

Abstract
Macrophages are becoming increasingly recognized as key agents in chronic diseases of aging, including cancer, metabolic disease, and, the topic of this review, atherosclerosis. In atherosclerosis, monocytes hone to focal area of the arterial subendothelium in response to matrix-retained apolipoprotein B-lipoproteins. Monocyte-derived macrophages then participate in a maladaptive, non-resolving inflammatory response that leads to subendothelial expansion with additional cells, lipid, and matrix. A few lesions undergo necrotic disruption, triggering acute thrombotic vascular disease, including myocardial infarction, stroke, and sudden cardiac death. This review discusses how macrophages contribute to both early and advanced atherosclerosis, with an emphasis on concepts supported by solid mechanistic and in-vivo data.
Macrophages, central effectors of innate immunity, are now recognized as key pathophysiologic agents in wide-spread disease processes associated with chronic inflammation and aging, including cancer, obesity-induced insulin resistance, and, as discussed in this review, atherosclerosis. Atherosclerosis underlies the leading cause of death in industrialized societies, soon to be world-wide (Lloyd-Jones et al., 2010). Atherosclerosis first involves a decades-long expansion of the arterial intima, a normally small area between the endothelium and the underlying smooth muscle cells of the media, with lipids, cells, and extracellular matrix (Figure 1). While this process itself rarely leads to major symptoms due to preservation of the arterial lumen, a few of these lesions undergo necrotic breakdown, which precipitates acute, occlusive lumenal thrombosis and its consequences: myocardial infarction ("heart attack"), unstable angina (accelerating chest pain due to ongoing heart muscle ischemia), and sudden cardiac death, and stroke (Virmani et al., 2002). As will be discussed in the sections below, monocyte-derived macrophages play key roles in both early atherogenesis and advanced plaque progression.
Figure 1. Progression of an atherosclerotic lesion.

Early fatty streak lesions are characterized by the accumulation of apoB-LPs in the subendothelial space which incites the recruitment of dendritic cells and macrophages. As the atherosclerotic lesion progresses, smooth muscle and T cells also infiltrate the intima, and apoB-LP retention is amplified. Vulnerable plaques are characterized by the accumulation of apoptotic cells and defective efferocytosis resulting in the lipid filled necrotic core. A thinning fibrous cap decreases lesion stability making these atherosclerotic plaques susceptible to rupture and the formation of a thrombus.
The Role of Monocyte-Derived Macrophages in Lesion Initiation and Early Progression
Monocytes and Monocyte Entry Into Lesions (Figure 2)
Figure 2. Retention of apoB-LPs incites monocyte recruitment in early atherosclerotic lesions.

ApoB-LPs enter the intima, bind to proteoglycans, and undergo various modifications, including oxidation and hydrolysis by sPLA2 and S-SMase. These modifications incite an inflammatory response characterized by chemokine secretion and altered expression of adhesion molecules by the overlying endothelial cells; cause lipoprotein aggregation; and further promote lipoprotein retention. The inflammatory signals lead to monocyte recruitment into the intima, where they differentiate into macrophages and internalize native and modified lipoproteins resulting in foam cell formation. Moreover, foam cells can contribute further to, and thus amplify, lipoprotein modifications and retention (dotted arrow).
Atherosclerosis is a focal disease process that occurs predominantly at sites of disturbed laminar flow, notably, arterial branch points and bifurcations. Careful morphological and functional studies of the earliest stages of atherogenesis in human and animal models indicate that the key initiating step is subendothelial accumulation of apolipoprotein B-containing lipoproteins (apoB-LPs) (Williams and Tabas, 1995). ApoB-LPs are made by liver and intestinal cells and consist of a core of neutral lipids, notably cholesteryl fatty acyl esters (CE) and triglycerides, surrounded by a monolayer of phospholipid and proteins. Hepatic apoB-LPs are secreted as very low-density lipoproteins (VLDL), which are converted in the circulation to atherogenic low-density lipoprotein (LDL), and intestinal apoB-LPs are secreted as chylomicrons, which are converted by lipolysis into atherogenic particles called remnant lipoproteins.
The key early inflammatory response to retained apoB-LPs, which may be enhanced by oxidative modification of the LPs, is activation of overlying endothelial cells in a manner that leads to recruitment of blood-borne monocytes (Glass and Witztum, 2001; Mestas and Ley, 2008). Activated endothelial cells secrete chemoattractants, or "chemokines," that interact with cognate chemokine receptors on monocytes and promote directional migration. Importantly, prevention of monocyte entry by blocking chemokines or their receptors, prevents or retards atherogenesis in mouse models of atherosclerosis (Mestas and Ley, 2008). It should be noted that although early apoB-LP retention precedes and triggers endothelial activation and monocyte entry, lesional monocyte-derived macrophages (below) may subsequently secrete apoB-LP-binding proteoglycans (Williams and Tabas, 1995). This mechanism likely plays an important role in the amplification of LP retention once lesions becomes established, which in turn can help explain why atherosclerotic lesions fail to undergo inflammation resolution (Tabas, 2010a).
Monocytes originate from bone marrow-derived progenitor cells and this early stage of monocyte development may be regulated by cellular cholesterol content in a manner that can affect atherogenesis. Mice whose monocyte progenitor cells have defective cholesterol efflux due to deficiency of ABCA1 and ABCG1 transporters (below) show an increase in circulating monocyte number ("monocytosis") and increased atherosclerosis (Yvan-Charvet et al., 2010a). Importantly, both the monocytosis and increased atherosclerosis are reversible by restoring cholesterol efflux to high-density lipoprotein (HDL) and there are correlations in humans among high HDL, lower blood monocyte counts, and decreased risk for atherosclerosis (Coller, 2005).
The role of monocyte subsets in atherogenesis has been a topic of great interest throughout the last decade. In mice, hypercholesterolemia is associated with an increase in an inflammatory monocyte subset referred to as Lychi, and these cells enter developing atherosclerotic lesions more readily than Lyclo monocytes. Data in other models of inflammation have suggested that Lychi monocytes participate in the acute response to inflammation, whereas Lyclo monocytes are associated with inflammation resolution (Arnold et al., 2007). However, this scenario may not be directly applicable to atherosclerosis because of the chronic nature of this inflammatory process, and maximal suppression of atherogenesis requires genetic manipulations that block entry of both subsets (Tacke et al., 2007). Moreover, there are important differences in the characterization and roles of monocyte subsets in humans vs. mice. Thus, the key issue as to whether monocyte subsets—and the macrophages and other cells (e.g., dendritic cells) derived from them—have different roles in atherogenesis or plaque progression remains unresolved.
Following chemokinesis, monocytes become tethered and roll on endothelial cells overlying retained apoB-LPs, notably through the interaction of monocyte P-selectin glycoprotein ligand-1 (PSGL-1) with endothelial selectins (Mestas and Ley, 2008). Monocytes then become firmly adhered to lesional endothelial cells through the interaction of monocyte integrins with endothelial cell ligands, and recent evidence has implicated monocyte type I interferon signaling in this process (Goossens et al., 2010). Immunohistochemical analysis of human lesions and molecular-genetic causation studies in gene-targeted mice suggest that the monocyte integrins very late antigen-4 (VLA-4) and lymphocyte function-associated antigen 1 (LFA-1) and their respective endothelial cell ligands vascular cell adhesion molecule (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1), may be particularly important in the setting of early atherogenesis. Moreover, it should be noted that platelet aggregation on endothelium overlying atherosclerotic lesions may also promote monocyte-endothelial interaction by activating NF-κB signaling and adhesion molecule expression, and via deposition of platelet-derived chemokines on activated endothelium (Mestas and Ley, 2008; Koenen et al., 2009). Finally, firm adhesion of monocytes is followed by their entry into the subendothelial space (diapedesis) (Kamei and Carman, 2010). However, while diapedesis has been studied extensively in other models of inflammation, work in atherogenesis is limited. In particular, molecules that have been implicated in diapedesis in vitro, such as tissue factor and PECAM-1, have not been shown to participate in diapedesis in the setting of atherogenesis per se (Goel et al., 2008; Harry et al., 2008). Part of this complexity may be due to the multiple functions of these molecules and the difficulty of assaying diapedesis in vivo.
Monocyte Differentiation into Macrophages
Driven by macrophage-colony stimulating factor (M-CSF) and probably other differentiation factors, the majority of monocytes in early atheromata become cells with macrophage- and/or dendritic cell-like features (Johnson and Newby, 2009; Paulson et al., 2009). There has been great interest in macrophage heterogeneity in atherosclerotic lesions, particularly those related to pro-inflammatory processes ("M1") vs. those involved in resolution and repair ("M2"), but no clear picture has yet emerged from these studies (Johnson and Newby, 2009). Much of the theory in this area has been driven by in vitro studies exploring gene/protein expression patterns and functional attributes in monocytes or macrophages subjected to different types of growth factors; Th1- vs. Th2-derived cytokines; transcription factors, notably PPARs; growth factors; and even atherogenic lipoproteins and lipids (Johnson and Newby, 2009; Kadl et al., 2010). However, the situation in the subendothelium is almost certainly more complex and there is a significant gap between the in vitro and in vivo observations. Future studies will likely need to focus on macrophage heterogeneity characterized by differential expression of specific molecules or molecular networks that have functional significance for atherogenesis rather than heterogeneity based on the aforementioned M1 vs. M2 paradigm (Kadl et al., 2010). Moreover, as discussed below, any discussion of "macrophage heterogeneity" must take into account the role of dendritic-like cells and the plasticity of myeloid-derived cells.
Foam Cells (Figure 2)
Even at very early stages of atherogenesis, many macrophages and dendritic-like cells are noted to have cytoplasmic, membrane-bound lipid droplets (foam cells). Foam cell formation begins when phagocytes ingest and process apoB-LPs. The mechanism of this uptake is a widely studied and hotly debated area. Early work suggested that uptake of oxidized LDL by scavenger receptors, notably the type A scavenger receptor (SRA) and a member of the type B family, CD36 (Kunjathoor et al., 2002), was a prominent mechanism. However, recent gene-targeting studies in Apoe−/− mice that have paid careful attention to genetic background indicate that additional mechanisms of foam cell formation are also operational in atherosclerosis (Moore et al., 2005; Manning-Tobin et al., 2009). In vitro work offers plausible mechanisms, including phagocytosis of matrix-retained and aggregated LPs and fluid phase pinocytosis of non-retained native LDL (Tabas et al., 1993; Kruth et al., 2005). Further mechanistic and in vivo studies are needed to fully assess the relative importance of these processes, taking into account stage and location of lesions and the particular model being investigated.
Once ingested, the cholesteryl esters (CE) of the LPs are hydrolyzed in late endosomes to cholesterol, often referred to as free cholesterol (FC), and fatty acids (Maxfield and Tabas, 2005). Late endosomal FC is trafficked to peripheral cellular sites by poorly understood mechanisms involving the late endosomal proteins NPC1 and NPC2 and the lipid lysobisphosphatidic acid. Recent work also points to a possible role of the ABC transporter protein ABCG1 in this process (Tarr and Edwards, 2008). FC delivery to the endoplasmic reticulum (ER) has important roles in down-regulating both LDL receptors and endogenous cholesterol synthesis by suppressing the sterol-regulatory element binding pathway (SREBP) (Brown and Goldstein, 1997). Moreover, the FC undergoes re-esterification to cholesteryl fatty acid esters (the "foam" of foam cells) by the ER enzyme acyl-CoA:cholesterol ester transferase (ACAT) (Brown et al., 1980), a process critical to preventing FC-induced cytotoxicity, which if defective, may promote macrophage death in advanced atheromata (below).
