

Abstract
Background/Aims
During the development of liver fibrosis, mediators are produced that stimulate cells in the liver to differentiate into myofibroblasts and to produce collagen. Recent studies demonstrated that the transcription factor, hypoxia-inducible factor-1α (HIF-1α), is critical for upregulation of profibrotic mediators, such as platelet-derived growth factor-A (PDGF-A), PDGF-B, and plasminogen activator inhibitor-1 (PAI-1) in the liver during the development of fibrosis. What remains unknown is the cell type-specific regulation of these genes by HIF-1α in liver cell types. Accordingly, the hypothesis was tested that HIF-1α is activated in hypoxic hepatocytes and regulates production of profibrotic mediators by these cells.
Methods
In this study, hepatocytes were isolated from the livers of control and HIF-1α or HIF-1β-Deficient mice and exposed to hypoxia.
Results
Exposure of primary mouse hepatocytes to 1% oxygen stimulated nuclear accumulation of HIF-1α and upregulated PAI-1, vascular endothelial cell growth factor, and the vasoactive peptides adrenomedullin-1 (ADM-1) and ADM-2. In contrast, levels of PDGF-A and PDGF-B mRNAs were unaffected in these cells by hypoxia. Exposure of HIF-1α-Deficient hepatocytes to 1% oxygen only partially prevented upregulation of these genes, suggesting that other hypoxia-regulated transcription factors, such as HIF-2α, may also regulate these genes. In support of this, HIF-2α was activated in hypoxic hepatocytes, and exposure of HIF-1β-Deficient hepatocytes to 1% oxygen completely prevented upregulation PAI-1, VEGF, and ADM-1, suggesting that HIF-2α may also contribute to upregulation of these genes in hypoxic hepatocytes.
Conclusions
Collectively, our results suggest that HIFs may be important regulators of profibrotic and vasoactive mediators by hypoxic hepatocytes.
Keywords: Hypoxia-inducible factor, hepatocytes, liver fibrosis
Hypoxia-inducible factors (HIFs) are a group of transcription factors rapidly activated in hypoxic cells. Once activated, these transcription factors regulate expression of genes that allow cells to adapt to a hypoxic environment. HIFs are composed of an α subunit, either HIF-1α or HIF-2α, and a β subunit, HIF-1β (1, 2). HIF-1α and HIF-2α protein subunits are constitutively produced in cells. In normoxic cells, these subunits are immediately targeted for proteosomal degradation. In cells that are hypoxic, however, the mechanisms that target HIFs for degradation are inhibited, allowing HIF-1α and HIF-2α to translocate to the nucleus (3). In the nucleus both HIF-1α and HIF-2α heterodimerize with HIF-1β and regulate expression of genes involved in angiogenesis, iron homeostasis, glycolysis, and others (2). Although HIFs are essential for survival, several studies have implicated HIFs in the pathogenesis of several disease processes, including liver fibrosis.
Liver fibrosis is characterized by excessive deposition of extracellular matrix in the liver during chronic injury. This disease has many causes including those of known etiology, such as chronic alcohol consumption, hepatitis virus, nonalcoholic steatohepatitis, and genetic disorders, and those of unknown etiology, such as primary biliary cirrhosis, sclerosing cholangitis, and others. Liver fibrosis is initiated when chronic liver injury stimulates numerous cells types, including hepatocytes, bile duct epithelial cells, Kupffer cells, and other inflammatory cells to produce protein and nonprotein mediators (e.g., growth factors, chemokines, reactive oxygen species). These mediators cause cells in the liver, such as hepatic stellate cells, peribiliary fibroblasts, hepatocytes, bile duct epithelial cells, and bone marrow-derived cells to differentiate into myofibroblasts (4). In addition, these mediators stimulate myofibroblast proliferation (5), and stimulate these cells to migrate to injured regions of the liver (i.e., chemotaxis) (6, 7). Once the myofibroblasts accumulate in these regions, they are stimulated to produce collagen and other components of extracellular matrix causing fibrosis.
Recent studies have shown that HIF-1α is an important regulator of profibrotic mediator production in the liver during the development of fibrosis (8). In this study, regions of hypoxia developed in the livers of bile duct-ligated mice prior to the development of liver fibrosis (8). Furthermore, HIF-1α was activated in hepatocytes and Kupffer cells within these hypoxic regions. Mice deficient in HIF-1α developed less fibrosis after bile duct ligation, compared to control mice with normal levels of HIF-1α, indicating that HIF-1α is important for the development of liver fibrosis. The mechanism by which HIF-1α promoted fibrosis appeared to be related to HIF-1α-dependent regulation of profibrotic mediators, such as platelet-derived growth factor-A (PDGF-A), PDGF-B, and plasminogen activator inhibitor-1 (PAI-1) (8). Although these studies demonstrated an important role for HIF-1α in regulating production of profibrotic mediators during the development of fibrosis, the cell type specific regulation of profibrotic genes by HIF-1α was not examined. Accordingly, in the present study, the hypothesis was tested that HIF-1α is activated in hypoxic hepatocytes and regulates expression of genes, such as PDGF-A, PDGF-B, and PAI-1 that promote fibrosis.
