

Abstract
2-Keto-3-deoxy-6-phosphogluconate (KDPG) aldolase catalyzes the reversible cleavage of KDPG to pyruvate and glyceraldehyde-3-phosphate. The enzyme is a class I aldolase whose reaction mechanism involves formation of Schiff base intermediates between Lys-133 and a keto substrate. A covalent adduct was trapped by flash freezing KDPG aldolase crystals soaked with 10 mM pyruvate in acidic conditions at pH 4.6. Structure determination to 1.95-Å resolution showed that pyruvate had undergone nucleophilic attack with Lys-133, forming a protonated carbinolamine intermediate, a functional Schiff base precursor, which was stabilized by hydrogen bonding with active site residues. Carbinolamine interaction with Glu-45 indicates general base catalysis of several rate steps. Stereospecific addition is ensured by aromatic interaction of Phe-135 with the pyruvate methyl group. In the native structure, Lys-133 donates all of its hydrogen bonds, indicating the presence of an ɛ-ammonium salt group. Nucleophilic activation is postulated to occur by proton transfer in the monoprotonated zwitterionic pair (Glu-45/Lys-133). Formation of the zwitterionic pair requires prior side chain rearrangement by protonated Lys-133 to displace a water molecule, hydrogen bonded to the zwitterionic residues.
Aldolases have been the subject of continuous interest because such enzymes provide a mechanism for carbon—carbon bond formation in living organisms. 2-Keto-3-deoxy-6-phosphogluconate (KDPG) aldolase (E.C. 4.1.2.14), is best known for its role in the Entner-Doudoroff pathway of bacteria, where it catalyzes the reversible cleavage of KDPG to pyruvate and glyceraldehyde-3-phosphate (1). Gene sequencing showed KDPG aldolase to be identical to 2-keto-4-hydroxyglutarate (KHG) aldolase (2), which has enzymatic specificity toward glyoxylate, forming KHG in the presence of pyruvate (3), and is capable of regulating glyoxylate levels in the glyoxylate bypass, an alternate pathway when bacteria are grown on acetate carbon sources (4). In mammals, KHG aldolase catalyzes a central reaction in hydroxyproline catabolism (5). The enzyme also efficiently catalyzes β-decarboxylation of oxaloacetate regardless of source (6). The reaction mechanism of KDPG aldolase involves Schiff base formation between a substrate carbonyl and lysine residue in the active site (7), making it a class I aldolase. Key intermediates are depicted in reaction Scheme S1.
Scheme 1.

In the condensation reaction shown in Scheme S1, Schiff base formation with pyruvate is thought to occur through transient formation of a dipolar carbinolamine, with the keto function resulting in the neutral carbinolamine species shown that is then dehydrated to the imine form of the Schiff base (8). Proton abstraction creates a reactive enamine, which, on condensation with d-glyceraldehyde-3-phosphate (G3P), yields a second Schiff base with KDPG as shown. Hydrolysis transforms the Schiff base into a neutral carbinolamine species that, on decomposition of the dipolar carbinolamine, releases KDPG from the active site, thereby regenerating the enzyme. In bacteria, the enzyme operates under kinetic control (9) to provide stereochemically pure products (10). The considerable substrate tolerance displayed by KDPG aldolase is useful in catalysis of stereospecific aldol addition of pyruvate to a range of unnatural electrophilic substrates at rates practical for large-scale preparative organic synthesis (11).
The structure of KDPG aldolase trimer from Pseudomonas putida was determined to 2.8-Å resolution (12), and the trace of polypeptide folding of each subunit corresponds to that of a β barrel. The P. putida enzyme is closely related to the Escherichia coli KDPG aldolase and shares 45% sequence identity. Structure determination of P. putida KDPG aldolase, however, presented interpretative difficulties such that location of the essential active site residues could not be assigned unambiguously.
