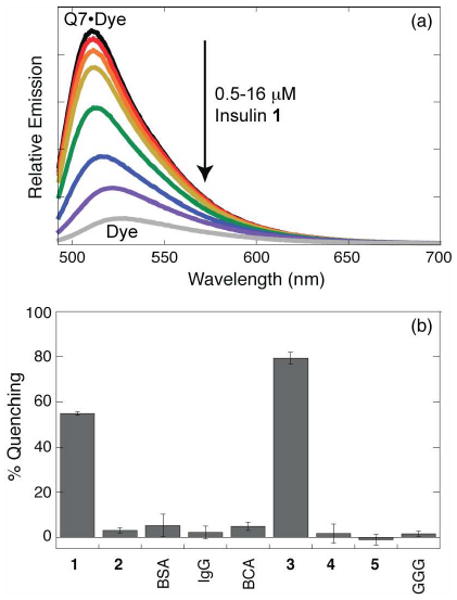
Fluorescent indicator displacement assay for insulin. (a) Decrease in emission intensity upon titrating insulin at 0, 0.5, 1.0, 2.0, 4.0, 8.0, and 16.0 μM into a mixture of Q7 (6 μM) and acridine orange (8 μM). (b) Fluorescence quenching for a series of proteins and peptides at 4 μM in a mixture of 4 μM Q7 and 6 μM acridine orange, showing selective binding of native insulin 1 and positive control peptide 3. Error bars are standard deviatations from at least three measurements. BSA, bovine serum albumin; IgG, immunoglobulin G; BCA, bovine carbonic anhydrase; GGG, peptide Gly-Gly-Gly.
The fluorescence response for insulin was compared to compounds 2-5, the peptide Gly-Gly-Gly, and a series of commercially available blood proteins, including bovine serum albumin (BSA), human immunoglobulin G (IgG), and bovine carbonic anhydrase (BCA) (Figure 4b). We observed substantial quenching (55-80%) with insulin 1 and peptide 3 (positive control), but insignificant quenching for the other analytes (1-5%), which lack an aromatic terminus. This result demonstrates that a single residue can enable the selective binding of insulin versus other proteins by at least 50-100-fold in Ka29 under the conditions used in this study. We note explicitly, however, that insulin recognition in vivo would be precluded by (i) its relatively high dissociation constant (0.67 μM) compared to the sub-nM concentrations of insulin in blood and saliva, (ii) the presence of weak binding proteins (e.g., albumin) at sufficiently high concentrations to effectively compete for binding,30-32 and (iii) the possibility of competition by other peptides and proteins with N-terminal aromatic groups. This paper does not claim to address these challenges pertaining to in vivo applications.
This paper describes an approach to protein recognition that exploits the unique properties of a protein terminus to achieve single-site binding that is predictable from the sequence of amino acids. Unlike small molecules that bind inside protein cavities,5 and unlike natural and synthetic receptors that bind to large areas on the surfaces of proteins,4 Q7 binds to insulin by incorporating the terminal residue within its cavity. When the terminus unravels from the surface it becomes fully solvent exposed, thus allowing Q7 to bind to insulin in the same manner as to a short peptide. This motif is extraordinarily minimal, comprising a small and structurally simple receptor, a single amino acid residue, and a binding interface of ∼200 Å2. This approach to protein recognition differs from existing methods, such as genetically engineering polyhistidine (His-tag) or glutathione S-transferase (GST) fusion proteins into target proteins for affinity chromatography,33 or chemically modifying proteins with biotin or aminomethylferrocene groups for capture using avidin or Q7,34-36 respectively. In the current study, Q7 binds directly to the native protein sequence, not an engineered or chemically modified sequence,37 although we consider these techniques to be complementary. We believe this approach will facilitate the design of receptors for other proteins as well as the development of techniques for single-site labeling of proteins and for sensing and separating proteins according to the identity of the terminal residue.