FC trafficking from lysosomes, as well as from re-hydrolyzed CE droplets, can also traffic to the plasma membrane and thus be available for efflux out of the cell. Cholesterol efflux is thought to be a major process involved in plaque regression when hypercholesterolemia is reversed. This expansive area has been covered in many reviews over the last decade, however, it is worth emphasizing several key points. The mechanisms and exact route of cholesterol transport to the plasma membrane are not fully known, although Golgi-to-plasma membrane vesicular transport may be involved (Chang et al., 2006; Maxfield and Tabas, 2005). Interestingly, trafficking of FC out of lysosomes may become defective in lesional macrophages, which would constitute a barrier to cholesterol efflux and lesion regression (Jerome, 2006). Once at the plasma membrane, cholesterol is transferred to the outer leaflet, where it is removed from the cells by ABCA1- and ABCG1-mediated transport to apolipoprotein A1 and HDL, respectively, or by "passive diffusion" to cholesterol-poor HDL (Tall et al., 2008; Rothblat and Phillips, 2010). As predicted, deletion of both ABCA1 and ABCG1 in macrophages enhances atherosclerosis in mice (Tall et al., 2008). Interestingly, deletion of ABCG1 alone in macrophages has the opposite effect, which has been ascribed to either compensatory induction of ABCA1 and apolipoprotein E or to depletion of early lesional macrophages by oxysterol-induced apoptosis (Ranalletta et al., 2006; Tarling et al., 2010).
Extensive in vitro and in vivo work has focused on how a sterol-regulated transcription factor, LXRα, induces ABCA1 and ABCG1 and, through this and other mechanisms, promotes regression of foam cell lesions (Calkin and Tontonoz, 2010). Evidence that ABC transporters, LXRα, and other molecules carry out cholesterol efflux and "reverse cholesterol transport" in vivo has come from studies in which wild-type and gene-targeted mice are injected i.p. with macrophages loaded with radiolabeled cholesterol, and then the appearance of label in blood and feces is followed (Degoma et al., 2008). As a therapeutic strategy to promote lesion regression, investigators have attempted to enhance macrophage cholesterol efflux by increasing HDL or HDL-like particles or by increasing ABC transporters (Tall et al., 2008; Rothblat and Phillips, 2010). Examples include cholesteryl ester transfer protein inhibitors, LXR activators, and HDL mimetics (cf. section below on Therapeutic Strategies). While no drugs have yet been approved for this purpose, this set of strategies continues to be a major focus in cardiovascular drug discovery.
Functional Significance of Macrophage Cholesterol Loading
A central question in atherogenesis is how the loading of macrophages with lipoprotein-derived cholesterol alters macrophage function, particularly with regard to specific pro-atherogenic processes. While markedly excessive FC loading in the face of defective esterification can have profound effects on macrophages in advanced lesions (below), the influence of CE storage is less certain, because CE is stored in membrane-bound neutral lipid droplets in the cytoplasm and is thus sequestered from cellular membranes (Maxfield and Tabas, 2005). However, even with normal esterification, macrophages exposed to atherogenic lipoproteins may have some degree of FC enrichment in the plasma membrane which may enhance inflammatory signaling through clustering-mediated activation of signaling receptors due to the increased order parameter of cholesterol-enriched membranes (Yvan-Charvet et al., 2007; Zhu et al., 2008; Tang et al., 2009). In addition, other pro-atherogenic lipids delivered to cells by lipoproteins, particularly modified lipoproteins, may have potent effects. An example includes apoptosis induced by oxysterols and oxidized phospholipids delivered through the uptake of oxidatively modified forms of lipoproteins (Seimon et al., 2010b).
As a new approach to the question of how lipoprotein loading affects macrophage biology, a recent study used liquid chromatography-electrospray ionization-tandem mass spectrometry (LC-ESI-MS/MS) to identify macrophage secreted proteins using an in vivo model of unloaded vs. cholesterol-loaded macrophages (Becker et al., 2010). Gene Ontology analysis of proteins differentially expressed by peritoneal macrophages obtained from chow- vs. fat-fed Ldlr−/− mice revealed networks involved in cytoskeletal regulation, vesicle-mediated transport, and lipid binding. Interestingly, a subset of the differential expression appeared to be mediated by a decrease in apolipoprotein E (apoE) expression in the cholesterol-loaded macrophages. In terms of relevance to atherosclerosis, a very high percentage of these proteins have been implicated in lesion development in various gene-targeted mouse models, and when the Ldlr−/− mice were administered two drugs that decrease atherosclerosis—a statin and a thiazolidinedione—the differences in macrophage protein profiles were markedly decreased. This study not only reveals potentially important information about how macrophages respond to lipoprotein loading in vivo but also provides an example of how system biology approaches may be applied to this area of research.
Inflammatory Activation of Lesional Macrophages (Figure 3)
Figure 3. Signaling pathways activated in the lesional macrophage.

Various pro- and anti-inflammatory forces act on the macrophage in atherosclerotic lesions leading to activation of downstream cascades, such as the inflammasome, scavenger receptor (SR)/Toll-like receptor (TLR) cooperative signaling, ER stress, expression of the sterol responsive network, and efflux of cholesterol via ABCA1 and ABCG1 transporters.
The role of cholesterol loading in macrophage inflammation raises the broader issue of mechanisms and consequences of macrophage-mediated inflammation in atherosclerosis. Immunohistochemical studies, and studies of RNA isolated from lesional macrophages by laser capture microdissection, have clearly identified markers of inflammation in macrophages in human and animal plaques (Shibata and Glass, 2009). Moreover, there have been hundreds of studies examining the effect of injected anti- or pro-inflammatory reagents or of gene targeting pro-or anti-inflammatory genes in animal models of atherosclerosis. In most cases, manipulations that decrease inflammation have a protective effect on lesion area, and vice versa. However, given the importance of inflammatory pathways in endothelial cells (above), the role of macrophage inflammation per se can only be determined through studies that specifically target this cell type.
In one such study in which the pathway involved in NF-κB activation was specifically targeted in macrophages using the “Cre-LoxP” system of gene recombination, surprising results were found. To selectively disable NF-κB signaling, the investigators first flanked a critical exon of the Ikk2 gene with loxP sites in mice and crossed these to mice that expressed the Cre recombinase under control of the LysM promoter, which directs expression in macrophages, as well as neutrophils (Kanters et al., 2003). The resultant Ikk2fl/flLysmcre or Ikk2fl/fl (control) mice were used as bone marrow donors to Ldlr−/− mice, which were then fed a Western-type diet for 10 weeks. Surprisingly, although macrophages isolated from the Ldlr−/−Ikk2fl/flLysmcre chimeric mice had an approximately 50% decrease in NF-κB and LPS-induced TNFα secretion, they showed an increase in lesion area compared with the control group (Ldlr−/−Ikk2fl/fl). As an illustration of the importance of cell type specificity in the investigation of atherosclerosis, a study in Apoe-/− mice showed that endothelial-targeted inhibition of NF-κB signaling results in the "expected" decease in lesion area, which was associated with a decrease in recruitment of macrophages to lesions (Gareus et al., 2008). Although no definitive mechanism linking increased lesion area to macrophage NF-κB suppression was elucidated in the Ikk2fl/flLysMCre study, macrophages from these mice showed a decreased IL-10 response, which is athero-protective in mice (Han et al., 2010).
In terms of pathways through which NF-kB becomes activated in lesional macrophages, there are many studies showing that global deletion of various TLR family members or their adaptors decrease lesion area in fat-fed Ldlr−/− or Apoe−/− mice (Curtiss and Tobias, 2008). One of the few studies to address the role to TLR signaling in macrophages showed that fat-fed Ldlr−/− mice transplanted with bone marrow from Tlr2−/− mice had decreased lesion area only when the mice were challenged with a TLR2 activator (Mullick et al., 2005). In addition, a newly described TLR heterodimer of TLR4/TLR6 has been indentified to cooperate with CD36 to activate NFKB in response to oxidized LDL (Stewart et al., 2009). Furthermore, stimulation of the macrophage CD40 receptor by its ligand, CD40L, may also trigger NF-kB activation, and fat-fed Ldlr−/− mice transplanted with bone marrow from Cd40−/− mice showed a reduction in lesion area and inflammation (Lutgens et al., 2010).
A fascinating new area is the role of macrophage inflammasome signaling in atherosclerosis (Figure 3). When macrophages are exposed to crystalline material, maturation and secretion of IL-1β and IL-18 is effected through an "inflammasome" complex involving multiple proteins, notably NLRP3, ASC, and caspase-1 (Duewell et al., 2010; Rajamaki et al., 2010). This pathway can be induced in cultured macrophages and in mouse peritoneal macrophages in vivo by cholesterol crystals, which have been shown to exist even in early atherosclerotic lesions. Cholesterol crystal-induced macrophage inflammation involves phagolysosomal disruption, inflammasome components, cathepsin B, and cathepsin L (Duewell et al., 2010; Rajamaki et al., 2010). Western diet-fed Ldlr−/− mice transplanted with marrow cells lacking NLRP3 or ASC had a decrease in lesion size and in levels of IL-18 (Duewell et al., 2010).
The Role Macrophages in Advanced Atherosclerosis (Figure 4)
Figure 4. Forces in the advanced lesion that destabilize the atherosclerotic plaque.
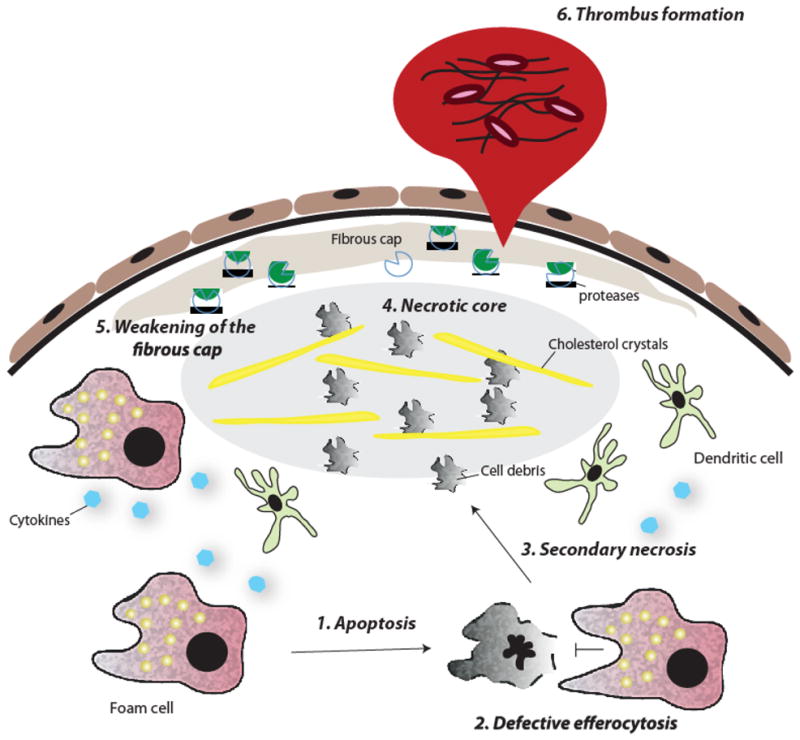
(1) Macrophage foam cells undergo apoptosis as a result of prolonged ER stress and other stimuli; (2) these apoptotic cells are not effectively cleared by advanced lesional macrophages (defective efferocytosis); (3) accumulation of apoptotic cells induces secondary necrosis and (4) contributes to the formation of a necrotic core. (5) Smooth muscle cell death and protease degradation of extracellular matrix weakens the fibrous cap, making it susceptible to rupture. (6) Exposure of the thrombogenic material of the lesion causes platelet aggregation and thrombus formation.