To investigate the reaction mechanism in pyruvate aldolases and the role of active site residues, a crystallographic study of the E. coli KDPG aldolase in complex with pyruvate was undertaken. Naturally occurring Schiff base intermediates however have not been observed in crystal structure determinations of class I aldolases at physiological pH, even in presence of excess substrate. Schiff base intermediates appear to be only transiently stable and can be trapped by acid quenching (13, 14) or by sodium borohydride reduction. Neither approach is very satisfactory in terms of resultant structural information. Trapping by quenching with acid destroys the active site geometry whereas sodium borohydride reduction results in formation of a covalent analogue that sterically does not resemble the Schiff base. Our approach exploited flash freezing techniques used in cryocrystallographic data collection to trap Schiff base intermediates. E. coli KDPG aldolase, contrary to P. putida KDPG aldolase, lends itself well to this technique because it is active over a wide pH range (10), allowing screening of a large number of crystallization conditions. From model studies, Schiff base formation at acidic pH is kinetically a very slow process (15), making it feasible to attempt intermediate trapping on a time scale compatible with crystallographic flash freezing techniques.
We have succeeded in trapping a carbinolamine intermediate at acidic pH and show here the structure of the active site of the KDPG aldolase containing this intermediate. To our knowledge, there have been no previous reports of a structure that contains a covalent carbinolamine intermediate and that is directly implicated in a reaction pathway.
Methods
Purification and Crystallization.
The coding region for the KDPG aldolase gene from E. coli (16) was amplified by PCR and cloned into the overexpression plasmid pKK-223 (17) and overexpressed in E. coli cells. Recombinant KDPG aldolase was purified by ion exchange chromatography and concentrated to 10 mg/ml for crystallization. Crystals were obtained by using the hanging drop method, after equilibration of the protein against 10 mM Tris⋅HCl (pH 8), in 18.5% polyethylene glycol (PEG) 3500, 0.2 M ammonium sulfate, and 0.1 M sodium acetate at pH 4.6.
Seleno-l-methionine (SeMet) isoform of KDPG aldolase was produced in E. coli strain BL21 (18) in the presence of high concentrations of isoleucine, lysine, and threonine to inhibit methionine biosynthesis (19). SeMet KDPG aldolase was purified and crystallized under identical conditions as the native enzyme, except for the presence of 1 mM DTT in purification buffers.
Data Collection.
Before data collection, crystals were cryoprotected by transfer to mother liquor made up with 20% glycerol, as well as 10 mM pyruvate in the case of pyruvate-soaked crystals. Crystals were soaked for 15 min and then flash frozen at −170°C. A Quantum4 (Area Detector Systems, Poway, CA) charge-coupled device (CCD) detector was used to collect data from a single crystal at National Synchrotron Light Source (NSLS) beam line X8-C. KDPG aldolase crystals have space group P212121, with unit cell dimensions a = 55.0 Å, b = 85.2 Å, and c = 133.7 Å and a trimer in the asymmetric unit cell. Unit cell dimensions (a = 54.7 Å, b = 96.4 Å, and c = 120.1 Å) but not space group differed for the SeMet derivative with respect to native crystals, and unit cell dimensions were similar in the presence or absence of pyruvate. The asymmetric unit cell contents were nevertheless consistent with a trimeric quaternary structure for the SeMet derivative. Data sets were collected at a crystal-to-detector distance of 160 mm and by using 0.66° oscillations per image. Anomalous pairs for the SeMet:pyruvate derivative were collected in a single pass by using inverse-normal beam geometry at three different wavelengths. An x-ray fluorescence spectrum was recorded and used to select the wavelength optima for multiple wavelength anomalous dispersion (MAD) data collection. Data were collected at 0.9791 Å (the inflection point of the fluorescence spectrum, f′ minimum), 0.9791 Å (f′′ maximum), and 0.9300 Å (remote high-energy wavelength). All data sets were processed independently by using the programs denzo and scalepack (20). Data statistics are shown in Table 1.
Table 1.