Materials and methods
Mice
Deletion of HIF-1α or HIF-1β in mice causes embryonic lethality (9, 10). To selectively reduce HIF-1α or HIF-1β levels in adult mice, HIF-1αfl/fl and HIF-1βfl/fl mice, described in detail previously (11, 12), were crossed with mice expressing Cre recombinase under control of the Mx interferon-inducible promoter (13)(Mx-Cre+/− mice; Jackson Laboratories, Bar Harbor, ME). Offspring of the HIF-1αfl/fl mouse breeding were HIF-1αfl/fl-Mx-Cre+ (i.e., HIF-1α-deletable) or HIF-1αfl/fl-Mx-Cre− (i.e., HIF-1α-nondeletable littermate controls). Offspring of the HIF-1βfl/fl mouse breeding were HIF-1βfl/fl-Mx-Cre+ (i.e., HIF-1β-deletable) or HIF-1βfl/fl-Mx-Cre− (i.e., HIF-1β-nondeletable littermate controls). PCR of genomic DNA was used to detect the floxed HIF-1α gene, floxed HIF-1β gene, and the Cre transgene as described previously (11, 12). To activate the MxCre promoter, HIF-1αfl/fl-Mx-Cre+, HIF-1αfl/fl-Mx-Cre−, HIF-1βfl/fl-Mx-Cre+, and HIF-1βfl/fl-Mx-Cre− mice were treated with 500 μg of polyinosinic–polycytidylic acid (pIpC, Sigma Chemical Company, St. Louis, MO) dissolved in sterile saline by intraperitoneal injection every three days for a total of three injections. In mice containing the MxCre transgene, this treatment causes a near complete deletion of loxP-containing genes in liver and immune organs and a partial deletion in other tissues (11, 13). In HIF-1αfl/fl-Mx-Cre+ mice, this treatment results in deletion of exons 13–15 in the HIF-1α gene which encode for the COOH-terminal transactivation domain and a portion of the nuclear localization sequence, both of which are essential for hypoxia responsiveness by HIF-1α (12). In HIF-1βfl/fl-Mx-Cre+ mice, this treatment results in deletion of exon 6 in the HIF-1β gene which encodes for the basic-helix-loop-helix domain (11). Deletion of this region completely abrogates HIF signaling in cells (11). Mice were used for experiments one week after the final pIpC injection.
All mice were maintained on a 12-h light/dark cycle under controlled temperature (18–21°C) and humidity. Food (Rodent Chow; Harlan-Teklad, Madison, WI) and tap water were allowed ad libitum. All procedures on animals were carried out in accordance with the Guide for the Care and Use of Laboratory Animals promulgated by the National Institutes of Health and were approved by the institutional IACUC committee at the University of Kansas Medical Center.
Hepatocyte Isolation
Hepatocytes were isolated from the livers of mice by collagenase perfusion as described in detail by us previously (14). The hepatocytes were cultured in Williams’ medium E containing 10% FBS and Penicillin-Streptomycin. After a 3 hour attachment period, the medium withunattached cells was removed, and fresh serum-free medium added. After 16 hours, the hepatocytes were cultured in an environment containing room air, 3% oxygen, or 1% oxygen in a NAPCO CO2 7000 cell culture incubator (NAPCO Precision, Winchester, VA). These environments also contained 5% CO2 and were balanced with nitrogen.
Analysis of HIF-1β and HIF-1α Deletion in Hepatocytes
Genomic DNA was isolated from hepatocytes, and PCR was used to determine the degree of deletion of the floxed exons in the HIF-1α or HIF-1β genes as described previously (11, 12, 15).
Real-time PCR
Total liver RNA was isolated using TRI reagent (Sigma Chemical Company, St. Louis, MO), and reverse transcribed into cDNA as described by us previously (14). Real-time PCR was used to quantify PAI-1, PDGF-A, PDGF-B, vascular endothelial cell growth factor (VEGF), adrenomedullin-1 (ADM-1), ADM-2, and 18S and performed on an Applied Biosystems Prism 7300 Real-time PCR Instrument (Applied Biosystems, Foster City, CA) using the SYBR green DNA PCR kit (Applied Biosystems) as described by us previously. The sequences of the primers were as follows: 18S Forward: 5′-TTG ACG GAA GGG CAC CAC CAG-3′; 18S Reverse: 5′-GCA CCA CCA CCC ACG GAA TCG-3′; PAI-1 Forward: 5′-AGT CTT TCC GAC CAA GAG CA-3′; PAI-1 Reverse: 5′-ATC ACT TGC CCC ATG AAG AG-3′; PDGF-A Forward: 5′-GAG ATA CCC CGG GAG TTG AT-3′; PDGF-A Reverse: 5′-AAA TGA CCG TCC TGG TCT TG-3′; PDGF-B Forward: 5′-CCC ACA GTG GCT TTT CAT TT-3′; PDGF-B Reverse: 5′-GTG GAG GAG CAG ACT GAA GG-3′; VEGF Forward: 5′-CAT CTT CAA GCC GTC CTG TGT-3′; VEGF Reverse: 5′-CTC CAG GGC TTC ATC GTT ACA-3′; ADM-1 Forward: 5′-GGA ACT ACA AGC ATC CAG CAG C-3′; ADM-1 Reverse: 5′-CAG TTG TGT TCT GCT CGT CCA G-3′; ADM-2 Forward: 5′-TTG CTA TGG TTC ATC TGC CTC A-3′; ADM-2 Reverse: 5′-TGG ACT TGG CAT GTA CCC AGT A-3′.
Western Blot Analysis
Nuclear extracts were isolated from hepatocytes. Briefly, the hepatocytes were scraped into 10 mM Hepes, pH 7.9 containing 100 mM KCl, 1.5 mM MgCl2, 0.1 mM EGTA, 0.5 mM DTT, 0.5% NP-40, and Halt Protease Inhibitor Cocktail (Pierce Biotechnology, Rockford, IL) and incubated on ice. After 15 minutes, the cells were vortexed and centrifuged for 10 seconds at 10,000 g. The pellet was resuspended in 10 mM Hepes, pH 7.9 containing 420 mM NaCl, 1.5 mM MgCl2, 0.1 mM EGTA, 0.5 mM DTT, 5% glycerol, and Halt Protease Inhibitor Cocktail mixed for 1 hour at 4°C. The samples were centrifuged at 10,000 g for 10 minutes at 4°C and the concentration of protein determined in the supernatant. For western blot analysis, aliquots(15 μg) of nuclear extracts were subjected to 10% SDS-polyacrylamidegel electrophoresis, and proteins were transferred toimmobilon polyvinylidene difluoride transfer membranes (Millipore Corporation, Bedford, MA). The membranes were then probed with rabbit polyclonal anti-HIF-1α antibody (NB100-449, Novus Biologicals, Littleton, CO) diluted 1:1000, rabbit polyclonal anti-HIF-2α (NB100-122, Novus BIologicals) diluted 1:1000, or mouse monoclonal anti-HIF-1β (Millipore) diluted 1:1000 followed by incubation with goatanti-rabbit antibody conjugated to horseradish peroxidase (SantaCruz Biotechnology) for HIF-1α and HIF-2α or goat anti-mouse antibody conjugated to horseradish peroxidase (Santa Cruz Biotechnology) for HIF-1β. Immunoreactive bands were visualizedusing the Immune-Star HRP Substrate Kit (Bio-Rad Laboratories, Hercules CA).