As described in the previous sections, atherosclerosis is a non-resolving inflammatory condition, and monocytes continue to enter plaques and differentiate into macrophages as atherosclerotic lesions progress. Importantly, macrophages contribute to size-independent changes in plaque morphology, notably necrotic core formation and fibrous cap thinning, that characterize the “vulnerable” plaque. Measures of these processes in advanced plaque progression may be particularly relevant to acute atherothrombotic cardiovascular disease in humans. Lumenal thrombosis requires communication between pro-coagulant and pro-thrombotic factors in the intima with coagulation factors and platelets in the lumen. In approximately 70% of autopsy specimens that successfully identify the lesion responsible for an acute vascular event ("culprit" lesion), this breach is manifested as a "rupture" site (Virmani et al., 2002). The rupture site, which is often located to the side ("shoulder") of raised lesions, is almost always in close proximity to areas of plaque necrosis ("necrotic core") and is associated with thinning of the protective scar ("fibrous cap") that normally covers inflamed atheromata. One of the key questions in the atherosclerosis field is how macrophages contribute to the features of such culprit lesions.
Possible Role of Advanced Lesional Macrophages in Fibrous Cap Thinning
Any process that decreases the synthesis of fibrous cap collagen by fibromyoblast-like smooth muscle cells (SMCs) in the intima and/or contributes to cap collagen degradation would be expected to promote the formation of rupture-prone plaques. Vulnerable plaques show evidence of SMC death and decreased numbers of SMCs, and in vitro data show that macrophages can trigger apoptosis in SMCs by activating their Fas apoptotic pathway and by secreting TNFα and nitric oxide (Boyle et al., 2003). Macrophages may also decrease collagen synthesis in intimal SMCs without actually killing the cells. For example, in regions of vulnerable plaques that have defective clearance of apoptotic cells (below), macrophage secretion of TGFβ is likely decreased (Fadok et al., 1998), which may deprive neighboring SMCs of this important inducer of collagen biosynthesis.
A widely studied topic in the atherosclerosis field is the potential role of macrophage-derived matrix metalloproteinases (MMPs) in fibrous cap thinning. MMPs are a family of protease-activated enzymes that can degrade various types of extracellular matrix (ECM) proteins, including collagens, collagen fragments, elastin, and other ECM molecules including laminin, fibronectin, and proteoglycan core proteins. The regulation of MMPs by athero-relevant factors is complex and is influenced by transcriptional inducers and repressors and by protease activators and inhibitors. Moreover, once activated, certain MMPs can activate other ones.
Studies showing a temporal and spatial correlation between the presence of macrophages in rupture-prone shoulder regions of plaques, thinning of the fibrous cap in these regions, and local accumulation of activated MMPs, especially MMP-2 and MMP-9, have stimulated great interest in the potential role of MMPs in plaque rupture (Galis et al., 1994). Unfortunately, because mouse models of atherosclerosis do not recapitulate plaque rupture, specific molecular hypotheses in advanced atherosclerosis have been difficult to test in vivo through gene-targeting studies, leading investigations to focus on lesion area or aneurysm development instead. The lesion area studies have yielded results that have been difficult to synthesize into a unified, mechanism-based hypothesis. However, when the endpoint of advanced plaque collagen content has been studied in mouse models lacking global or macrophage-derived MMP-13 and-14, respectively, increases in plaque interstitial collagen were observed (Deguchi et al., 2005; Schneider et al., 2008). In an MMP overexpression study that attempted to look directly at plaque disruption, macrophage overexpression of wild type MMP-9 had little effect in Apoe−/− mice due to a lack of MMP activation in plaques, but overexpression of a constitutively active mutant form of MMP-9 resulted in plaque fissuring (Gough et al., 2006).
In summary, observational data in vulnerable human plaques and the known biology of MMPs have generated plausible hypotheses regarding their role of certain MMPs in fibrous cap thinning and plaque rupture. Moreover, there are other in vitro and in vivo data implicating roles for macrophage-derived serine proteases, such as neutrophil elastase, and cysteine proteases, such as cathepsins S, K, and L, in the degradation of elastin and collagen in advanced lesions (Liu et al., 2004). However, the few molecular causation studies in mice addressing matrix protease hypotheses have had to rely on the endpoints of plaque collagen or elastin content, fibrous cap thinning, and matrix fissuring. New mouse models of frank plaque rupture and ideally acute lumenal thrombosis are needed to validate these ideas and to provide the impetus for testing MMP inhibitor strategies in humans.
Advanced Lesional Macrophages and Plaque Necrosis
A second critical feature of dangerous plaques is the necrotic core, which contributes to inflammation, thrombosis, proteolytic plaque breakdown, and physical stress on the fibrous cap (Virmani et al., 2002). Necrotic cores arise from the combination of apoptosis of advanced lesional macrophages and defective phagocytic clearance, or "efferocytosis," of the apoptotic macrophages in advanced plaques (Tabas, 2010a). This combination is critical: macrophage death also occurs in early atherosclerotic lesions, but here efferocytic clearance is efficient, leading to decreases in lesion cellularity, inflammation, and plaque progression rather than an increase in plaque necrosis.
A number of processes in advanced lesions may trigger macrophage death, and it is almost certain that a combination of factors and processes play a role in vivo. Examples include growth factor deprivation, oxidative stress, and death receptor activation by ligands that exist in advanced atheromata. A potential role for these and other processes is supported primarily by studies with cultured macrophages, but, in most cases, evidence of relevance in vivo through molecular-genetic causation studies is lacking. In this regard, several studies stand out in terms of their effects on macrophage-induced plaque necrosis, including the detrimental effect of NF-κB suppression (Kanters et al., 2003) and the beneficial effects of interrupting type I interferon signaling (Goossens et al., 2010) or TLR2/4 signaling (Seimon et al., 2010b).
Another emerging concept that has gained in vivo support is that prolonged activation of endoplasmic reticulum (ER) stress pathways, primarily the Unfolded Protein Response (UPR), contributes to macrophage death and subsequent plaque necrosis in advanced atheromata (Tabas, 2010b). The UPR functions in normal physiology to correct disequilibria in ER function, but when ER stress is abnormally prolonged, persistent expression of the UPR effector CHOP can trigger apoptosis (Tabas and Ron, 2010). There are many molecules and processes in advanced lesions that can lead to UPR activation, and markers of ER stress are elevated in these lesions. Importantly, CHOP expression and apoptosis show a strong correlation with stage of human coronary artery lesions, with both parameters markedly increased in plaques with vulnerable morphology (Myoishi et al., 2007), and deletion of CHOP in mouse models suppresses lesional macrophage death and plaque necrosis (Thorp et al., 2009; Tsukano et al., 2010). Several downstream mediators of CHOP have been implicated in this process including the Ca2+/calmodulin-dependent protein kinase II (CaMKII), which is a central integrator of the apoptotic execution pathway through activation of Fas, outer mitochondrial membrane permeabilization, STAT1, and NADPH oxidase (NOX) (Timmins et al., 2009). In this context, a recent study implicated oxidative stress and NOX in plaque necrosis in aged Ldlr−/− mice (Collins et al., 2009).
A potent inducer of prolonged ER stress in macrophages is insulin resistance, and advanced plaques of type 2 diabetics are characterized by very large necrotic cores independent of lesion size (Tabas et al., 2010). Macrophages with defective insulin signaling, such as occurs in type 2 diabetes, have an amplified ER stress-apoptosis response, leading to increased macrophage apoptosis and plaque necrosis in a mouse model of atherosclerosis (Han et al., 2006). Another potential inducer of ER stress in the settings of insulin resistance and obesity are elevated saturated fatty acids (SFAs). These ER stress-induced macrophage apoptosis pathways likely complement other consequences of diabetes on advanced lesional macrophages, such as dyslipidemia-mediated pro-inflammatory effects, to promote advanced plaque progression.
Two key concepts related to the mechanism and in vivo relevance of ER stress-induced macrophage death are the presence of compensatory cell survival pathways and the role of a "two-hit" apoptotic process. In vitro and in vivo data indicate that before macrophages subjected to prolonged ER stress undergo apoptosis, there is activation of a number of compensatory cell-survival pathways, including those involving NF-κB, Akt, p38α, and autophagy (Tabas, 2010b). Importantly, the onset of apoptosis correlates with eventual suppression of these pathways, and apoptosis can be accelerated by experimentally interfering with the pathways. The two-hit concept is based upon the likelihood that ER stress in vivo may be more subtle than is often modeled in vitro, where one can induce macrophage apoptosis by high concentrations of ER stress-inducing agents. In this context, sub-apoptotic, prolonged ER stress in macrophages can set the stage for apoptosis induction by other sub-apoptotic factors, leading to a synergistic apoptotic response. Remarkably, a potent and athero-relevant "second hit" in ER-stressed macrophages is activation of innate immune pattern recognition receptors (PRRs), which may have evolved as a host defense response against invasive organisms that require living macrophages to survive (Seimon et al., 2010a). Two major classes of PRRs, scavenger receptors (SRs) and TLRs, have been implicated in atherosclerosis and are likely to be most relevant to apoptosis in ER-stressed macrophages. These PRR pathways trigger apoptosis in ER-stressed macrophages by suppressing compensatory cell-survival pathways that are induced by prolonged ER stress and by adding to the burden of oxidative stress. Importantly, atherosclerotic mouse models lacking two SRs, SR-A and CD36, or two TLRs, TLR4 and TLR2, are partially protected against advanced lesional macrophage death and plaque necrosis in a manner that cannot be explained simply by other effects on early plaque progression. In addition, a lipoprotein in humans called lipoprotein(a) [Lp(a)], which is highly associated with atherothrombotic vascular disease and a carrier of oxidized phospholipids (oxPLs) (Bergmark et al., 2008), is a potent trigger of apoptosis in ER-stressed macrophages through oxPL-mediated activation of a CD36/TLR2 "2nd-hit" pathway (Seimon et al., 2010b). Notably, a study in a transgenic rabbit model showed that increased plasma Lp(a) led to a marked increase in plaque necrosis without altering signs of early lesion development (Sun et al., 2002).
Defective efferocytosis
Macrophage apoptosis by itself will not trigger plaque necrosis. Rather, plaque necrosis results when apoptotic macrophages are not sufficiently cleared by phagocytes (efferocytosis) (Tabas, 2010a). Efficient efferocytosis of apoptotic macrophages results in at least three critical protective effects: (a) it clears the cells before membrane damage leads to extracellular leakage of toxic intracellular material; (b) it triggers an IL-10- and TGFβ-mediated anti-inflammatory response in the efferocytes; and (c) it promotes survival of the efferocytes themselves so that they do not succumb to potentially toxic factors in apoptotic cells. This cell survival effect involves robust esterification and efflux of cholesterol, efflux of pro-apoptotic oxidized lipids, and activation of Akt and NF-κB cell-survival pathways.