Data reduction and refinement statistics for KDPG aldolase
| Data set | Wavelength, Å | Resolution, Å | Completeness, % | Rsym*, % | Redundancy | Figure of merit | Refinement
statistics
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. of reflections | No. of protein atoms | No. of water molecules | Rcryst†, % | Rfree‡, % | rms bond length, Å | rms bond angle, degrees | |||||||
| Native:pyruvate | 1.00 | 1.95 | 99.4 | 8.7 | 11.1 | — | 44,409 | 4,698 | 615 | 20.1 | 24.3 | 0.006 | 1.30 |
| Native | 0.9787 | 2.00 | 94.8 | 7.5 | 4.97 | — | 39,315 | 4,698 | 749 | 20.2 | 25.9 | 0.006 | 1.30 |
| SeMet:pyruvate | |||||||||||||
| Remote | 0.9400 | 2.00 | 85.9 | 9.8 | 3.76 | — | 37,390 | 4,698 | 825 | 19.4 | 25.4 | 0.006 | 1.40 |
| Inflexion | 0.9790 | 2.00 | 84.4 | 10.9 | 3.78 | 0.82§ | — | — | — | — | — | — | — |
| Peak | 0.9788 | 2.00 | 84.6 | 10.5 | 3.81 | — | — | — | — | — | — | — | — |
Rsym =
∑hkl ∑i|Ii(hkl)
−
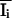 (hkl)|/∑hkl ∑iIi(hkl)
with i running over the number of independent observations
of reflection hkl.
(hkl)|/∑hkl ∑iIi(hkl)
with i running over the number of independent observations
of reflection hkl.
Rcryst = ∑hkl∥Fo(hkl)| − |Fc(hkl)∥/∑hkl|Fo(hkl)|.
Rfree = ∑hkl∈T∥Fo(hkl)| − |Fc(hkl)∥/∑hkl∈T|Fo(hkl)|, where T is a test data set randomly selected from the observed reflections prior to refinement. Test data set contained 5% of the total observed data and was not used throughout refinement.
Figure of merit calculated for 2.8 Å resolution data by program solve.
Structure Solution and Refinement.
The MAD data (Table 1) were scaled by the program solve (21), and, using data in 15.0- to 3.0-Å resolution data, 11 of 12 selenium sites, including the N terminus substituted methionines, were obtained for the SeMet:pyruvate derivative. Three-fold noncrystallographic symmetry (NCS) was deduced from SeMet positions. Initial phases were improved by the dm program (22) and included NCS averaging at a 3-Å resolution and yielded a traceable calculated electron density map corresponding to 80% of the structure. Model building was initiated by manual fitting of a KDPG aldolase model (kindly provided by A. Tulinsky, Michigan State University, East Lansing, MI) into the MAD density using o (23) for one subunit. The KDPG aldolase trimer was constructed by applying noncrystallographic symmetry to the manually fitted subunit. The trimer was then subjected to the rigid body refinement with NCS constraints by using the cns program (24). Although resultant R factors were high (Rcryst = 0.54, Rfree = 0.55), the subunits were reasonably placed into electron density. Manual rebuilding with o (23), including mutation and baton option, was performed on one subunit, and the trimer was recreated from the NCS symmetry. The model was subjected to another round of rigid body refinement and to the slow cooling procedure against amplitude-based maximum likelihood target. In all subsequent rounds of refinement, NCS constraints were discarded. Iterative rounds of model building and refinement led to an R factor of 0.19 by using all data in the resolution range between 37.6 and 2.0 Å and with Rfree of 0.25.
The structure of the native crystal was determined by molecular replacement using the SeMet:pyruvate trimer as a model. Combination of model rebuilding and molecular dynamics refined the native structure to 2.0-Å resolution. Isomorphism of native:pyruvate soaked crystals with native crystals allowed refinement of the native:pyruvate structure. Protomer polypeptide folding of SeMet:pyruvate, native, and native:pyruvate crystals was identical; however, protomer assembly into trimers differed slightly between SeMet and native quaternary structures (rms Cα < 0.8 Å). The observed unit cell differences between SeMet and native structures may be ascribed to N terminus conformational differences because of seleno-methionine substitution that perturbed trimer packing. Conformational stability of the penultimate N-terminal residues involves hydrophobic interaction between Met-1 and Trp-4. Substitution of SeMet modified this interaction geometry and destabilized the packing interaction existing in the native structure between Met-1 of subunit B in one trimer and Leu-213, the C-terminal residue of the same subunit in an adjacent trimer.