HIF-1α and HIF-2α Immunohistochemistry
For HIF-1α and HIF-2α immunostaining, hepatocytes were fixed in 4% formalin in phosphate-buffered saline (PBS) for 10 minutes at room temperature. Cells were incubated with rabbit polyclonal anti-HIF-1α (NB100-449, Novus BIologicals, Littleton, CO) or rabbit polyclonal anti-HIF-2α (NB100-122, Novus BIologicals) diluted 1:50 in PBS containing 3% goat serum at room temperature for 3 hours. The sections were washed with PBS, and then incubated with secondary antibody conjugated to Alexa 488 (green staining; Molecular Probes, Eugene, OR). The sections were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) to stain DNA.
Assessment of Hepatocyte Necrosis
Hepatocyte necrosis was evaluated by measuring the release of alanine aminotransferase (ALT) into the medium as described previously (16).
Statistical Analysis
Results are presented as the mean ± SEM. Data were analyzed by Analysis of Variance (ANOVA). ANOVAs were performed on log X-transformed data in instances in which variances were not homogenous. Comparisons among group means were made using the Student-Newman-Keuls test. The criterion for significance was p < 0.05 for all studies.
Results
Activation of HIF-1α in Hypoxic Hepatocytes
To determine whether HIF-1α is activated in hypoxic hepatocytes, primary mouse hepatocytes were isolated and exposed to room air, 3% oxygen, or 1% oxygen. Western blot analysis of nuclear extracts from these cells showed increased levels of HIF-1α protein following 3% and 1% oxygen (Fig. 1A). Quantification of the western blot showed an increase in HIF-1α protein by 15 minutes, which increased further at 1 hour (Fig. 1B).
Fig. 1.

Hepatocytes were isolated from mice and exposed to room air, 3% oxygen, or 1% oxygen for either 15 minutes or 1 hour. HIF-1α was then detected in nuclear extracts by western blot. (A) Representative western blot of an n=3. (B) Intensity of the HIF-1α band was quantified, and the fold change relative to room air was determined. Data are expressed as means ± SEM; n = 3, where each n represents hepatocytes isolated from a different mouse.
Immunohistochemistry was next used to confirm nuclear accumulation of HIF-1α in hypoxic hepatocytes. Exposure of hepatocytes to 1% and 3% oxygen increased nuclear levels of HIF-1α by 1 hour after exposure (Fig. 2C and 2E). Nuclear accumulation of HIFs was confirmed by colocalization with 4′,6-diamidino-2-phenylindole (DAPI) which stains DNA (Fig. 2D and 2F). These results show that HIF-1α is activated in hypoxic mouse hepatocytes.
Fig. 2.
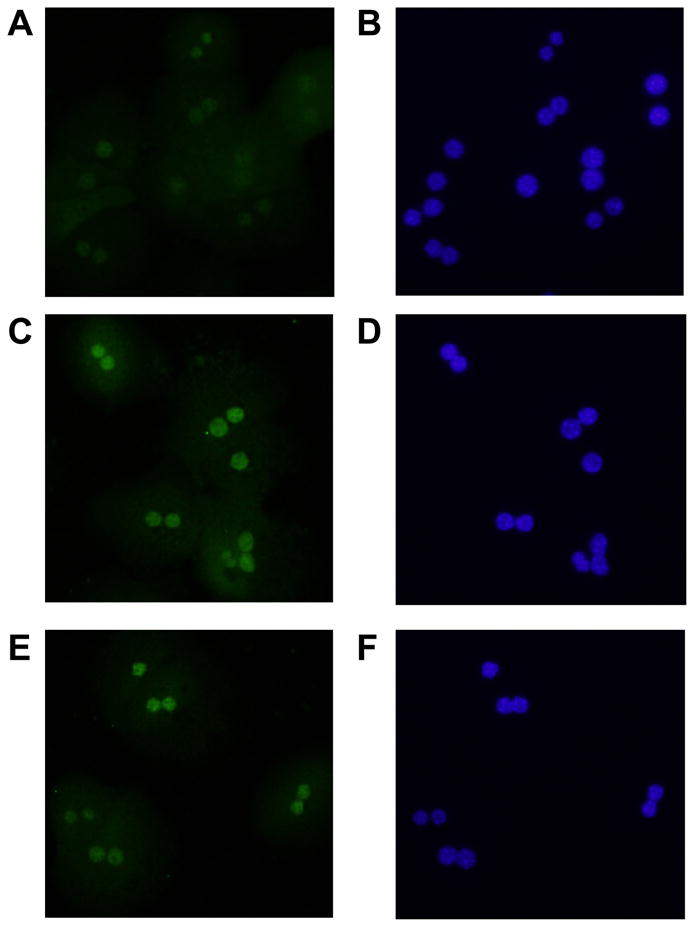
Hepatocytes were isolated from mice and exposed to room air (A and B), 3% oxygen (C and D), or 1% oxygen (E and F). One hour later, immunohistochemistry was used to detect HIF-1α (green fluorescence; A, C, and E). The same cells were counterstained with DAPI (blue fluorescence; B, D, and F) to identify the nuclei and show colocalization of HIF-1α immunostaining with nuclear staining. Representative of an n=3.
Inhibition of HIF-1α Signaling in Hypoxic Mouse Hepatocytes
To evaluate the role of HIF-1α in regulation of gene expression in hypoxic hepatocytes, HIF-1αfl/fl mice, containing loxP sites flanking Exons 13–15 of the HIF-1α gene, were crossed with Mx-Cre+/− mice. Exons 13–15 of the HIF-1α gene encode for the COOH-terminal transactivation domain and a portion of the nuclear localization sequence, both of which are essential for hypoxia responsiveness by HIF-1α (12). Treatment of HIF-1αfl/fl-Mx-Cre+ mice with pIpC upregulates Cre recombinase in most cell types in these mice and deletes Exons 13–15 in the HIF-1α gene. For the present studies, we first determined the efficiency of deletion of Exons 13–15 of the HIF-1α gene in hepatocytes isolated from HIF-1αfl/fl-Mx-Cre+ mice and HIF-1αfl/fl-Mx-Cre− mice treated with pIpC. One week after the final pIpC injection, hepatocytes were isolated from the mice and the HIF-1α gene was analyzed using PCR of genomic DNA. As shown in Figure 3A, there was a complete deletion of Exons 13–15 of the HIF-1α gene from hepatocytes isolated from HIF-1αfl/fl-Mx-Cre+ mice. Furthermore, HIF-1α protein levels were substantially reduced in hepatocytes isolated from HIF-1αfl/fl-Mx-Cre+ and exposed to 1% oxygen when compared to hepatocytes isolated from HIF-1αfl/fl-Mx-Cre− mice and exposed to 1% oxygen (Fig. 3B). For the remainder of this manuscript, hepatocytes isolated from HIF-1αfl/fl-Mx-Cre+ mice treated with pIpC will be referred to as HIF-1α-Deficient hepatocytes, and hepatocytes isolated from HIF-1αfl/fl-Mx-Cre− mice treated with pIpC will be referred to as HIF-1α-Control hepatocytes.