Efferocytosis is effected through phagocytic receptors (e.g., MerTK, LRP-1), apoptotic cell ligands (e.g., phosphatidylserine, C1q), and bridging molecules (e.g., Gas6) that enable recognition of apoptotic cells by the phagocytes (Henson et al., 2001). In advanced atherosclerosis, the combination of apoptotic macrophages and tissue necrosis implies that efferocytosis is defective (Henson et al., 2001). Direct support for this concept came from a study that carefully quantified apoptotic cell-phagocyte association in human advanced plaques and control tonsillar tissue (Schrijvers et al., 2005). In tonsils, as expected, almost all of the apoptotic cells were associated with phagocytes, whereas in advanced atheromata there were many free apoptotic cells. Similar results have been found in mouse models of advanced atherosclerosis, and when efferocytosis is decreased further through gene targeting of effector molecules like MerTK, LRP-1, transglutaminase-2, C1q, and MFG-E8, there is increased presence of uningested apoptotic cells, inflammation, and plaque necrosis (Tabas, 2010a). Normal functioning of MerTK and LRP-1 may be particularly important in preventing plaque necrosis, because in addition to their roles in efferocytosis, MerTK suppresses macrophage inflammation, and LRP-1 promotes macrophage survival {4682}.
The mechanisms of defective efferocytosis in advanced plaques are not known. Overwhelming apoptosis is not likely to be a major factor, because efferocytosis is a high-capacity process (Henson et al., 2001), and when apoptosis is increased through genetic manipulations in early atherosclerotic lesions, where efferocytosis is not defective, apoptotic cells are efficiently cleared (Tabas, 2010a). Rather, it is likely that efferocytosis becomes defective in advanced lesions. Several possible mechanisms might contribute to defective efferocytosis, including oxidative stress-induced efferocyte death resulting from defective cholesterol efflux after apoptotic cell engulfment (Yvan-Charvet et al., 2010b); LP-associated PLA2-mediated hydrolysis of oxidized PS on the surface of apoptotic cells, which is a ligand for efferocytes (Wilensky and Macphee, 2009); and protease-mediated cleavage of the efferocytosis receptor MerTK (Sather et al., 2007). Another mechanism that may be relevant to obesity is based on in vitro and in vivo studies showing that macrophages exposed to saturated fatty acids have defective engulfment of apoptotic cells, perhaps due to changes in plasma membrane structure (Li et al., 2010). These and other ideas will require careful assessment in vulnerable plaques and then genetic causation testing in mouse models of advanced atherosclerosis.
Dendritic Cells and Atherosclerosis
The distinction between macrophages and dendritic cells (DCs) is controversial topic, and it is further compounded by the likelihood of plasticity between these two types of cells (Leon et al., 2005). For the purpose of this section, DCs will be defined as immune cells that internalize, process, and present antigen, leading to activation or suppression of T cells. Both pre-atherosclerotic susceptible regions of arteries and established atheromata are populated with cells that have DC-like properties (Paulson et al., 2009; Choi et al., 2009). The exact origins of arterial-wall and lesional DCs have not yet been definitively delineated. A study in fat-fed Apoe−/− mice suggests that a population of DCs in atherosclerotic lesions originates from Ly-6Clo monocytes (Tacke et al., 2007). In terms of functional significance, there is some evidence that lesional DCs present antigen to and activate lesional T cells (Bobryshev, 2005). Moreover, early lesional DCs show two fundamental characteristics that were previously ascribed to early lesional macrophages: proliferation and foam cell formation (Paulson et al., 2009). Attempts to probe these events through in vivo causation studies have yielded mixed results. For example, while acute ablation of DCs in Ldlr−/− mice fat a high−fat diet for 5 days resulted in a 55% decrease in foam cell lesion area (Paulson et al., 2010), lesion area was only modestly decreased in 10-week fat-fed Gmcsf−/−Ldlr−/− mice, which is a model of chronic DC deficiency (Shaposhnik et al., 2007). Finally, in the setting of rapid and robust plasma cholesterol unloading in a mouse model of atherosclerosis, DC-like cells emigrated from lesions in a CCR7-dependent manner, leading to plaque regression (Feig et al., 2010). These intriguing studies warrant further study of the role of DCs in atherosclerosis.
Methods of Investigation and Their Limitations
The reliability of the data cited in this review are only as good as the methods used to generate and analyze the data. Thus, it is important to understand the limitations of both mechanistic studies using cultured macrophages and in vivo studies using animal models. There are many different types of cultured macrophage models, with important differences among them and actual lesional macrophages. In general, studies with macrophages differentiated and activated in vivo, such as those isolated from the peritoneum of mice treated under various conditions, are probably the most relevant to atherosclerosis, particularly in the setting of hypercholesterolemia, which promotes loading with lipoprotein-derived lipid (Becker et al., 2010). However, the only way to determine the relevance of findings with any cultured macrophage model is to test the findings in animal models of atherosclerosis and to show links with human studies.
In that context, there are several important points to consider with regard to animal studies that attempt to explore macrophage-based hypotheses of atherosclerosis. In an attempt to improve specificity for macrophages, many studies use bone marrow transplantation (BMT) of irradiated mice with donor cells derived from bone marrow or fetal liver. However, BMT studies may suffer from the effects of aortic irradiation and from the fact that all bone marrow-derived cells are affected, including neutrophils, T cells, B cells, and mast cells (Schiller et al., 2001). Approaches to improve macrophage specificity in this model include viral transduction of the donor cells with macrophage promoter-driven genes (Gough et al., 2006) and the use of various cre-lox models, as in the aforementioned study using Ikk2fl/flLysmcre mice (Kanters et al., 2003). Although, the latter approach has proven to be very useful, it is occasionally hampered by heterogeneity in cre-mediated recombination among individual lesional macrophages, and LysMCre also targets neutrophils, which are becoming increasingly recognized as participants in atherosclerosis (Baetta and Corsini, 2009).
In all cases, the greatest limitation is that animal models of atherosclerosis may only partially reflect processes in human atherosclerosis. As is apparent from the previous sections, most atherosclerosis studies are carried out in mice, and the two most widely used models are high-fat ("Western-type diet (WD)")-fed Ldlr−/− mice and chow-fed or WD-fed Apoe−/− mice (Breslow, 1996). Ldlr−/− mice are a model of the rare human disease, homozygous familial hypercholesterolemia, although LDL-induced atherosclerosis is, in general, a very common process in humans. Apoe−/− mice have a form of remnant lipoproteinemia that is rare in humans, but post-prandial remnant lipoproteins likely contribute to atherosclerosis in most humans. Influences of other systemic risk factors in these mice, such as levels of plasma HDL and diet-induced insulin resistance, may or may not mimic certain features of humans at risk for atherosclerotic vascular disease. Moreover, most studies use mice on the atherosclerosis-susceptible C57BL6/J background, which has peculiar features that likely affect macrophage biology and atherosclerosis, including a pro-atherogenic increase in the Th1/Th2 balance (Schulte et al., 2008). Of note, the number of mice used in many atherosclerosis studies is far below that required by power calculations and studies in which pure backgrounds are not used may be confounded by major differences in atherosclerosis susceptibility among different strains of mice (Koschinsky and Marcovina, 2004). The latter problem is not always solved by using littermate controls due to the influence of "passenger" genes clustered near the targeted locus (Lusis et al., 2007), and the use of pure C57BL6/J ES cells for gene targeting studies, while not as efficient as other types of ES cells, may be the only way to avoid this problem.
Finally, the choice of atherosclerosis endpoints may influence the usefulness of the data obtained from animal studies. Most studies use cross-sectional lesional area at the aortic root and/or en face lipid staining of the whole aorta. These endpoints are relevant for early atherogenesis and for processes related to early-to-mid-stage lesion progression. While the pathological features of early-to-mid-stage lesions in mice share many common properties with those in humans, lesion area is a poor predictor of clinical disease in humans (Virmani et al., 2002). There has been increasing interest in measuring certain advanced plaque features in mouse models of atherosclerosis, including necrotic core formation, thinning of the fibrous cap, breaches in the subendothelial matrix, and measures of intimal cell apoptosis and apoptotic cells clearance by lesional phagocytes (efferocytosis). Although these features mimic those in human advanced plaques, mouse atherosclerosis is not a good model of true plaque rupture or acute lumenal thrombosis (Rosenfeld et al., 2002). There are claims that close inspection of the innominate (brachiocephalic) artery in hypercholesterolemic mice or the use of wire-induced injury in various animal models can partially overcome this problem, but true plaque rupture and acute lumenal thrombosis are rarely seen, thus necessitating the development of new models of these processes.
The litany of problems with methods of investigation in this area of research may lead one to be skeptical about studies in the atherosclerosis field. Indeed, the literature on cultured macrophages and gene targeting studies in this area is overwhelming, with many genes being reported to have "major" effects, all in the face of a significant number of studies showing opposite results in different laboratories. Moreover, many studies elucidate a mechanism in vitro, show an overall effect on lesion area in vivo, but fail to link the two, i.e., by demonstrating that the mechanism gleaned from in vitro experiments is operational and causative for the atherosclerosis changes in vivo. However, technical and conceptual advances on many fronts have recently increased the quality of work in this are, and consistent results are apparent in the most carefully conducted and comprehensive investigations. In the end, the true test of validity will be the demonstration that the results are consistent with the results of human genetic studies and with the outcomes of mechanism-based clinical trials.
Therapeutic Strategies Based on the Pathobiology of Macrophages in Atherosclerosis
General Principles
The most effective and direct way to prevent or treat atherosclerosis is to decrease subendothelial apoB-LP retention by lowering apoB-LPs in the blood through lifestyle changes and drugs (Steinberg et al., 2008). Studies have shown that this both decreases monocyte entry (Potteaux et al., 2010) and promotes macrophage egress from lesions (Feig et al., 2010). Indeed, if circulating apoB-LPs could be lowered below the threshold level required for subendothelial lipoprotein retention prior to lesion initiation, which in Western society occurs in the early teens, atherosclerosis could be completely prevented. However, until this goal can be achieved in a safe manner with universal application and compliance, atherosclerotic disease will remain a major cause of death, particularly in view of expanding risk factors, notably obesity, physical inactivity, and insulin resistance world-wide and smoking in emerging countries. Thus, translational researchers in this area have considered the concept that therapy directed at the arterial wall in general, and macrophages in particular, could be additive or synergistic with realistic goals of apoB-LP lowering.
Studies in mice have shown that prevention of monocyte entry into lesions by antibody or gene-targeted neutralization of chemokines, chemokine receptors, or adhesion molecules lessens atherosclerosis (Mestas and Ley, 2008). However, by the time most subjects come under clinical care, many lesions are already far advanced, and so simply blocking monocyte entry at this stage may not be able to reverse key aspects of plaque progression, such as necrotic core formation. As mentioned above, reduction of early lesional macrophages through forced apoptosis and subsequent efferocytosis lessens lesion progression (Liu et al., 2005; Arai et al., 2005; Babaev et al., 2008), however attempts to increase macrophage apoptosis in lesions with defective efferocytosis may cause the untoward effect of plaque necrosis and lead to lesion vulnerability. Finally, the macrophage-related mechanisms involved in atherosclerosis and host defense are not easily distinguishable, and so many therapeutic approaches along these lines will likely result in increased susceptibility to infectious organisms. These problems could possibly be lessened by local delivery systems, such as atherosclerosis-directed nanoparticles (Chan et al., 2010), encapsulated macrophage-targeted siRNA (Aouadi et al., 2009), and stent-based delivery systems (Nakano et al., 2007), or by identifying and then targeting mechanisms that are more operational in atherosclerosis than in host defense. Although promising, these avenues will require future development.