Electron density was contiguous for all residues in both native and native:pyruvate structures. Table 1 shows the overall crystallographic R factor and the free R factor for all models and all observed reflections within the indicated resolution range. A Ramachadran plot analysis by the program procheck (25) indicates that, for native and native:pyruvate structures, 92.3% of all residues lie in the most favorable regions and 7.5% in additional allowable regions. The analysis showed that, for native and native:pyruvate structures, all stereochemical parameters are better than expected at their given resolution.
Results and Discussion
KDPG aldolase protomer displays an (α/β)8 fold (subunit molecular mass of 22 kDa) found in other class I aldolases. The trimeric subunit assembly mimics KDPG aldolase from P. putida (12) (rms deviation Cα coordinates 1.5 Å) and resembles a ship's propeller, with identical protomer structures approximating elongated ellipsoids. The N-terminal α-helix in each protomer caps the N-terminal end of the barrel. As in other (α/β)8 barrel structures, the active site was located in the depression of the C-terminal end of the barrel. The active site is freely accessible to solvent, consistent with sulfate ions present in each protomer in pyruvate-free KDPG aldolase crystals, one of which is bound in the active site cavity, shown in Fig. 1, whereas a second binds at the active site periphery ≈8 Å away. The active site sulfate interacts through hydrogen bonding with conserved active site residues, Glu-45, Arg-49, Thr-73, and Lys-133, shown in Fig. 2. Spatial proximity of Glu-45, Arg-49, and Lys-133, which forms the Schiff base with substrate, corroborates previous identification of essential active site residues (26–29). A water molecule, Wat93, stabilizes the arrangement of active site residues, Glu-45 and Lys-133, through hydrogen bonding with both residues. Hydrogen bonding by the sulfate oxyanion, O1, with Glu-45 carboxylate oxygen requires Glu-45 to be protonated in the crystal structure. Other than a change in Val-118 rotamer conformation, which may be a consequence of Pro-94 pyrridoline ring isomerization on pyruvate binding, superposition of native and native:pyruvate structures showed no evidence for subunit conformational changes (rms deviation Cα < 0.2 Å).
Figure 1.

Stereo views of the active site in a native KDPG aldolase protomer. Electron density from a 2-Å annealed omit map encompassing Lys-133 and sulfate ion was contoured at 3σ. Sulfate binding and associated water molecule positions are identical in all subunits. Contacts between non-bonded atoms within hydrogen bonding distance are shown by dotted lines. Lys-133 Nz amine participates in hydrogen bonds with sulfate oxyanions O1 and O2, as well as water molecules Wat13 and Wat93; O1, O2, and Wat13 accept hydrogen bonds consistent with Lys-133 protonated. The Lys-133 side chain shown is oriented roughly perpendicular to the β-barrel axis and is located near the carboxyl end of the β strands comprising the β-barrel. Drawing was made with the program bobscript (38) and presented by using raster3d (39).
Figure 2.

Non-bonded interactions shown in active site of KDPG aldolase. Hydrogen bonds shown by dotted lines correspond to distances between 2.5 and 2.9 Å. Hydrogen bonding by Glu-45 with sulfate oxyanion O1 requires Glu-45 to be protonated.
Continuous electron density, extending beyond Lys-133 Nz in the native:pyruvate structure of each protomer, shown in Fig. 3a, indicates formation of a stable covalent adduct. The shape of the electron density in Fig. 3b is inconsistent with Schiff base or enamine adducts, because these intermediates imply a trigonal bonding geometry about the pyruvate C2, which was not observed. Refinement of the native:pyruvate structure showed the electron density seen in Fig. 3b to indicate tetrahedral bonding geometry about the pyruvate C2 carbon and consistent with a carbinolamine intermediate covalently bound in each aldolase subunit.
Figure 3.

Stereoview of the active site of a KDPG aldolase protomer corresponding to native crystals soaked with 10 mM pyruvate and showing electron density from a 1.95-Å annealed omit map contoured at 5σ. The orientation shown in a is similar to the view in Fig. 1. The continuous electron density extending from Lys-133 Nz was interpreted as a pyruvate molecule covalently bound to Lys-133. Electron density in b clearly shows the tetrahedral bonding geometry about pyruvate C2. View in b corresponds to looking from the pyruvate carboxylate toward Lys-133 Nz, with the plane of the drawing oriented roughly perpendicular with respect to a. The conformation of the carbinolamine intermediate is identical in all subunits. Hydrogen bonding contacts are shown as dotted lines. Hydrogen bonding of pyruvate with Glu-45, Arg-49, and Thr-73 stabilizes the carbinolamine intermediate. Wat35 in subunit A, shown here, is favorably oriented, making hydrogen bonds with both Glu-45 and O3, to catalyze proton transfers at the level of the carbinolamine intermediate. In the other subunits, Wat35 hydrogen bonds only to Glu-45. Wat35 was also omitted in the annealed difference map calculations, and, because of the smaller O3 B factor, the difference electron density is slightly elongated toward Wat35 at this contour level.