Fig. 3.

(A) Hepatocytes were isolated from HIF-1αfl/fl-Mx-Cre− and HIF-1α fl/fl-Mx-Cre+ mice treated with pIpC. PCR was used to estimate the extent of deletion in the HIF-1α gene in hepatocytes. Representative of an n=3. The floxed, undeleted HIF-1α gene produces a 441 bp PCR fragment, whereas the floxed, deleted HIF-1α gene produces a 355 bp PCR fragment as described previously. (B) Hepatocytes were isolated from HIF-1αfl/fl-Mx-Cre− and HIF-1α fl/fl-Mx-Cre+ mice treated with pIpC and exposed to room air or 1% oxygen. One hour later HIF-1α was detected in nuclear extracts by western blot analysis.
HIF-1α-dependent Regulation of Gene Expression in Hypoxic Hepatocytes
Previous studies showed that PAI-1, PDGF-A, and PDGF-B, which have been implicated in the development of liver fibrosis, are upregulated in the livers of bile duct-ligated mice in a HIF-1α-dependent manner (8). These studies did not, however, investigate the cell type specific regulation of these genes by HIF-1α. Accordingly, in the present study it was determined whether these genes are upregulated in hepatocytes by hypoxia and whether this occurs in a HIF-1α-dependent manner. Exposure of HIF-1α-Control hepatocytes to 1% and 3% oxygen increased PAI-1 mRNA levels (Fig. 4A). Loss of HIF-1α signaling in HIF-1α-Deficient hepatocytes prevented upregulation of PAI-1 by approximately 50% at both 3% and 1% oxygen (Fig 4A). In contrast to PAI-1, neither PDGF-A or PDGF-B mRNA levels were increased by hypoxia in hepatocytes (Fig. 4B and 4C).
Fig. 4.

Hepatocytes were isolated from HIF-1α-Control and HIF-1α-Deficient mice, and exposed to room air, 3% oxygen, or 1% oxygen. Sixteen hours later, (A) PAI-1, (B) PDGF-A, and (C) PDGF-B mRNA levels were quantified by real-time PCR. aSignificantly different from hepatocytes exposed to room air (p<0.05). bSignificantly different from HIF-1α-Deficient hepatocytes exposed to the same concentration of oxygen (p<0.05). Data are expressed as means ± SEM; n = 3.
Recent studies suggest that VEGF is important for the development of fibrosis by promoting angiogenesis and by stimulating hepatic stellate cell activation and migration (7, 17). Whether VEGF is upregulated in hypoxic hepatocytes in a HIF-1α-dependent manner, however, is not known. Exposure of HIF-1α-Control hepatocytes to 1% and 3% oxygen increased VEGF mRNA levels (Fig. 5). Loss of HIF-1α signaling in HIF-1α-Deficient hepatocytes prevented upregulation of VEGF by 3% and 1% oxygen by approximately 80% and 70% respectively (Fig 5).
Fig. 5.

Hepatocytes were isolated from HIF-1α-Control and HIF-1α-Deficient mice, and exposed to room air, 3% oxygen, or 1% oxygen. Sixteen hours later, VEGF mRNA levels were quantified by real-time PCR. aSignificantly different from hepatocytes exposed to room air (p<0.05). bSignificantly different from HIF-1α-Deficient hepatocytes exposed to the same concentration of oxygen (p<0.05). Data are expressed as means ± SEM; n = 3.
Adrenomedullin levels are increased in patients with cirrhosis (18). It has been suggested that this mediator may contribute to cardiocirculatory abnormalities that occur in these patients. Studies suggest that adrenomedullin may be a hypoxia-regulated gene (19). Whether adrenomedullin is upregulated in hypoxic hepatocytes in a HIF-1α-dependent manner, however, is not known. Exposure of HIF-1α-Control hepatocytes to 1% and 3% oxygen increased ADM-1 and ADM-2 mRNA levels (Fig. 6A and 6B). Loss of HIF-1α signaling in HIF-1α-Deficient hepatocytes prevented upregulation of ADM-1 by 3% and 1% oxygen by approximately 85% and 60% respectively (Fig 6A). Loss of HIF-1α signaling in HIF-1α-Deficient hepatocytes completely prevented upregulation of ADM-2 by 3% and 1% oxygen (Fig. 6B).
Fig. 6.

Hepatocytes were isolated from HIF-1α-Control and HIF-1α-Deficient mice, and exposed to room air, 3% oxygen, or 1% oxygen. Sixteen hours later, (A) ADM-1 and (B) ADM-2 mRNA levels were quantified by real-time PCR. aSignificantly different from hepatocytes exposed to room air (p<0.05). bSignificantly different from HIF-1α-Deficient hepatocytes exposed to the same concentration of oxygen (p<0.05). Data are expressed as means ± SEM; n = 3.
Cell Viability in HIF-1α-Control and HIF-1α-Deficient Hepatocytes Exposed to Hypoxia
Exposure of HIF-1α-Control hepatocytes to 3% and 1% oxygen for 16 hours did not stimulate release of ALT into the medium, a marker of cell death (Figure 7). This was not affected by loss of HIF-1α signaling in HIF-1α-Deficient hepatocytes.
Fig. 7.