Therapeutically inducing cholesterol efflux in macrophages or macrophage precursors by raising HDL and/or cholesterol efflux transporters is likely to have several beneficial, athero-specific effects, including decreasing inflammation related to monocytosis; suppressing activation of inflammatory signaling pathways in macrophages; and increasing efferocytosis by preventing cholesterol- and oxysterol-induced efferocyte death (above) (Tall et al., 2008). Moreover, HDL may have benefits beyond those related to macrophage cholesterol efflux, including prevention of subendothelial apoB-LP retention, decreasing activation of endothelial cells, and reducing LDL oxidation. Animal studies in which HDL-like particles are administered or in which HDL is raised through genetic techniques show beneficial effects on atherosclerosis. In humans, clinical trials are ongoing to assess the effect of cholesteryl ester transfer protein (CETP) inhibitors, which raise HDL and, importantly, also lower LDL. Although an initial trial in this area showed disappointing results, possibly due to off-target effects, the early results of a second CETP inhibitor trial appear promising (Cannon et al., 2010). Similarly, boosting macrophage ABC transporters through LXR agonists is also beneficial in animal models, and LXR agonists are being developed for trials in humans (Tall et al., 2008) (www.bms.com/research/pipeline/Pages/default.aspx). LXR agonists also have anti-inflammatory effects, and they increase expression of CCR7, which promotes macrophage egress from lesions, and MerTK, which mediates efferocytosis (Feig et al., 2010; Gonzalez et al., 2009). PPAR agonists, like LXR agonists, have been shown to have potentially beneficial effects on cholesterol metabolism and inflammation in macrophages in vitro and in atherosclerosis in animal models (Rigamonti et al., 2008). However, their actions are complex, multiple cell types are affected, and currently available drugs have off-target effects, some of which may actually exacerbate CAD.
Anti-oxidant therapy has been considered as a way to prevent atherosclerosis, in part through effects on macrophage processes (Witztum and Steinberg, 2001). The hope was that anti-oxidants would prevent foam cell formation and subsequent inflammatory changes by suppressing the formation of oxidized lipoproteins and lipids and by decreasing oxidative stress (Witztum and Steinberg, 2001). To the extent that scavenger receptors mediate oxidized lipoprotein-induced foam cell formation, the aforementioned studies in mice in lacking these receptors (Moore et al., 2005; Manning-Tobin et al., 2009) have raised uncertainties about this particular mechanism. Moreover, the results of vitamin E trials in human heart disease have been disappointing (Williams and Fisher, 2005). Nonetheless, this overall strategy should not necessarily be abandoned, because oxidized lipoproteins and lipids exist in human atheromata; there are enough in vitro mechanistic studies and pre-clinical studies to support certain aspects of the oxidative stress hypothesis, including roles in endothelial activation and advanced plaque progression; and it is likely that vitamin E was a poor choice to test this concept in humans. Thus, as we gain more knowledge about specific oxidative processes that contribute to atherosclerosis, such as possible roles for members of the NADPH oxidase family, new therapeutic opportunities in this area may become evident.
Targeting Macrophages in Advanced Lesions
Because only 2-3% of atherosclerotic lesions cause acute cardiovascular syndromes (Virmani et al., 2002), an efficient strategy may be to therapeutically alter those macrophage processes that are involved in advanced plaque progression. As mentioned above, HDL/sterol efflux-based therapy and well-conceived anti-oxidant approaches may be relevant to this topic. More direct attempts to block advanced plaque macrophage inflammation may be too non-specific or subject to adverse effects. For example, blocking TLRs would likely suppress host defense mechanisms, and blocking NF-κB may block an important cell-survival pathway in macrophages. A targeted approach that has been considered is prevention of secretion or activation of MMPs, or inhibition of their activity. However, as noted above, this is a complex area with poor understanding of mechanism and lack of definitive causation data in gene-targeting studies. Moreover, the interpretation of pre-clinical MMP inhibitor drug studies are hindered by the possibility of off-target effects, lack of specificity with regard to members of the MMP family, and lack of documentation of inhibition of MMP activity in plaques.
Our increasing understanding of how macrophages die in advanced lesions has provided new targets that could be the basis for drugs that prevent plaque necrosis, particularly inhibitors of pro-apoptotic ER stress pathways (above) (Tabas, 2010b). However, a concern is that inhibition of macrophage apoptosis would increase the number of living macrophages in advanced plaques, which might cause damage even in the face of decreased necrotic core formation. A more effective strategy may be to enhance the phagocytic clearance of the dead macrophages, which would result not only in decreased necrotic core formation but also in less macrophages in advanced lesions (Tabas, 2010a). Strategies being explored include pro-inflammation resolution mediators such as lipoxins, resolvins and interleukin-10 receptor activators (Serhan et al., 2008); inhibitors of MerTK cleavage (Sather et al., 2007); LXR activators, which increase MerTK expression, increase secretion of the efferocytosis effector apolipoprotein E, and protect efferocytes through ABCA1/G1-mediated efflux of toxic sterols {4386, 4682, 4631}; and PPARδ activators, which enhance efferocytosis in vitro (Mukundan et al., 2009). As with early inflammatory processes (above), those involved in advanced plaque progression may require local delivery systems.
Lessons from Human Genetic Studies
The above discussion has considered a relatively large number of potential macrophage targets for therapeutic development in the area of atherothrombosis. To what extent might the results of human genetic studies on atherothrombotic coronary artery disease (CAD) guide the choice of which targets to pursue? There are several caveats that need to be considered in this area: the genes identified thus far each account for only a small percentage of risk, as expected for a complex disorder; most findings to date are associated with systemic risk factors, such as high LDL or low HDL; and for many genome association findings, the actual gene responsible for the effect on CAD is not known. More specifically with regard to this review, no genes associated with human CAD have been directly tied to macrophage biology per se. However, several studies have shown that polymorphisms associated with increased levels of Lp(a), a carrier of oxidized phospholipids, are associated with CAD risk, which is consistent with many clinical studies linking high levels of Lp(a) with acute cardiovascular events (Tregouet et al., 2009). Another CAD locus is situated near the gene encoding CXCL12 (SDF-1), which is expressed in atherosclerotic lesional cells, including macrophages (Abi-Younes et al., 2000). However, it is unclear whether the CXCL12 gene per se is affected and, if so, whether CXCL12 is protective or detrimental for CAD. The locus with the strongest and most consistent association with coronary and peripheral atherosclerosis, but not necessarily acute thrombotic vascular events, is located on chromosome 9p21.3 (Lotta, 2010). This locus is associated with aneurysms in addition to CAD, potentially indicating a mechanism related to arterial wall remodeling, but the mechanism is not known. The locus contains an antisense non-coding RNA in the INK4 locus (ANRIL) as well as the cell cycle genes CDKN2B, which encodes p15INK4B; CDKN2A, which encodes p14ARF and p16INK4A; and MTAP. Several studies have begun to study this genomic region in depth, but much more work is needed to elucidate the mechanism of its association with atherosclerosis and whether it might be associated with macrophage biology.
Macrophage-Based Atherosclerosis Therapy and Clinical Trials: The Need for Improved Imaging Techniques
How would one test the effectiveness of an anti-atherosclerosis drug targeted at lesional macrophages in humans? Unlike the situation with LDL lowering therapy, there are no easily obtainable surrogate endpoints. The only sure sign of effectiveness would be clinical trials showing protection against acute atherothrombotic vascular events, but such trials are extremely expensive owing to the large number of patients required and the relatively long duration of the observation period. Thus, the drug industry has a taken a guarded approach toward the overall concept of arterial-based strategies. The eventual answer may lie in defining intermediate endpoints that are proven to be reliable for predicting the ability of a drug to decrease acute vascular events. While circulating biomarkers would be ideal for this purpose, their track record for this type of endeavor has not yet been encouraging. However, given the unique morphology of the plaques that actually cause acute coronary syndromes, imaging modalities focused in this area may offer the best hope. Indeed, this is a highly active area, and new advances in fluorodeoxyglucose-based PET-CT to image activated lesional macrophages together with novel high-resolution MRI techniques to assess plaque necrosis are examples of advances in this area (Sadeghi et al., 2010). Functional imaging based, for example, on detecting MMP activation in lesions is another area in development at the pre-clinical level (Deguchi et al., 2006). In all likelihood, success will depend on a combination of approaches. Moreover, there will be a significant lag period before such methods can be used routinely to evaluate new drugs, because the first step will be to demonstrate that they can accurately predict acute vascular events, followed by repeated demonstrations that changes in these imaging endpoints with known athero-protective drugs are directly associated with improvement in clinical outcome.
Conclusions and Perspectives
There are scores of thousands of paper in the literature on the cell-based mechanisms and in vivo processes operative in atherosclerosis—which is appropriate for the leading cause of death in the industrialized world—and many of these papers are focused on macrophages. The emphasis in this review has been to highlight major concepts of how macrophages affect plaque initiation and progression, all in the background of understanding the limitations of our methods to investigate these areas. In this regard, it is instructive to compare this current state of knowledge with that reported in a classic and highly cited review article by Russell Ross in 1986 (Ross, 1986). As expected, progress over the last 25 years has been built upon earlier findings, as illustrated by the appreciation in 1986 that monocyte-endothelial interactions give rise to macrophage-derived foam cells in lesions, which "may represent a form of inflammatory response." However, most emphasis was placed on endothelial "injury" and growth factor-induced smooth muscle cell proliferation rather than the critical role of subendothelial apoB-LP retention in triggering lesion initiation and then a maladaptive inflammatory response leading to dangerous intimal necrosis. The last 25 years has witnessed major advances in understanding the molecular-cellular mechanisms and in-vivo relevance of processes involved in plaque development and advanced plaque progression, such as macrophage inflammatory and apoptotic signaling pathways, as well as in monocyte biology and macrophage cholesterol efflux.
There are several principles reviewed herein that are worth reiterating. First, the roles of macrophages in atherosclerosis, and atherosclerosis itself, would be a mute topic if we had safe and effective ways to markedly lower apoB-LPs in every human before they reached their teen years, the time at which lesion initiation begins. Unfortunately, we are far from this theoretical goal. Second, most if not all of the processes by which macrophage promote atherothrombotic vascular disease have evolved to optimize our chances of survival to reproductive age. Thus, for example, macrophage mechanisms for killing infectious organisms through inflammatory and oxidative processes are two of the most important means by which macrophages promote atherosclerosis, and ER stress signaling pathways active in advanced plaque progression are utterly essential for the survival of the macrophage during host defense (Martinon et al., 2010). Third, despite the intense effort in the area of how macrophages affect atherosclerosis, there a remarkable number of fundamental issues that remain mysterious. For example, we are far from understanding how the most obvious and distinguishing feature of lesional macrophages, lipid loading, precisely affects the macrophage biology and atherosclerotic processes. Solving these mysteries will require a combination of innovative ideas and new and improved methods of investigation. Finally, investigators in the field must continually evaluate how work in this area can be translated into new and useful therapeutic strategies. Even as we continue to attack the unsolved mysteries in this area, there are already an abundance of plausible targets. However, there are two rate-limiting steps in this area: specificity issues, particularly with regard to avoiding compromises in host defense; and the feasibility of evaluating the effectiveness of arterial-wall based therapy in humans without the requirement for long and costly clinical trials that rely on delayed disease endpoints. The former area is likely to benefit from innovative drug delivery systems, such as arterial-wall targeted nanoparticles, and the latter from new imaging procedures focused on the features of the vulnerable plaque. As is the case with the identification of the drug targets themselves, both of these challenges in drug development will benefit from continued mechanistic and in vivo studies on the cellular biology of macrophages in atherosclerosis.