The tetrahedral adduct observed can be represented by any one of three carbinolamine species or a combination of these. Shown in Scheme S2 are the following: a neutral carbinolamine species (T0), a protonated species (T+), wherein the Lys-133 Nz (R) is positively charged, and a zwitterionic form (T±), where Lys-133 Nz and carbinolamine hydroxyl group O3 are charged positive and negative, respectively. The pyruvate carboxylate in the carbinolamine adducts is taken to be fully ionized at pH 4.6 in accordance with its pKa of 2.5 in free pyruvate (30).
Scheme 2.

Carbinolamine formation in model compounds at neutral or acidic pH values indicates a pKa of 8–9 in T+ and 13–14 in T± for the carbinolamine hydroxyl group O3 (31); hence, the dipolar ion cannot be present at acidic pH. Perturbation of the O3 oxygen pKa by the active site microenvironment is unlikely, given hydrogen bonding of water molecules with O3. Model studies in aqueous solutions suggest a pKa for the protonated amine in T+ of ≈8 (15), and solvent effects on the protonated amine are small, amounting to a reduction by ≈1 pKa unit in 50% aqueous dioxane (32). The protonated carbinolamine species, T+, should thus be the tetrahedral adduct observed in the active site of KDPG aldolase crystals. Schiff base species were not observed, consistent with inhibition of carbinolamine precursor formation. In acidic media, formation of the neutral carbinolamine T0 species, requisite Schiff base precursor, is kinetically disfavored from the T+ species (33).
Active Site Residues.
Carbinolamine adduct formation involves negligible conformational changes in the molecular architecture of the KDPG aldolase active site. The carbinolamine intermediate is stabilized by numerous hydrogen bonds, shown in Fig. 4, and implicating the same residues used to bind the sulfate ion in the active site. Glu-45 carboxylate interacts with the carbinolamine hydroxyl group O3 whereas the pyruvate carboxylate moiety interacts with Thr-73 backbone amide as well as Arg-49 and Thr-73 side chains. The van der Waals contact between the pyruvate C3 methyl group and aromatic ring of Phe-135 contributes not only to stability of the intermediate through hydrophobic interactions but also to stereospecific alignment of the pyruvate in the active site.
Figure 4.

Non-bonded interactions in active site of KDPG aldolase in complex with pyruvate. Hydrogen bonds shown by dotted lines correspond to distances between 2.5 and 2.9 Å. Interactions with Wat-35 are subunit dependent; distances to Glu-45 OE2 are 2.8, 3.0, and 3.0 Å and are 2.9, 3.5, and 3.7 Å to pyruvate O3 in subunits A, B, and C, respectively. Distance between pyruvate C3 and the center of the ring of Phe-135 is 3.7 Å.
The only residue capable of acting as a general base and that is proximal to the carbinolamine complex is Glu-45, suggesting a multifunctional role by this residue in the KDPG aldolase reaction mechanism. The Glu-45 carboxylate stabilizes a mutual hydrogen bonding arrangement with Wat35 and O3 in the T+ complex, shown in Fig. 4. This arrangement would permit Glu-45 to facilitate proton exchange with O3 either directly and/or indirectly and thereby catalyze dehydration of the T0 complex to the cationic Schiff base. Microscopic reversibility would dictate a similar role for Glu-45 in Schiff base hydrolysis.