Supplementary Material
Acknowledgments
This paper is dedicated to Prof. Jonathan L. Sessler on the occasion of his 55th birthday. Support for this work from the National Science Foundation (CHE-0748483, ARU) and Welch Foundation (W-1640, ARU and AQ-1399, PJH) is gratefully acknowledged. We thank Novo Nordisk for generous donation of the GluB1, GluB27 variant insulin 2. The circular dichroism spectropolarimeter was provided by a grant from the National Science Foundation (DBI-0718766, ARU). The fluorescence spectrometer and titration calorimeter were provided by a grant to Trinity University by the W. M. Keck Foundation. LMR had a Jean Dreyfus Boissevain Undergraduate Scholarship for Excellence in Chemistry from the Camille and Henry Dreyfus Foundation. This work is based on research conducted at the Advanced Photon Source on the Northeastern Collaborative Access Team (NE-CAT) beamlines, which are supported by award RR-15301 from the National Center for Research Resources at the National Institutes of Health. Use of the Advanced Photon Source is supported by the U. S. Department of Energy, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. We thank Dr. Jonathan P. Schuermann of NE-CAT for collecting the data for the Q7•1 complex. Support for the X–ray Crystallography Core Laboratory at the University of Texas Health Science Center by the Institutional Executive Research Council and the San Antonio Cancer Institute is gratefully acknowledged.
Footnotes
Supporting Information Available: Experimental details, thermodynamic binding data, X-ray crystallographic details, circular dichroism spectra.
References
- 1.Schneider HJ. Angew Chem Intl Ed. 2009;48:3924–3977. doi: 10.1002/anie.200802947. [DOI] [PubMed] [Google Scholar]
- 2.Williams DH, Stephens E, O'Brien DP, Zhou M. Angew Chem Intl Ed. 2004;48:6596–6616. doi: 10.1002/anie.200300644. [DOI] [PubMed] [Google Scholar]
- 3.Meyer EA, Castellano RK, Diederich F. Angew Chem Intl Ed. 2003;42:1210–1250. doi: 10.1002/anie.200390319. [DOI] [PubMed] [Google Scholar]
- 4.Giralt E, Peczuh M, Salvatella X, et al., editors. Protein surface recognition approaches for drug design. John Wiley & Sons; West Sussex: 2001. [Google Scholar]
- 5.Böhm HJ, Schneider G, et al., editors. Protein-ligand interactions: from molecular recognition to drug design. Wiley-VCH; Weinheim: [Google Scholar]
- 6.Krishnamurthy VM, et al. Chem Rev. 2008;108:946–1051. doi: 10.1021/cr050262p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peczuh MW, Hamilton AD. Chem Rev. 2000;100:2479–2494. doi: 10.1021/cr9900026. [DOI] [PubMed] [Google Scholar]
- 8.Breslow R, Yang Z, Ching R, Trojandt G, Odobel F. J Am Chem Soc. 1998;120:3536–3537. [Google Scholar]
- 9.Hossain MA, Schneider HJ. J Am Chem Soc. 1998;120:11208–11209. [Google Scholar]
- 10.Wagner H, Still WC, Chen CT. Science. 1998;279:851–853. doi: 10.1126/science.279.5352.851. [DOI] [PubMed] [Google Scholar]
- 11.Tashiro S, Tominaga M, Kawano M, Therrien B, Ozeki T, Fujita M. J Am Chem Soc. 2005;127:4546–4547. doi: 10.1021/ja044782y. [DOI] [PubMed] [Google Scholar]
- 12.Schmuck C. Coord Chem Rev. 2006;250:3053–3067. [Google Scholar]
- 13.Wehner M, Janssen D, Schafer G, Schrader T. Eur J Org Chem. 2006:138–153. [Google Scholar]
- 14.Bush ME, Bouley ND, Urbach AR. J Am Chem Soc. 2005 doi: 10.1021/ja0548440. [DOI] [PubMed] [Google Scholar]
- 15.Heitmann LM, Taylor AB, Hart PJ, Urbach AR. J Am Chem Soc. 2006;128:12574–12581. doi: 10.1021/ja064323s. [DOI] [PubMed] [Google Scholar]