Supplementary Material
Acknowledgments
This work was supported by NIH grants P01-HL054591, R01-HL075662, R01-HL057560, R01-AG20255, and P01-HL087123. The authors acknowledge invaluable discussions with members of their laboratories and colleagues in the field.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abi-Younes S, Sauty A, Mach F, Sukhova GK, Libby P, Luster AD. The stromal cell-derived factor-1 chemokine is a potent platelet agonist highly expressed in atherosclerotic plaques. Circ Res. 2000;86:131–138. doi: 10.1161/01.res.86.2.131. [DOI] [PubMed] [Google Scholar]
- Aouadi M, Tesz GJ, Nicoloro SM, Wang M, Chouinard M, Soto E, Ostroff GR, Czech MP. Orally delivered siRNA targeting macrophage Map4k4 suppresses systemic inflammation. Nature. 2009;458:1180–1184. doi: 10.1038/nature07774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai S, Shelton JM, Chen M, Bradley MN, Castrillo A, Bookout AL, Mak PA, Edwards PA, Mangelsdorf DJ, Tontonoz P, Miyazaki T. A role for the apoptosis inhibitory factor AIM/Spα/Api6 in atherosclerosis development. Cell Metabolism. 2005;1:201–213. doi: 10.1016/j.cmet.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Arnold L, Henry A, Poron F, Baba-Amer Y, Van RN, Plonquet A, Gherardi RK, Chazaud B. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J Exp Med. 2007;204:1057–1069. doi: 10.1084/jem.20070075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babaev VR, Chew JD, Ding L, Davis S, Breyer MD, Breyer RM, Oates JA, Fazio S, Linton MF. Macrophage EP4 deficiency increases apoptosis and suppresses early atherosclerosis. Cell Metab. 2008;8:492–501. doi: 10.1016/j.cmet.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baetta R, Corsini A. Role of polymorphonuclear neutrophils in atherosclerosis: Current state and future perspectives. Atherosclerosis. 2009 doi: 10.1016/j.atherosclerosis.2009.10.028. [DOI] [PubMed] [Google Scholar]
- Becker L, Gharib SA, Irwin AD, Wijsman E, Vaisar T, Oram JF, Heinecke JW. A macrophage sterol-responsive network linked to atherogenesis. Cell Metab. 2010;11:125–135. doi: 10.1016/j.cmet.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmark C, Dewan A, Orsoni A, Merki E, Miller ER, Shin MJ, Binder CJ, Horkko S, Krauss RM, Chapman MJ, Witztum JL, Tsimikas S. A novel function of lipoprotein [a] as a preferential carrier of oxidized phospholipids in human plasma. J Lipid Res. 2008;49:2230–2239. doi: 10.1194/jlr.M800174-JLR200. [DOI] [PubMed] [Google Scholar]
- Bobryshev YV. Dendritic cells in atherosclerosis: current status of the problem and clinical relevance. Eur Heart J. 2005;26:1700–1704. doi: 10.1093/eurheartj/ehi282. [DOI] [PubMed] [Google Scholar]
- Boyle JJ, Weissberg PL, Bennett MR. Tumor necrosis factor-alpha promotes macrophage-induced vascular smooth muscle cell apoptosis by direct and autocrine mechanisms. Arterioscler Thromb Vasc Biol. 2003;23:1553–1558. doi: 10.1161/01.ATV.0000086961.44581.B7. [DOI] [PubMed] [Google Scholar]
- Breslow JL. Mouse models of atherosclerosis. Science. 1996;272:685–688. doi: 10.1126/science.272.5262.685. [DOI] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331–340. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- Brown MS, Ho YK, Goldstein JL. The cholesteryl ester cycle in macrophage foam cells: Continual hydrolysis and re-esterification of cytoplasmic cholesteryl esters. J Biol Chem. 1980;255:9344–9352. [PubMed] [Google Scholar]
- Calkin AC, Tontonoz P. Liver x receptor signaling pathways and atherosclerosis. Arterioscler Thromb Vasc Biol. 2010;30:1513–1518. doi: 10.1161/ATVBAHA.109.191197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon CP, Shah S, Dansky HM, Davidson M, Brinton EA, Gotto AM, Stepanavage M, Liu SX, Gibbons P, Ashraf TB, Zafarino J, Mitchel Y, Barter P. Safety of Anacetrapib in Patients with or at High Risk for Coronary Heart Disease. N Engl J Med. 2010 doi: 10.1056/NEJMoa1009744. [DOI] [PubMed] [Google Scholar]
- Chan JM, Zhang L, Tong R, Ghosh D, Gao W, Liao G, Yuet KP, Gray D, Rhee JW, Cheng J, Golomb G, Libby P, Langer R, Farokhzad OC. Spatiotemporal controlled delivery of nanoparticles to injured vasculature. Proc Natl Acad Sci U S A. 2010;107:2213–2218. doi: 10.1073/pnas.0914585107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TY, Chang CC, Ohgami N, Yamauchi Y. Cholesterol sensing, trafficking, and esterification. Annu Rev Cell Dev Biol. 2006;22:129–157. doi: 10.1146/annurev.cellbio.22.010305.104656. [DOI] [PubMed] [Google Scholar]
- Choi JH, Do Y, Cheong C, Koh H, Boscardin SB, Oh YS, Bozzacco L, Trumpfheller C, Park CG, Steinman RM. Identification of antigen-presenting dendritic cells in mouse aorta and cardiac valves. J Exp Med. 2009;206:497–505. doi: 10.1084/jem.20082129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coller BS. Leukocytosis and ischemic vascular disease morbidity and mortality: is it time to intervene? Arterioscler Thromb Vasc Biol. 2005;25:658–670. doi: 10.1161/01.ATV.0000156877.94472.a5. [DOI] [PubMed] [Google Scholar]
- Collins AR, Lyon CJ, Xia X, Liu JZ, Tangirala RK, Yin F, Boyadjian R, Bikineyeva A, Pratico D, Harrison DG, Hsueh WA. Age-accelerated atherosclerosis correlates with failure to upregulate antioxidant genes. Circ Res. 2009;104:e42–e54. doi: 10.1161/CIRCRESAHA.108.188771. [DOI] [PubMed] [Google Scholar]
- Curtiss LK, Tobias PS. Emerging role of toll-like receptors in atherosclerosis. J Lipid Res. 2008 doi: 10.1194/jlr.R800056-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degoma EM, deGoma RL, Rader DJ. Beyond high-density lipoprotein cholesterol levels evaluating high-density lipoprotein function as influenced by novel therapeutic approaches. J Am Coll Cardiol. 2008;51:2199–2211. doi: 10.1016/j.jacc.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deguchi JO, Aikawa E, Libby P, Vachon JR, Inada M, Krane SM, Whittaker P, Aikawa M. Matrix metalloproteinase-13/collagenase-3 deletion promotes collagen accumulation and organization in mouse atherosclerotic plaques. Circulation. 2005;112:2708–2715. doi: 10.1161/CIRCULATIONAHA.105.562041. [DOI] [PubMed] [Google Scholar]
- Deguchi JO, Aikawa M, Tung CH, Aikawa E, Kim DE, Ntziachristos V, Weissleder R, Libby P. Inflammation in atherosclerosis: visualizing matrix metalloproteinase action in macrophages in vivo. Circulation. 2006;114:55–62. doi: 10.1161/CIRCULATIONAHA.106.619056. [DOI] [PubMed] [Google Scholar]
- Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-β, PGE2, and PAF. J Clin Invest. 1998;101:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feig JE, Pineda-Torra I, Sanson M, Vengrenyuk Y, Gautier EL, Rubinstein D, Bradley M, Hong C, Liu J, van Rooijen N, Bogunvic D, Bhardwaj N, Garabedian MJ, Tontonoz P, Fisher EA. LXR promotes maximal egress of monocyte-derived cells from mouse aortic plaques during atherosclerosis regression. J Clin Invest. 2010;120:4415–4424. doi: 10.1172/JCI38911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galis ZS, Sukhova GK, Lark MW, Libby P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest. 1994;94:2493–2503. doi: 10.1172/JCI117619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gareus R, Kotsaki E, Xanthoulea S, van dMI, Gijbels MJ, Kardakaris R, Polykratis A, Kollias G, de Winther MP, Pasparakis M. Endothelial cell-specific NF-kappaB inhibition protects mice from atherosclerosis. Cell Metab. 2008;8:372–383. doi: 10.1016/j.cmet.2008.08.016. [DOI] [PubMed] [Google Scholar]
- Glass CK, Witztum JL. Atherosclerosis. the road ahead. Cell. 2001;104:503–516. doi: 10.1016/s0092-8674(01)00238-0. [DOI] [PubMed] [Google Scholar]
- Goel R, Schrank BR, Arora S, Boylan B, Fleming B, Miura H, Newman PJ, Molthen RC, Newman DK. Site-specific effects of PECAM-1 on atherosclerosis in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2008;28:1996–2002. doi: 10.1161/ATVBAHA.108.172270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez N, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N, Deniz J, Ramirez C, Diaz M, Gallardo G, de Galarreta CR, Salazar J, Lopez F, Edwards P, Parks J, Andujar M, Tontonoz P, Castrillo A. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity. 2009;31:245–258. doi: 10.1016/j.immuni.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goossens P, Gijbels MJ, Zernecke A, Eijgelaar W, Vergouwe MN, van dMI, Vanderlocht J, Beckers L, Buurman WA, Daemen MJ, Kalinke U, Weber C, Lutgens E, de Winther MP. Myeloid type I interferon signaling promotes atherosclerosis by stimulating macrophage recruitment to lesions. Cell Metab. 2010;12:142–153. doi: 10.1016/j.cmet.2010.06.008. [DOI] [PubMed] [Google Scholar]
- Gough PJ, Gomez IG, Wille PT, Raines EW. Macrophage expression of active MMP-9 induces acute plaque disruption in apoE-deficient mice. J Clin Invest. 2006;116:59–69. doi: 10.1172/JCI25074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Liang CP, DeVries-Seimon T, Ranalletta M, Welch CL, Collins-Fletcher K, Accili D, Tabas I, Tall AR. Macrophage insulin receptor deficiency increases ER stress-induced apoptosis and necrotic core formation in advanced atherosclerotic lesions. Cell Metabolism. 2006;3:257–266. doi: 10.1016/j.cmet.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Han X, Kitamoto S, Wang H, Boisvert WA. Interleukin-10 overexpression in macrophages suppresses atherosclerosis in hyperlipidemic mice. FASEB J. 2010;24:2869–2880. doi: 10.1096/fj.09-148155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harry BL, Sanders JM, Feaver RE, Lansey M, Deem TL, Zarbock A, Bruce AC, Pryor AW, Gelfand BD, Blackman BR, Schwartz MA, Ley K. Endothelial cell PECAM-1 promotes atherosclerotic lesions in areas of disturbed flow in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 2008;28:2003–2008. doi: 10.1161/ATVBAHA.108.164707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henson PM, Bratton DL, Fadok VA. Apoptotic cell removal. Curr Biol. 2001;11:R795–R805. doi: 10.1016/s0960-9822(01)00474-2. [DOI] [PubMed] [Google Scholar]