As a general base and because of its proximity to Lys-133, Glu-45 could also catalyze proton abstraction of a pyruvate methyl proton during enamine formation. However, to do so, prior rotation must occur to orient the methyl group in the Schiff base complex proximal to Glu-45. Cationic Schiff base formation imposes a planar geometry about the pyruvate C2 carbon that would result in an unfavorable eclipsed configuration by the methyl group with respect to the pyruvate carboxylate orientation observed in the T+ complex if the methyl group were oriented in the direction of Phe-135. Relief of the intramolecular steric strain would provide the driving force for methyl group reorientation. Substrate rotation differs from a previously proposed model to explain KDPG aldolase catalysis where single base-mediated proton activation involved side chain rotation of an active site glutamate residue (34). Enzyme inactivation, a consequence of 3-bromopyruvate esterifying Glu-45 carboxylate (26), is also consistent with substrate rotation in the active site placing the 3-bromomethyl in spatial proximity of Glu-45. Wat35 within hydrogen bonding distance of Glu-45 in the T+ complex suggests a potential binding site for the O4 hydroxyl in KDPG/KHG substrates that would allow proton abstraction of the O4 hydroxyl for C3—C4 bond cleavage. A multifunctional role for a carboxylate ion acting as a general base is supported by model studies on Schiff base formation and hydrolysis (32). Protonation of Glu-45 at acidic pH would inhibit its function, as a general base, in catalyzing T0 carbinolamine dehydration consistent with trapping of the T+ carbinolamine intermediate.
Dibasic sulfate ion binding in each subunit active site of native KDPG aldolase at acidic pH is consistent with both Arg-49 and Lys-133 being positively charged. Structural evidence for Lys-133 being protonated is supported by contacts made with sulfate oxyanions O1 and O2 and water molecules Wat13 and Wat93 that are all within hydrogen bonding distance, as seen in Fig. 2. The two sulfate oxyanions are hydrogen bond acceptors whereas Wat13, by hydrogen bonding to both oxyanions, must accept a hydrogen bond from Lys-133, indicating a fully protonated lysine residue. These arguments are consistent with Lys-133 existing in a predominantly protonated state in the native structure.
Formation of the T+ carbinolamine complex in KDPG aldolase structure requires that Lys-133 be uncharged to be competent as a nucleophile. Enzymes have evolved strategies to modify the reactivity of a lysine residue in the active site microenvironment that includes presence of an adjacent positive charged residue (35), a highly hydrophobic environment about the lysine residue (36), and activation through proton transfer to water or a base such as a glutamate residue (37). Alternatively, the neutral form of Lys-133 may be present as a minor species, yet in sufficient concentrations at acidic pH to catalyze observed carbinolamine formation. Of these strategies, the first two are unlikely. Although Arg-49 does contribute a positive charge in the active site, it is distant from Lys-133 and is neutralized through interaction with pyruvate. Interaction of water and sulfate molecules with the active site suggests that the active site microenvironment is solvent accessible rather than hydrophobic. Activation by proton transfer via Glu-45 is feasible because Lys-133 is proximal to Glu-45 and slower turnover at acidic pH because of decrease in Lys-133 amine species would not be inconsistent with the observed pH activity profile (10). Lysine reactivity would be pKa dependent in both instances, with concentrations of Glu-45 as conjugate base and Lys-133 as neutral species determining the relative importance of these reaction mechanisms in carbinolamine formation.
At pH 4.6, concentration of the neutral form of the conserved Lys-133 would be reduced 1000-fold from that at pH 7.5. Assuming carbinolamine formation on Lys-133 to be rate limiting at pH 4.6, reduction in kcat, 8820 min−1 at pH 7.5 (10), would correspond to a turnover of 9 min−1, which is sufficiently fast to account for the observed carbinolamine complex to have been formed during 15 min soaking of native crystals with pyruvate. The pH activity profile of E. coli KDPG aldolase, however, shows a single transition corresponding to less than a 2.5-fold reduction in activity between pH 4.5 and 7.5 and having an apparent macroscopic ionization constant of ≈5 (10). Based on both native and complex structures, the identity of the residue would most likely be conserved Glu-45 because there are no other carboxylate residues in the active site. The comparatively slight activity reduction at acidic pH and the identification of Glu-45 as the putative ionized residue would suggest that the protonation state of Lys-133 has little effect on enzymatic activity; hence, at pH 4.6, Lys-133, to act as a nucleophile, would require either proton transfer from Lys-133 to a water molecule, unlikely at acidic pH, or to the conjugate base of Glu-45, whose relative concentration would be sufficient under experimental conditions. Proton transfer to Glu-45 from Lys-133 is sterically feasible and would require only side chain rearrangement by Lys-133, displacing Wat93 hydrogen bonded to both residues. The resultant monoprotonated zwitterionic pair (Lys-133/Glu-45) could also isomerize to the neutral form (free base Lys-133, protonated Glu-45) by proton transfer. Whether activation of Lys-133 for nucleophilic attack occurs by a concerted general base catalysis involving Glu-45 or by preequilibrium proton transfer in the zwitterionic complex depends on the dielectric constant of the enzyme active site. A decrease in the effective dielectric constant of the active site with respect to water could favor the latter possibility.