Hepatocytes were isolated from HIF-1α-Control and HIF-1α-Deficient mice, and exposed to room air, 3% oxygen, or 1% oxygen for 16 hours. ALT activity was measured in the media and cell lysates. The percentage of ALT activity in the medium was then calculated. Data are expressed as means ± SEM; n = 3.
Activation of HIF-2α in Hypoxic Hepatocytes
Since loss of HIF-1α signaling did not completely prevent upregulation of PAI-1, VEGF, and ADM-1 by hypoxia (Figs. 4–6), this suggests that other hypoxia-activated transcription factors also regulate these genes in hepatocytes under hypoxic conditions. One possibility is HIF-2α. To determine whether HIF-2α is activated in hypoxic hepatocytes, primary mouse hepatocytes were isolated and exposed to room air, 3% oxygen, or 1% oxygen. Low levels of HIF-2α protein were detected in hepatocytes exposed to room air (Fig. 8A). HIF-2α protein levels increased following exposure to 3% and 1% oxygen. Quantification of the western blot showed an increase in HIF-2α protein following 15 minutes of hypoxia (Fig. 8B).
Fig. 8.

Hepatocytes were isolated from mice and exposed to room air, 3% oxygen, or 1% oxygen for either 15 minutes or 1 hour. HIF-2α was then detected in nuclear extracts by western blot. (A) Representative western blot of an n=3. (B) Intensity of the HIF-2α band was quantified, and the fold change relative to room air was determined. Data are expressed as means ± SEM; n = 3, where each n represents hepatocytes isolated from a different mouse.
Immunohistochemistry was next used to confirm nuclear accumulation of HIF-2α in hypoxic hepatocytes. Exposure of hepatocytes to 1% and 3% oxygen increased nuclear levels of HIF-2α by 1 hour after exposure (Fig. 9C and 9E). Nuclear accumulation of HIFs was confirmed by colocalization with DAPI (Fig. 9D and 9F). These results show that HIF-2α is activated in hypoxic mouse hepatocytes.
Fig. 9.

Hepatocytes were isolated from mice and exposed to room air (A and B), 3% oxygen (C and D), or 1% oxygen (E and F). One hour later, immunohistochemistry was used to detect HIF-2α (green fluorescence; A, C, and E). The same cells were counterstained with DAPI (blue fluorescence; B, D, and F) to identify the nuclei and show colocalization of HIF-2α immunostaining with nuclear staining. Representative of an n=3.
Inhibition of HIF-1β Signaling in Hypoxic Mouse Hepatocytes
To determine whether HIF-2α contributes to upregulation of PAI-1, VEGF, and ADM-1 in hypoxic hepatocytes, cells were isolated from HIF-1β-Deficient mice. When HIF-1α and HIF-2α are activated in hypoxic cells, they translocate to the nucleus and heterodimerize with HIF-1β (20, 21). Heterodimerization with HIF-1β is required for upregulation of genes by HIF-1α and HIF-2α (20, 22). Since loss of HIF-1α signaling in hepatocytes only partially prevented upregulation of PAI-1, VEGF, and ADM-1 (Figs. 4–6), loss of HIF-1β signaling should completely prevent upregulation of these genes if HIF-2α also contributes to upregulation of these genes by hypoxia in hepatocytes. This approach was taken, because HIF-2αfl/fl mice are not currently available.
For these studies, HIF-1βfl/fl mice, containing loxP sites flanking Exon 6 of the HIF-1β gene, were crossed with Mx-Cre+/− mice. Exon 6 in the HIF-1β gene encodes for the basic-helix-loop-helix domain. Deletion of this region completely abrogates HIF signaling in cells (11). Treatment of HIF-1βfl/fl-Mx-Cre+ mice with pIpC upregulates Cre recombinase as described above and deletes Exon 6 in the HIF-1β gene. For the present studies, we first determined the efficiency of deletion of Exon 6 of the HIF-1β gene in hepatocytes isolated from HIF-1βfl/fl-Mx-Cre+ mice and HIF-1βfl/fl-Mx-Cre− mice treated with pIpC. One week after the final pIpC injection, hepatocytes were isolated from the mice and the HIF-1β gene was analyzed using PCR of genomic DNA. As shown in Figure 10A, there was a complete deletion of Exon 6 of the HIF-1β gene from hepatocytes isolated from these mice. Furthermore, HIF-1β protein levels were substantially reduced in hepatocytes isolated from HIF-1βfl/fl-Mx-Cre+ when compared to hepatocytes isolated from HIF-1βfl/fl-Mx-Cre− mice (Fig. 10B). For the remainder of this manuscript, hepatocytes isolated from HIF-1βfl/fl-Mx-Cre+ mice treated with pIpC will be referred to as HIF-1β-Deficient hepatocytes, and hepatocytes isolated from HIF-1βfl/fl-Mx-Cre− mice treated with pIpC will be referred to as HIF-1β-Control hepatocytes.
Fig. 10.

Hepatocytes were isolated from HIF-1βfl/fl-Mx-Cre− and HIF-1β fl/fl-Mx-Cre+ mice treated with pIpC. (A) PCR was used to estimate the extent of deletion in the HIF-1β gene in hepatocytes. Representative of an n=3. The floxed, undeleted HIF-1β gene produces a 359 bp PCR fragment, whereas the floxed, deleted HIF-1β gene produces a 317 bp PCR fragment as described previously. (B) HIF-1β was detected in nuclear extracts by western blot analysis.
HIF-1β-dependent Regulation of Gene Expression in Hypoxic Hepatocytes
Exposure of HIF-1β-Control hepatocytes to 1% and 3% oxygen increased PAI-1, VEGF, ADM-1, and ADM-2 mRNA levels (Fig. 11). Loss of HIF-1β signaling in HIF-1β-Deficient hepatocytes completely prevented upregulation of these genes by hypoxia (Fig 11).
Fig. 11.

Hepatocytes were isolated from HIF-1β-Control and HIF-1β-Deficient mice and exposed to room air, 3% oxygen, or 1% oxygen. Sixteen hours later, (A) PAI-1, (B) VEGF, (C) ADM-1, and (D) ADM-2 mRNA levels were quantified by real-time PCR. aSignificantly different from hepatocytes exposed to room air (p<0.05). bSignificantly different from HIF-1α-Deficient hepatocytes exposed to the same concentration of oxygen (p<0.05). Data are expressed as means ± SEM; n = 3.