- Jerome WG. Advanced atherosclerotic foam cell formation has features of an acquired lysosomal storage disorder. Rejuvenation Res. 2006;9:245–255. doi: 10.1089/rej.2006.9.245. [DOI] [PubMed] [Google Scholar]
- Johnson JL, Newby AC. Macrophage heterogeneity in atherosclerotic plaques. Curr Opin Lipidol. 2009;20:370–378. doi: 10.1097/MOL.0b013e3283309848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadl A, Meher AK, Sharma PR, Lee MY, Doran AC, Johnstone SR, Elliott MR, Gruber F, Han J, Chen W, Kensler T, Ravichandran KS, Isakson BE, Wamhoff BR, Leitinger N. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ Res. 2010;107:737–746. doi: 10.1161/CIRCRESAHA.109.215715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamei M, Carman CV. New observations on the trafficking and diapedesis of monocytes. Curr Opin Hematol. 2010;17:43–52. doi: 10.1097/MOH.0b013e3283333949. [DOI] [PubMed] [Google Scholar]
- Kanters E, Pasparakis M, Gijbels MJ, Vergouwe MN, Partouns-Hendriks I, Fijneman RJ, Clausen BE, Forster I, Kockx MM, Rajewsky K, Kraal G, Hofker MH, de Winther MP. Inhibition of NF-kappaB activation in macrophages increases atherosclerosis in LDL receptor-deficient mice. J Clin Invest. 2003;112:1176–1185. doi: 10.1172/JCI18580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenen RR, von HP, Nesmelova IV, Zernecke A, Liehn EA, Sarabi A, Kramp BK, Piccinini AM, Paludan SR, Kowalska MA, Kungl AJ, Hackeng TM, Mayo KH, Weber C. Disrupting functional interactions between platelet chemokines inhibits atherosclerosis in hyperlipidemic mice. Nat Med. 2009;15:97–103. doi: 10.1038/nm.1898. [DOI] [PubMed] [Google Scholar]
- Koschinsky ML, Marcovina SM. Structure-function relationships in apolipoprotein(a): insights into lipoprotein(a) assembly and pathogenicity. Curr Opin Lipidol. 2004;15:167–174. doi: 10.1097/00041433-200404000-00009. [DOI] [PubMed] [Google Scholar]
- Kruth HS, Jones NL, Huang W, Zhao B, Ishii I, Chang J, Combs CA, Malide D, Zhang WY. Macropinocytosis is the endocytic pathway that mediates macrophage foam cell formation with native low density lipoprotein. J Biol Chem. 2005;280:2352–2360. doi: 10.1074/jbc.M407167200. [DOI] [PubMed] [Google Scholar]
- Kunjathoor VV, Febbraio M, Podrez EA, Moore KJ, Andersson L, Koehn S, Rhee JS, Silverstein R, Hoff HF, Freeman MW. Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J Biol Chem. 2002;277:49982–49988. doi: 10.1074/jbc.M209649200. [DOI] [PubMed] [Google Scholar]
- Leon B, Lopez-Bravo M, Ardavin C. Monocyte-derived dendritic cells. Semin Immunol. 2005;17:313–318. doi: 10.1016/j.smim.2005.05.013. [DOI] [PubMed] [Google Scholar]
- Li S, Sun Y, Thorp E, Liang CP, Han S, Jehle A, Viswanathan S, Kanter J, Li R, Welch CL, Hasty AH, Bornfeldt KE, Tabas I, Tall AR. Defective phagocytosis of apoptotic cells by macrophages in atherosclerotic lesions of ob/ob mice and its reversal by a fish oil diet. Circulation Research. 2010 doi: 10.1161/CIRCRESAHA.109.199570. Ref Type: In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Sukhova GK, Sun JS, Xu WH, Libby P, Shi GP. Lysosomal cysteine proteases in atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24:1359–1366. doi: 10.1161/01.ATV.0000134530.27208.41. [DOI] [PubMed] [Google Scholar]
- Liu J, Thewke DP, Su YR, Linton MF, Fazio S, Sinensky MS. Reduced macrophage apoptosis is associated with accelerated atherosclerosis in low-density lipoprotein receptor-null mice. Arterioscler Thromb Vasc Biol. 2005;25:174–179. doi: 10.1161/01.ATV.0000148548.47755.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De SG, Ferguson TB, Ford E, Furie K, Gillespie C, Go A, Greenlund K, Haase N, Hailpern S, Ho PM, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott MM, Meigs J, Mozaffarian D, Mussolino M, Nichol G, Roger VL, Rosamond W, Sacco R, Sorlie P, Stafford R, Thom T, Wasserthiel-Smoller S, Wong ND, Wylie-Rosett J. Executive summary: heart disease and stroke statistics--2010 update: a report from the American Heart Association. Circulation. 2010;121:948–954. doi: 10.1161/CIRCULATIONAHA.109.192666. [DOI] [PubMed] [Google Scholar]
- Lotta LA. Genome-wide association studies in atherothrombosis. Eur J Intern Med. 2010;21:74–78. doi: 10.1016/j.ejim.2009.11.003. [DOI] [PubMed] [Google Scholar]
- Lusis AJ, Yu J, Wang SS. The problem of passenger genes in transgenic mice. Arterioscler Thromb Vasc Biol. 2007;27:2100–2103. doi: 10.1161/ATVBAHA.107.147918. [DOI] [PubMed] [Google Scholar]
- Lutgens E, Lievens D, Beckers L, Wijnands E, Soehnlein O, Zernecke A, Seijkens T, Engel D, Cleutjens J, Keller AM, Naik SH, Boon L, Oufella HA, Mallat Z, Ahonen CL, Noelle RJ, de Winther MP, Daemen MJ, Biessen EA, Weber C. Deficient CD40-TRAF6 signaling in leukocytes prevents atherosclerosis by skewing the immune response toward an antiinflammatory profile. J Exp Med. 2010;207:391–404. doi: 10.1084/jem.20091293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning-Tobin JJ, Moore KJ, Seimon TA, Bell SA, Sharuk M, varez-Leite JI, de Winther MP, Tabas I, Freeman MW. Loss of SR-A and CD36 activity reduces atherosclerotic lesion complexity without abrogating foam cell formation in hyperlipidemic mice. Arterioscler Thromb Vasc Biol. 2009;29:19–26. doi: 10.1161/ATVBAHA.108.176644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F, Chen X, Lee AH, Glimcher LH. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol. 2010;11:411–418. doi: 10.1038/ni.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxfield FR, Tabas I. Role of cholesterol and lipid organization in disease. Nature. 2005;438:612–621. doi: 10.1038/nature04399. [DOI] [PubMed] [Google Scholar]
- Mestas J, Ley K. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc Med. 2008;18:228–232. doi: 10.1016/j.tcm.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KJ, Kunjathoor VV, Koehn SL, Manning JJ, Tseng AA, Silver JM, McKee M, Freeman MW. Loss of receptor-mediated lipid uptake via scavenger receptor A or CD36 pathways does not ameliorate atherosclerosis in hyperlipidemic mice. J Clin Invest. 2005;115:2192–2201. doi: 10.1172/JCI24061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukundan L, Odegaard JI, Morel CR, Heredia JE, Mwangi JW, Ricardo-Gonzalez RR, Goh YPS, Red Eagle A, Dunn SE, Awakuni JUH, Nguyen KD, Steinman L, Michie SA, Chawla A. PPARδ senses and orchestrates clearance of apoptotic cells to promote tolerance. Nature Medicine. 2009 doi: 10.1038/nm.2048. Ref Type: In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullick AE, Tobias PS, Curtiss LK. Modulation of atherosclerosis in mice by Toll-like receptor 2. J Clin Invest. 2005;115:3149–3156. doi: 10.1172/JCI25482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myoishi M, Hao H, Minamino T, Watanabe K, Nishihira K, Hatakeyama K, Asada Y, Okada K, Ishibashi-Ueda H, Gabbiani G, Bochaton-Piallat ML, Mochizuki N, Kitakaze M. Increased endoplasmic reticulum stress in atherosclerotic plaques associated with acute coronary syndrome. Circulation. 2007;116:1226–1233. doi: 10.1161/CIRCULATIONAHA.106.682054. [DOI] [PubMed] [Google Scholar]
- Nakano K, Egashira K, Ohtani K, Zhao G, Funakoshi K, Ihara Y, Sunagawa K. Catheter-based adenovirus-mediated anti-monocyte chemoattractant gene therapy attenuates in-stent neointima formation in cynomolgus monkeys. Atherosclerosis. 2007;194:309–316. doi: 10.1016/j.atherosclerosis.2006.10.029. [DOI] [PubMed] [Google Scholar]
- Paulson KE, Zhu SN, Chen M, Nurmohamed S, Jongstra-Bilen J, Cybulsky MI. Resident intimal dendritic Cells accumulate lipid and contribute to the initiation of atherosclerosis. Circ Res. 2009 doi: 10.1161/CIRCRESAHA.109.210781. [DOI] [PubMed] [Google Scholar]
- Paulson KE, Zhu SN, Chen M, Nurmohamed S, Jongstra-Bilen J, Cybulsky MI. Resident intimal dendritic cells accumulate lipid and contribute to the initiation of atherosclerosis. Circ Res. 2010;106:383–390. doi: 10.1161/CIRCRESAHA.109.210781. [DOI] [PubMed] [Google Scholar]
- Potteaux S, Gautier EL, Hutchinson SB, van Rooijen N, Rader DJ, Randolph GJ. Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of apoE−/− mice following apoE complementation. J Clin Invest. 2010 doi: 10.1172/JCI43802. Ref Type: In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajamaki K, Lappalainen J, Oorni K, Valimaki E, Matikainen S, Kovanen PT, Eklund KK. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS ONE. 2010;5:e11765. doi: 10.1371/journal.pone.0011765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranalletta M, Wang N, Han S, Yvan-Charvet L, Welch C, Tall AR. Decreased atherosclerosis in low-density lipoprotein receptor knockout mice transplanted with Abcg1−/− bone marrow. Arterioscler Thromb Vasc Biol. 2006;26:2308–2315. doi: 10.1161/01.ATV.0000242275.92915.43. [DOI] [PubMed] [Google Scholar]
- Rigamonti E, Chinetti-Gbaguidi G, Staels B. Regulation of macrophage functions by PPAR-alpha, PPAR-gamma, and LXRs in mice and men. Arterioscler Thromb Vasc Biol. 2008;28:1050–1059. doi: 10.1161/ATVBAHA.107.158998. [DOI] [PubMed] [Google Scholar]
- Rosenfeld ME, Carson KG, Johnson JL, Williams H, Jackson CL, Schwartz SM. Animal models of spontaneous plaque rupture: the holy grail of experimental atherosclerosis research. Curr Atheroscler Rep. 2002;4:238–242. doi: 10.1007/s11883-002-0025-3. [DOI] [PubMed] [Google Scholar]
- Ross R. The pathogenesis of atherosclerosis--an update. N Engl J Med. 1986;314:488–500. doi: 10.1056/NEJM198602203140806. [DOI] [PubMed] [Google Scholar]
- Rothblat GH, Phillips MC. High-density lipoprotein heterogeneity and function in reverse cholesterol transport. Curr Opin Lipidol. 2010;21:229–238. doi: 10.1097/mol.0b013e328338472d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadeghi MM, Glover DK, Lanza GM, Fayad ZA, Johnson LL. Imaging atherosclerosis and vulnerable plaque. J Nucl Med. 2010;51(Suppl 1):51S–65S. doi: 10.2967/jnumed.109.068163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sather S, Kenyon KD, Lefkowitz JB, Liang X, Varnum BC, Henson PM, Graham DK. A soluble form of the Mer receptor tyrosine kinase inhibits macrophage clearance of apoptotic cells and platelet aggregation. Blood. 2007;109:1026–1033. doi: 10.1182/blood-2006-05-021634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller NK, Kubo N, Boisvert WA, Curtiss LK. Effect of gamma-irradiation and bone marrow transplantation on atherosclerosis in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2001;21:1674–1680. doi: 10.1161/hq1001.096724. [DOI] [PubMed] [Google Scholar]