Nucleophilic activation leading to carbinolamine formation needs further investigation in KDPG aldolases. Nevertheless, it may represent an evolutionary paradigm for Schiff base-mediated catalysis in class I aldolases where, in the crystal structures, a conserved glutamate residue is systematically found within hydrogen bonding distance of the conserved lysine residue implicated in Schiff base formation.
Acknowledgments
The assistance of Dr. Tae-Sung Yoon in data collection and reduction is gratefully acknowledged. We thank Dr. D. Gravel (Université de Montréal) for critical review of the manuscript. Assistance by X8-C beam line personnel, Dr. L. Flaks, was appreciated. This research was supported by funding from the Medical Research Council (Canada) MT-14768. Work was carried out in part at the National Synchrotron Light Source, Brookhaven National Laboratory, which is supported by the U.S. Department of Energy, Division of Materials Sciences and Division of Chemical Sciences under contract number DE-AC02-98CH10886.
Abbreviations
- KDPG
2-keto-3-deoxy-6-phosphogluconate
- KHG
2-keto-4-hydroxyglutarate
- SeMet
seleno-l-methionine
- NCS
noncrystallographic symmetry
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: The atomic coordinates for both native and covalent intermediate E. coli KDPG aldolases have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID codes 1EUN and 1EUA, respectively).
References
- 1.Conway T. FEMS Microbiol Rev. 1992;9:1–27. doi: 10.1111/j.1574-6968.1992.tb05822.x. [DOI] [PubMed] [Google Scholar]
- 2.Egan S E, Fliege R, Tong S, Shibata A, Wolf R E, Conway T. J Bacteriol. 1992;174:4638–4646. doi: 10.1128/jb.174.14.4638-4646.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang J K, Dekker E E, Lewinski N D, Winter H C. J Biol Chem. 1981;256:1793–1800. [PubMed] [Google Scholar]
- 4.Gupta S C, Dekker E E. J Biol Chem. 1984;259:10012–10019. [PubMed] [Google Scholar]
- 5.Maitra U, Dekker E E. J Biol Chem. 1963;238:3660–3669. [PubMed] [Google Scholar]
- 6.Nishihara H, Dekker E E. J Biol Chem. 1972;247:5079–5087. [PubMed] [Google Scholar]
- 7.Meloche H P. Biochemistry. 1967;6:2273–2280. doi: 10.1021/bi00860a002. [DOI] [PubMed] [Google Scholar]
- 8.Jencks, W. P. Catalysis in Chemistry and Enzymology (1969) (McGraw–Hill, New York), pp. 490–496.