Cell Viability in HIF-1β-Control and HIF-1β-Deficient Hepatocytes Exposed to Hypoxia
Exposure of either HIF-1β-Control hepatocytes or HIF-1β-Deficient hepatocytes to 3% and 1% oxygen for 16 hours did not stimulate release of ALT into the medium (Figure 12).
Fig. 12.

Hepatocytes were isolated from HIF-1β-Control and HIF-1β-Deficient mice and exposed to room air, 3% oxygen, or 1% oxygen for 16 hours. ALT activity was measured in the media and cell lysates. The percentage of ALT activity in the medium was then calculated. Data are expressed as means ± SEM; n = 3.
Discussion
Previous studies have shown that HIF-1α is activated in the livers of mice with fibrosis (8). Furthermore, these studies demonstrated that loss of HIF-1α signaling in these mice partially prevented upregulation of profibrotic mediators, such as PDGF-A, PDGF-B, and PAI-1. These studies did not, however, investigate the cell type-specific regulation of profibrotic mediators by HIFs. Our studies demonstrate that both HIF-1α and HIF-2α are activated in hypoxic hepatocytes (Figs. 1, 2, 8 and 9), and that they both contribute to upregulation of profibrotic mediators in these cells. Although previous studies demonstrated that HIF-1α is activated in hypoxic hepatocytes (23), studies have not shown previously that HIF-2α is activated in these cells.
Studies have shown that PAI-1 is important for the development of liver fibrosis and that PAI-1 is upregulated to a lesser extent in the livers of bile duct-ligated HIF-1α-Deficient mice (8, 24, 25). Studies have also demonstrated that HIF-1α regulates PAI-1 in hypoxic hepatocytes (23). Our studies confirm these results, and suggest further that HIF-2α is an important regulator of PAI-1 in hypoxic hepatocytes, a result not reported previously. As discussed, HIFs are composed of an α subunit, either HIF-1α or HIF-2α, and a β subunit, HIF-1β (1, 2). HIF-1α and HIF-2α protein subunits are constitutively produced in cells and immediately targeted for proteosomal degradation in normoxic cells. In cells that are hypoxic, however, the mechanisms that target HIFs for degradation are inhibited, allowing HIF-1α and HIF-2α to translocate to the nucleus (3). In the nucleus both HIF-1α and HIF-2α heterodimerize with HIF-1β and regulate expression of genes (2). In the present studies, upregulation of PAI-1 was only prevented by approximately 50% in hepatocytes deficient in HIF-1α (Fig. 4), whereas upregulation of PAI-1 was completely prevented in hepatocytes isolated from HIF-1β-Deficient mice (Fig. 11). These results strongly suggest that HIF-2α is an important regulator of PAI-1 in hepatocytes. It remains possible that HIF-1β may interact with transcription factors other than HIF-2α to regulate PAI-1; however, this is very unlikely. HIF-1β interacts with the aryl hydrocarbon receptor to regulate gene expression (26); however, to the best of our knowledge, there is no evidence that the aryl hydrocarbon receptor is activated by hypoxia. A third HIF-α protein has been identified, HIF-3α, however studies have shown that it acts as a dominant-negative protein and prevents upregulation of genes by HIF-1α and HIF-2α (27). Therefore, our studies suggest that both HIF-1α and HIF-2α are important regulators of PAI-1 in hypoxic hepatocytes. These results may explain why deletion of HIF-1α in mice only partially prevents upregulation of PAI-1 in the liver after bile duct ligation. In these studies, upregulation of PAI-1 was prevented by less than 50% in the livers of HIF-1α-Deficient mice (8). Considering our in vitro results, it is possible that HIF-2α may also regulate PAI-1 in the livers of bile duct-ligated mice. Further studies are needed, however, to evaluate this possibility.
Interestingly, although PDGF-A and PDGF-B were upregulated in the livers of bile duct-ligated mice in a HIF-1α-dependent manner (8), these genes were not upregulated in hypoxic hepatocytes. This suggests that HIF-1α regulates production of these growth factors in other cell types in the liver during the development of fibrosis. One possibility is macrophages. Immunohistochemical studies and in situ hybridization techniques have shown that macrophages express PDGFs in the liver during the genesis of fibrosis, whereas hepatocytes do not (28, 29). Furthermore, studies have shown that hypoxic macrophages release PDGF (30). Therefore, during the development of fibrosis, hepatic macrophages may synthesize and release PDGF in a HIF-dependent manner. Further studies are needed, however, to test this possibility.
Studies suggest that proangiogenic mediators, such as VEGF, may promote liver fibrosis by stimulating hepatic stellate cell proliferation, chemotaxis, and collagen production (7, 17). Furthermore, studies have shown that VEGF is upregulated in hepatocytes in the livers of rats with fibrosis, and that VEGF expression colocalizes with regions of hypoxia (31). It is not known, however, whether VEGF levels are increased in hypoxic hepatocytes in a HIF-dependent manner. Our results indicate that VEGF is upregulated in hypoxic hepatocytes and that inhibition of HIF-1α signaling nearly completely prevented upregulation of VEGF in these cells (Fig. 5). This suggests that HIF-1α is the predominant regulator of HIF-1α in hypoxic hepatocytes. Deletion of HIF-1β further reduced VEGF levels in hypoxic hepatocytes, suggesting that HIF-2α may also be a minor regulator of VEGF production in these cells (Fig. 11).
One major complication that occurs in cirrhotic patients is arteriolar vasodilation that leads to cardiocirculatory dysfunction (32). Studies suggest that adrenomedullin-1 (ADM-1) may be an important mediator in this process. ADM-1 is a vasoactive peptide that stimulates vasodilation (33). Levels of this peptide are increased in patients with cirrhosis (18). The molecular mechanisms that regulate production of this peptide during fibrosis, however, are not completely understood. Studies suggest that ADM-1 may be a HIF-regulated gene (19). Therefore, we determined whether ADM-1 levels are increased in hypoxic hepatocytes in a HIF-dependent manner. Our results show that ADM-1 was increased in hypoxic hepatocytes, and that loss of HIF-1α signaling nearly completely prevented upregulation of ADM-1 (Fig. 6). This suggests that in cirrhotic patients, hypoxic hepatocytes may be an important source of ADM-1 in the circulation. In addition to ADM-1, we also investigated whether ADM-2 levels were increased in hypoxic hepatocytes in a HIF-dependent manner. ADM-2, also called intermedin, is also a potent vasodilator that binds to the same receptors as ADM-1, however, with different potency (34, 35). Our results show that this vasoactive peptide is also upregulated in hypoxic hepatocytes in a HIF-1α-dependent manner (Fig. 6). Our studies are the first to indicate that ADM-2 is upregulated by hypoxia in a HIF-dependent manner. No studies have yet investigated whether ADM-2 levels are increased in patients with cirrhosis. Our studies suggest that in addition to ADM-1, ADM-2 may be another important vasoactive substance produced in these patients.