- 16.Rekharsky MV, Yamamura H, Inoue C, Kawai M, Osaka I, Arakawa R, Shiba K, Sato A, Ko YH, Selvapalam N, Kim K, Inoue Y. J Am Chem Soc. 2006;128:14871–14880. doi: 10.1021/ja063323p. [DOI] [PubMed] [Google Scholar]
- 17.Rekharsky MV, Yamamura H, Ko YH, Selvapalam N, Kim K, Inoue Y. Chem Comm. 2008:2236–2238. doi: 10.1039/b719902c. [DOI] [PubMed] [Google Scholar]
- 18.Lee JW, Samal S, Selvapalam N, Kim HJ, Kim K. Acc Chem Res. 2003;36:621–630. doi: 10.1021/ar020254k. [DOI] [PubMed] [Google Scholar]
- 19.Lagona L, Mukhopadhyay P, Chakrabarti S, Isaacs L. Angew Chem Int Ed. 2005;44:4844–4870. doi: 10.1002/anie.200460675. [DOI] [PubMed] [Google Scholar]
- 20.Isaacs L. Chem Comm. 2009:619–629. doi: 10.1039/b814897j. [DOI] [PubMed] [Google Scholar]
- 21.Tian F, Cziferszky M, Jiao D, Wahlström K, Geng J, Scherman OA. Langmuir. 2011;27:1387–1390. doi: 10.1021/la104346k. [DOI] [PubMed] [Google Scholar]
- 22.Federwisch M, Dieken ML, De Meyts P, et al., editors. Insulin and Related Proteins – Structure to Function and Pharmacology. Kluwer Academic Publishers; Dordrecht, The Netherlands: 2002. [Google Scholar]
- 23.Uversky VN, Garriques LN, Millett IS, Frokjaer S, Brange J, Doniach S, Fink AL. J Pharmaceut Sci. 2003;92:847–858. doi: 10.1002/jps.10355. [DOI] [PubMed] [Google Scholar]
- 24.We expect the additional mutation of ThrB27 to Glu to have no impact on Q7 binding because prior work has shown that Qn's do not bind to Thr residues.38,39 Moreover, the circular dichroism (CD) spectrum of 2 is essentially identical to that of 1 (see Supporting Information), which shows that the mutations do not influence the folded structure of insulin.
- 25.Brunger AT et al. Acta Cryst D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 26.Jacob E, Unger R. Bioinformatics. 2006;23:e225–e230. doi: 10.1093/bioinformatics/btl318. [DOI] [PubMed] [Google Scholar]
- 27.Nguyen BT, Anslyn EV. Coord Chem Rev. 2006;250:3118–3127. [Google Scholar]
- 28.Shaikh M, Mohanty J, Singh PK, Nau WM, Pal H. Photochem Photobiol Sci. 2008;7:408–414. doi: 10.1039/b715815g. [DOI] [PubMed] [Google Scholar]
- 29.Calculated from the fraction of fluorescence quenching observed under these competitive equilibrium conditions.
- 30.Bhasikuttan AC, Mohanty J, Nau WM, Pal H. Angew Chem Int Ed. 2007;46:4120–4122. doi: 10.1002/anie.200604757. [DOI] [PubMed] [Google Scholar]
- 31.Shaikh M, Mohanty J, Bhasikuttan AC, Uzunova VD, Nau WM, Pal H. Chem Comm. 2008:3681–3683. doi: 10.1039/b804381g. [DOI] [PubMed] [Google Scholar]
- 32.Lei W, Jiang G, Zhou Q, Zhang B, Wang X. Phys Chem Chem Phys. 2010;12:13255–13260. doi: 10.1039/c001013h. [DOI] [PubMed] [Google Scholar]
- 33.Hochuli E, Bannwarth W, Döbeli H, Gentz R, Stüber D. Nature Biotech. 1988;6:1321–1325. [Google Scholar]
- 34.Hwang I, Baek K, Jung M, Kim Y, Park KM, Lee DW, Selvapalam N, Kim K. J Am Chem Soc. 2007;129:4170–4171. doi: 10.1021/ja071130b. [DOI] [PubMed] [Google Scholar]
- 35.Young JF, Nguyen HD, Yang L, Huskens J, Jonkheijm P, Brunsveld L. Chembiochem. 2010;11:180–183. doi: 10.1002/cbic.200900599. [DOI] [PubMed] [Google Scholar]
- 36.Lee DW, Park KM, Banerjee M, Ha SH, Lee T, Suh K, Paul S, Jung H, Kim J, Selvapalam N, Ryu SH, Kim K. Nature Chem. 2011;3:154–159. doi: 10.1038/nchem.928. [DOI] [PubMed] [Google Scholar]
- 37.Nguyen HD, Dang DT, van Dongen JLJ, Bruns-veld L. Angew Chem Int Ed. 2010;49:895–898. doi: 10.1002/anie.200904413. [DOI] [PubMed] [Google Scholar]
- 38.Rajgariah P, Urbach AR. J Incl Phenom Macrocycl Chem. 2008;62:251–254. [Google Scholar]
- 39.Cong H, Tao LL, Yu YH, Yang F, Du Y, Xue SF, Tao Z. Acta Chim Sin. 2006;64:989–996. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.