- 9.Meloche H P, Monti C T, Dekker E E. Biochem Biophys Commun. 1975;65:1033–1039. doi: 10.1016/s0006-291x(75)80489-x. [DOI] [PubMed] [Google Scholar]
- 10.Shelton M C, Cotterill I C, Novak S T A, Poonawala R M, Sudarshan S, Toone E C. J Am Chem Soc. 1996;118:2117–2125. [Google Scholar]
- 11.Fessner W D, Walter C. Top Curr Chem. 1998;184:98–194. [Google Scholar]
- 12.Mavridis I M, Hatada M H, Tulinsky A, Lebodia L. J Mol Biol. 1982;162:419–444. doi: 10.1016/0022-2836(82)90536-8. [DOI] [PubMed] [Google Scholar]
- 13.Kuo D J, Rose I A. Biochemistry. 1985;24:3947–3952. doi: 10.1021/bi00336a022. [DOI] [PubMed] [Google Scholar]
- 14.Rose I A, Warms J V. Biochemistry. 1985;24:3952–3957. doi: 10.1021/bi00336a023. [DOI] [PubMed] [Google Scholar]
- 15.Kayser R H, Pollack R M. J Am Chem Soc. 1977;99:3379–3386. [Google Scholar]
- 16.Egan S E, Fliege R, Tong S, Shibata A, Wolf R E, Conway T. J Bacteriol. 1992;174:4638–4646. doi: 10.1128/jb.174.14.4638-4646.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brosius J, Holy A. Proc Natl Acad Sci USA. 1984;81:6929–6933. doi: 10.1073/pnas.81.22.6929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Studier F W, Moffatt B A. J Mol Biol. 1986;189:113–130. doi: 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]
- 19.Doublié S. Methods Enzymol. 1997;276:523–532. [PubMed] [Google Scholar]
- 20.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–319. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 21.Terwilliger T C, Berendzen J. Acta Crystallogr D Biol Crystallogr. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cowtan K, Main P. Acta Crystallogr D Biol Crystallogr. 1998;54:487–493. doi: 10.1107/s0907444997011980. [DOI] [PubMed] [Google Scholar]
- 23.Jones T A, Zou J Y, Cowan S W, Kjeldgaard T. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 24.Brunger A T, Adams P D, Clore G M, DeLano W L, Gros P, Grosse-Kunstleve R W, Jiang J S, Kuszewski J, Nilges M, Pannu N S, et al. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 25.Laskowski R A, MacArthur M W, Moss D S, Thornton J M. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 26.Vlahos C J, Dekker E E. J Biol Chem. 1990;265:20384–20389. [PubMed] [Google Scholar]
- 27.Vlahos C J, Ghalambor M A, Dekker E E. J Biol Chem. 1985;260:5480–5485. [PubMed] [Google Scholar]
- 28.Vhalos C J, Dekker E E. J Biol Chem. 1986;261:11049–11055. [PubMed] [Google Scholar]
- 29.Vhalos C J, Dekker E E. J Biol Chem. 1988;263:11683–11691. [PubMed] [Google Scholar]
- 30.Sober H A, editor. Handbook of Biochemistry: Selected Data for Molecular Biology. Cleveland, OH: Chemical Rubber Co.; 1968. pp. J–119. [Google Scholar]
- 31.Jencks W P. Catalysis in Chemistry and Enzymology. New York: McGraw–Hill; 1969. p. 463. [Google Scholar]
- 32.Pollack R M, Kayser R H, Damewood J R. J Am Chem Soc. 1977;99:8232–8237. [Google Scholar]
- 33.Sayer J M, Pinsky B, Schonbrunn A, Washtien W. J Am Chem Soc. 1974;96:7998–8009. [Google Scholar]
- 34.Meloche H P, Glusker J P. Science. 1973;181:350–352. doi: 10.1126/science.181.4097.350. [DOI] [PubMed] [Google Scholar]
- 35.Highbarger L A, Gerlt J A, Kenyon G L. Biochemistry. 1996;35:41–46. doi: 10.1021/bi9518306. [DOI] [PubMed] [Google Scholar]
- 36.Barbas C F, 3rd, Heine A, Zhong G, Hoffmann T, Gramatikova S, Bjornestedt R, List B, Anderson J, Stura E A, Wilson I, et al. Science. 1997;278:2085–2092. doi: 10.1126/science.278.5346.2085. [DOI] [PubMed] [Google Scholar]
- 37.Dickopf S, Mielke T, Heyn M P. Biochemistry. 1998;37:16888–16897. doi: 10.1021/bi981879m. [DOI] [PubMed] [Google Scholar]
- 38.Esnouf R M. J Mol Graph. 1997;15:133–138. doi: 10.1016/S1093-3263(97)00021-1. [DOI] [PubMed] [Google Scholar]
- 39.Merritt E A, Murphy M E P. Acta Crystallogr D. 1994;50:869–873. doi: 10.1107/S0907444994006396. [DOI] [PubMed] [Google Scholar]