Collectively, our results indicate that HIFs may be important regulators of profibrotic mediators by hypoxic hepatocytes. Our results also suggest that hypoxic hepatocytes may be an important source of vasoactive mediators in patients with cirrhosis. Therefore, therapeutic inhibition of both HIF-1α and HIF-2α may be beneficial for patients with fibrosis by inhibiting both the progression of fibrosis and by limiting cardiovascular complications that develop in these patients. Furthermore, considering the potential importance of HIFs in tumor angiogenesis, inhibition of HIFs may also prevent development of hepatocellular carcinoma in patients with severe cirrhosis.
Acknowledgments
This study was supported by National Institutes of Health grants DK073566 (B.L.C.) and COBRE (Center of Biomedical Research Excellence) P20 RR021940 as well as the Molecular Biology Core and the Histology Core supported by the COBRE grant. In addition, this work was supported by a Lied Endowed Basic Science Pilot Research Grant (B.L.C.). The authors thank Dr. Frank J. Gonzalez at the National Cancer Institute for providing the HIF-1βfl/fl-Mx-Cre+, HIF-1βfl/fl-Mx-Cre−, HIF-1αfl/fl-Mx-Cre+, and HIF-1αfl/fl-Mx-Cre− mice for these studies.
References
- 1.Coleman ML, Ratcliffe PJ. Oxygen sensing and hypoxia-induced responses. Essays Biochem. 2007;43:1–15. doi: 10.1042/BSE0430001. [DOI] [PubMed] [Google Scholar]
- 2.Gaber T, Dziurla R, Tripmacher R, Burmester GR, Buttgereit F. Hypoxia inducible factor (HIF) in rheumatology: low O2! See what HIF can do! Ann Rheum Dis. 2005;64(7):971–80. doi: 10.1136/ard.2004.031641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cash TP, Pan Y, Simon MC. Reactive oxygen species and cellular oxygen sensing. Free Radic Biol Med. 2007;43(9):1219–25. doi: 10.1016/j.freeradbiomed.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134(6):1655–69. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Friedman SL, Arthur MJ. Activation of cultured rat hepatic lipocytes by Kupffer cell conditioned medium. Direct enhancement of matrix synthesis and stimulation of cell proliferation via induction of platelet-derived growth factor receptors. J Clin Invest. 1989;84 (6):1780–5. doi: 10.1172/JCI114362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marra F, Romanelli RG, Giannini C, Failli P, Pastacaldi S, Arrighi MC, et al. Monocyte chemotactic protein-1 as a chemoattractant for human hepatic stellate cells. Hepatology. 1999;29(1):140–8. doi: 10.1002/hep.510290107. [DOI] [PubMed] [Google Scholar]
- 7.Novo E, Cannito S, Zamara E, Valfre Di Bonzo L, Caligiuri A, Cravanzola C, et al. Proangiogenic cytokines as hypoxia-dependent factors stimulating migration of human hepatic stellate cells. Am J Pathol. 2007;170(6):1942–53. doi: 10.2353/ajpath.2007.060887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moon J-O, Welch TP, Gonzalez FJ, Copple BL. Role of hypoxia-inducible factor-1α in the development of liver fibrosis. Am J Physiol Gastrointest Liver Physiol. 2009;296 doi: 10.1152/ajpgi.90368.2008. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kozak KR, Abbott B, Hankinson O. ARNT-deficient mice and placental differentiation. Developmental Biology. 1997;191(2):297–305. doi: 10.1006/dbio.1997.8758. [DOI] [PubMed] [Google Scholar]
- 10.Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998;12(2):149–62. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tomita S, Sinal CJ, Yim SH, Gonzalez FJ. Conditional disruption of the aryl hydrocarbon receptor nuclear translocator (Arnt) gene leads to loss of target gene induction by the aryl hydrocarbon receptor and hypoxia-inducible factor 1alpha. Molecular Endocrinology. 2000;14(10):1674–81. doi: 10.1210/mend.14.10.0533. [DOI] [PubMed] [Google Scholar]
- 12.Tomita S, Ueno M, Sakamoto M, Kitahama Y, Ueki M, Maekawa N, et al. Defective brain development in mice lacking the Hif-1alpha gene in neural cells. Mol Cell Biol. 2003;23(19):6739–49. doi: 10.1128/MCB.23.19.6739-6749.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuhn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269(5229):1427–9. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- 14.Kim ND, Moon JO, Slitt AL, Copple BL. Early growth response factor-1 is critical for cholestatic liver injury. Toxicol Sci. 2006;90(2):586–95. doi: 10.1093/toxsci/kfj111. [DOI] [PubMed] [Google Scholar]
- 15.Le Provost F, Riedlinger G, Hee Yim S, Benedict J, Gonzalez FJ, Flaws J, et al. The aryl hydrocarbon receptor (AhR) and its nuclear translocator (Arnt) are dispensable for normal mammary gland development but are required for fertility. Genesis. 2002;32 (3):231–9. doi: 10.1002/gene.10037. [DOI] [PubMed] [Google Scholar]
- 16.Ganey PE, Bailie MB, Vancise S, Colligan ME, Madhukar BV, Robinson JP, et al. Activated neutrophils from rat injured isolated hepatocytes. Lab Invest. 1994;70(1):53–60. [PubMed] [Google Scholar]
- 17.Yoshiji H, Kuriyama S, Yoshii J, Ikenaka Y, Noguchi R, Hicklin DJ, et al. Vascular endothelial growth factor and receptor interaction is a prerequisite for murine hepatic fibrogenesis. Gut. 2003;52(9):1347–54. doi: 10.1136/gut.52.9.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fabrega E, Casafont F, Crespo J, De La Pena J, San Miguel G, De Las Heras G, et al. Plasma adrenomedullin levels in patients with hepatic cirrhosis. The American Journal of Gastroenterology. 1997;92(10):1901–4. [PubMed] [Google Scholar]