- Schneider F, Sukhova GK, Aikawa M, Canner J, Gerdes N, Tang SM, Shi GP, Apte SS, Libby P. Matrix-metalloproteinase-14 deficiency in bone-marrow-derived cells promotes collagen accumulation in mouse atherosclerotic plaques. Circulation. 2008;117:931–939. doi: 10.1161/CIRCULATIONAHA.107.707448. [DOI] [PubMed] [Google Scholar]
- Schrijvers DM, De Meyer GR, Kockx MM, Herman AG, Martinet W. Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arterioscler Thromb Vasc Biol. 2005;25:1256–1261. doi: 10.1161/01.ATV.0000166517.18801.a7. [DOI] [PubMed] [Google Scholar]
- Schulte S, Sukhova GK, Libby P. Genetically programmed biases in Th1 and Th2 immune responses modulate atherogenesis. Am J Pathol. 2008;172:1500–1508. doi: 10.2353/ajpath.2008.070776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seimon TA, Kim M, Blumenthal A, Koo J, Ehrt S, Wainwright H, Bekker LG, Kaplan G, Nathan C, Tabas I, Russell DG. Induction of ER stress in macrophages of tuberculosis granulomas. PLoS ONE. 2010a;5:e12772. doi: 10.1371/journal.pone.0012772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seimon TA, Liao X, Magallon J, Moore KJ, Witztum JL, Tsimikas S, Golenbock DT, Webb NR, Tabas I. Atherogenic lipids and lipoproteins trigger CD36-TLR2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell Metabolism. 2010b;12:467–482. doi: 10.1016/j.cmet.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8:349–361. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaposhnik Z, Wang X, Weinstein M, Bennett BJ, Lusis AJ. Granulocyte macrophage colony-stimulating factor regulates dendritic cell content of atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2007;27:621–627. doi: 10.1161/01.ATV.0000254673.55431.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata N, Glass CK. Regulation of macrophage function in inflammation and atherosclerosis. J Lipid Res. 2009;50(Suppl):S277–S281. doi: 10.1194/jlr.R800063-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg D, Glass CK, Witztum JL. Evidence mandating earlier and more aggressive treatment of hypercholesterolemia. Circulation. 2008;118:672–677. doi: 10.1161/CIRCULATIONAHA.107.753152. [DOI] [PubMed] [Google Scholar]
- Stewart CL, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, Rayner KJ, Boyer L, Zhong R, Frazier WA, Lacy-Hulbert A, El Khoury J, Golenbock DT, Moore KJ. CD36 ligands promote sterile inflammation though assembly of a Toll-like receptor 4 and 6 heterodimer. Nature Immuno. 2009;11:155–162. doi: 10.1038/ni.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Unoki H, Wang X, Liang J, Ichikawa T, Arai Y, Shiomi M, Marcovina SM, Watanabe T, Fan J. Lipoprotein(a) enhances advanced atherosclerosis and vascular calcification in WHHL transgenic rabbits expressing human apolipoprotein(a) J Biol Chem. 2002;277:47486–47492. doi: 10.1074/jbc.M205814200. [DOI] [PubMed] [Google Scholar]
- Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nature Rev Immunol. 2010a;10:36–46. doi: 10.1038/nri2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ Res. 2010b;107:839–850. doi: 10.1161/CIRCRESAHA.110.224766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I, Li Y, Brocia RW, Wu SW, Swenson TL, Williams KJ. Lipoprotein lipase and sphingomyelinase synergistically enhance the association of atherogenic lipoproteins with smooth muscle cells and extracellular matrix. A possible mechanism for low density lipoprotein and lipoprotein(a) retention and macrophage foam cell formation. J Biol Chem. 1993;268:20419–20432. [PubMed] [Google Scholar]
- Tabas I, Ron D. Molecular mechanisms integrating pathways of endoplasmic reticulum stress-induced apoptosis. Nat Cell Biol. 2010 doi: 10.1038/ncb0311-184. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I, Tall AR, Accili D. The impact of macrophage insulin resistance on advanced atherosclerotic plaque progression. Circ Res. 2010;106:58–67. doi: 10.1161/CIRCRESAHA.109.208488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, Garin A, Liu J, Mack M, Van RN, Lira SA, Habenicht AJ, Randolph GJ. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007;117:185–194. doi: 10.1172/JCI28549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tall AR, Yvan-Charvet L, Terasaka N, Pagler T, Wang N. HDL, ABC transporters, and cholesterol efflux: implications for the treatment of atherosclerosis. Cell Metab. 2008;7:365–375. doi: 10.1016/j.cmet.2008.03.001. [DOI] [PubMed] [Google Scholar]
- Tang C, Liu Y, Kessler PS, Vaughan AM, Oram JF. The macrophage cholesterol exporter ABCA1 functions as an anti-inflammatory receptor. J Biol Chem. 2009;284:32336–32343. doi: 10.1074/jbc.M109.047472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarling EJ, Bojanic DD, Tangirala RK, Wang X, Lovgren-Sandblom A, Lusis AJ, Bjorkhem I, Edwards PA. Impaired development of atherosclerosis in Abcg1−/− Apoe−/− mice: identification of specific oxysterols that both accumulate in Abcg1−/− Apoe−/− tissues and induce apoptosis. Arterioscler Thromb Vasc Biol. 2010;30:1174–1180. doi: 10.1161/ATVBAHA.110.205617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarr PT, Edwards PA. ABCG1 and ABCG4 are coexpressed in neurons and astrocytes of the CNS and regulate cholesterol homeostasis through SREBP–2. J Lipid Res. 2008;49:169–182. doi: 10.1194/jlr.M700364-JLR200. [DOI] [PubMed] [Google Scholar]
- Thorp E, Li G, Seimon TA, Kuriakose G, Ron D, Tabas I. Reduced apoptosis and plaque necrosis in advanced atherosclerotic lesions of Apoe−/− and Ldlr−/− mice lacking CHOP. Cell Metabolism. 2009;9:474–481. doi: 10.1016/j.cmet.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmins JM, Ozcan L, Seimon TA, Li G, Malagelada C, Backs J, Backs T, Bassel-Duby R, Olson EN, Anderson ME, Tabas I. Calcium/calmodulin-dependent protein kinase II links endoplasmic reticulum stress with Fas and mitochondrial apoptosis pathways. J Clin Invest. 2009;119:2925–2941. doi: 10.1172/JCI38857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tregouet DA, Konig IR, Erdmann J, Munteanu A, Braund PS, Hall AS, Grosshennig A, Linsel-Nitschke P, Perret C, DeSuremain M, Meitinger T, Wright BJ, Preuss M, Balmforth AJ, Ball SG, Meisinger C, Germain C, Evans A, Arveiler D, Luc G, Ruidavets JB, Morrison C, van der HP, Schreiber S, Neureuther K, Schafer A, Bugert P, El Mokhtari NE, Schrezenmeir J, Stark K, Rubin D, Wichmann HE, Hengstenberg C, Ouwehand W, Ziegler A, Tiret L, Thompson JR, Cambien F, Schunkert H, Samani NJ. Genome-wide haplotype association study identifies the SLC22A3-LPAL2-LPA gene cluster as a risk locus for coronary artery disease. Nat Genet. 2009;41:283–285. doi: 10.1038/ng.314. [DOI] [PubMed] [Google Scholar]
- Tsukano H, Gotoh T, Endo M, Miyata K, Tazume H, Kadomatsu T, Yano M, Iwawaki T, Kohno K, Araki K, Mizuta H, Oike Y. The endoplasmic reticulum stress-CHOP pathway-mediated apoptosis in macrophages contributes to the instability of atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 2010 doi: 10.1161/ATVBAHA.110.206094. [DOI] [PubMed] [Google Scholar]
- Virmani R, Burke AP, Kolodgie FD, Farb A. Vulnerable plaque: the pathology of unstable coronary lesions. J Interv Cardiol. 2002;15:439–446. doi: 10.1111/j.1540-8183.2002.tb01087.x. [DOI] [PubMed] [Google Scholar]
- Wilensky RL, Macphee CH. Lipoprotein-associated phospholipase A(2) and atherosclerosis. Curr Opin Lipidol. 2009;20:415–420. doi: 10.1097/MOL.0b013e3283307c16. [DOI] [PubMed] [Google Scholar]
- Williams KJ, Fisher EA. Oxidation, lipoproteins, and atherosclerosis: which is wrong, the antioxidants or the theory? Curr Opin Clin Nutr Metab Care. 2005;8:139–146. doi: 10.1097/00075197-200503000-00006. [DOI] [PubMed] [Google Scholar]
- Williams KJ, Tabas I. The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol. 1995;15:551–561. doi: 10.1161/01.atv.15.5.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witztum JL, Steinberg D. The oxidative modification hypothesis of atherosclerosis: does it hold for humans? Trends Cardiovasc Med. 2001;11:93–102. doi: 10.1016/s1050-1738(01)00111-6. [DOI] [PubMed] [Google Scholar]
- Yancey PG, Blakemore J, Ding L, Fan D, Overton CD, Zhang Y, Linton MF, Fazio S. Macrophage LRP-1 controls plaque cellularity by regulating efferocytosis and AKT activation. Arterioscler Thromb Vasc Biol. 2010 doi: 10.1161/ATVBAHA.109.202051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yvan-Charvet L, Pagler T, Gautier EL, Avagyan S, Siry RL, Han S, Welch CL, Wang N, Randolph GJ, Snoeck HW, Tall AR. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science. 2010a;328:1689–1693. doi: 10.1126/science.1189731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yvan-Charvet L, Pagler TA, Seimon TA, Thorp E, Tabas I, Tall AR. ABCA1 and ABCG1 protect against oxidative stress-induced macrophage apoptosis during efferocytosis. Circ Res. 2010b;106:1861–1869. doi: 10.1161/CIRCRESAHA.110.217281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yvan-Charvet L, Ranalletta M, Wang N, Han S, Terasaka N, Li R, Welch C, Tall AR. Combined deficiency of ABCA1 and ABCG1 promotes foam cell accumulation and accelerates atherosclerosis in mice. J Clin Invest. 2007;117:3900–3908. doi: 10.1172/JCI33372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Lee JY, Timmins JM, Brown JM, Boudyguina E, Mulya A, Gebre AK, Willingham MC, Hiltbold EM, Mishra N, Maeda N, Parks JS. Increased cellular free cholesterol in macrophage-specific Abca1 knock-out mice enhances pro-inflammatory response of macrophages. J Biol Chem. 2008;283:22930–22941. doi: 10.1074/jbc.M801408200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.