- 19.Garayoa M, Martinez A, Lee S, Pio R, An WG, Neckers L, et al. Hypoxia-inducible factor-1 (HIF-1) up-regulates adrenomedullin expression in human tumor cell lines during oxygen deprivation: a possible promotion mechanism of carcinogenesis. Molecular Endocrinology. 2000;14(6):848–62. doi: 10.1210/mend.14.6.0473. [DOI] [PubMed] [Google Scholar]
- 20.Jiang BH, Rue E, Wang GL, Roe R, Semenza GL. Dimerization, DNA binding, and transactivation properties of hypoxia-inducible factor 1. J Biol Chem. 1996;271(30):17771–8. doi: 10.1074/jbc.271.30.17771. [DOI] [PubMed] [Google Scholar]
- 21.Erbel PJ, Card PB, Karakuzu O, Bruick RK, Gardner KH. Structural basis for PAS domain heterodimerization in the basic helix--loop--helix-PAS transcription factor hypoxia-inducible factor. Proc Natl Acad Sci U S A. 2003;100(26):15504–9. doi: 10.1073/pnas.2533374100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wood SM, Gleadle JM, Pugh CW, Hankinson O, Ratcliffe PJ. The role of the aryl hydrocarbon receptor nuclear translocator (ARNT) in hypoxic induction of gene expression. Studies in ARNT-deficient cells. J Biol Chem. 1996;271(25):15117–23. doi: 10.1074/jbc.271.25.15117. [DOI] [PubMed] [Google Scholar]
- 23.Kietzmann T, Roth U, Jungermann K. Induction of the plasminogen activator inhibitor-1 gene expression by mild hypoxia via a hypoxia response element binding the hypoxia-inducible factor-1 in rat hepatocytes. Blood. 1999;94(12):4177–85. [PubMed] [Google Scholar]
- 24.Bergheim I, Guo L, Davis MA, Duveau I, Arteel GE. Critical role of plasminogen activator inhibitor-1 in cholestatic liver injury and fibrosis. J Pharmacol Exp Ther. 2006;316(2):592–600. doi: 10.1124/jpet.105.095042. [DOI] [PubMed] [Google Scholar]
- 25.Wang H, Vohra BP, Zhang Y, Heuckeroth RO. Transcriptional profiling after bile duct ligation identifies PAI-1 as a contributor to cholestatic injury in mice. Hepatology. 2005;42(5):1099–108. doi: 10.1002/hep.20903. [DOI] [PubMed] [Google Scholar]
- 26.Reyes H, Reisz-Porszasz S, Hankinson O. Identification of the Ah receptor nuclear translocator protein (Arnt) as a component of the DNA binding form of the Ah receptor. Science. 1992;256(5060):1193–5. doi: 10.1126/science.256.5060.1193. [DOI] [PubMed] [Google Scholar]
- 27.Makino Y, Kanopka A, Wilson WJ, Tanaka H, Poellinger L. Inhibitory PAS domain protein (IPAS) is a hypoxia-inducible splicing variant of the hypoxia-inducible factor-3alpha locus. J Biol Chem. 2002;277(36):32405–8. doi: 10.1074/jbc.C200328200. [DOI] [PubMed] [Google Scholar]
- 28.Faiz Kabir Uddin Ahmed A, Ohtani H, Nio M, Funaki N, Iwami D, Kumagai S, et al. In situ expression of fibrogenic growth factors and their receptors in biliary atresia: comparison between early and late stages. The Journal of Pathology. 2000;192(1):73–80. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH657>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 29.Nakatsukasa H, Nagy P, Evarts RP, Hsia CC, Marsden E, Thorgeirsson SS. Cellular distribution of transforming growth factor-beta 1 and procollagen types I, III, and IV transcripts in carbon tetrachloride-induced rat liver fibrosis. J Clin Invest. 1990;85(6):1833–43. doi: 10.1172/JCI114643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuwabara K, Ogawa S, Matsumoto M, Koga S, Clauss M, Pinsky DJ, et al. Hypoxia-mediated induction of acidic/basic fibroblast growth factor and platelet-derived growth factor in mononuclear phagocytes stimulates growth of hypoxic endothelial cells. Proc Natl Acad Sci U S A. 1995;92(10):4606–10. doi: 10.1073/pnas.92.10.4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rosmorduc O, Wendum D, Corpechot C, Galy B, Sebbagh N, Raleigh J, et al. Hepatocellular hypoxia-induced vascular endothelial growth factor expression and angiogenesis in experimental biliary cirrhosis. Am J Pathol. 1999;155(4):1065–73. doi: 10.1016/S0002-9440(10)65209-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arroyo V, Jimenez W. Complications of cirrhosis. II. Renal and circulatory dysfunction. Lights and shadows in an important clinical problem. J Hepatol. 2000;32 (Suppl):157–70. doi: 10.1016/s0168-8278(00)80423-7. [DOI] [PubMed] [Google Scholar]
- 33.Kato J, Tsuruda T, Kita T, Kitamura K, Eto T. Adrenomedullin: a protective factor for blood vessels. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005;25(12):2480–7. doi: 10.1161/01.ATV.0000184759.91369.f8. [DOI] [PubMed] [Google Scholar]
- 34.Bell D, Mcdermott BJ. Intermedin (adrenomedullin-2): a novel counter-regulatory peptide in the cardiovascular and renal systems. British Journal of Pharmacology. 2008;153 (Suppl 1):S247–62. doi: 10.1038/sj.bjp.0707494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fujisawa Y, Nagai Y, Miyatake A, Miura K, Shokoji T, Nishiyama A, et al. Roles of adrenomedullin 2 in regulating the cardiovascular and sympathetic nervous systems in conscious rats. Am J Physiol Heart Circ Physiol. 2006;290(3):H1120–7. doi: 10.1152/ajpheart.00461.2005. [DOI] [PubMed] [Google Scholar]