

Abstract
An artificial neural network (ANN) solution is described for the recognition of domains in protein sequences. A query sequence is first compared to a reference database of domain sequences by use of BLAST and the output data, encoded in the form of six parameters, are forwarded to feed-forward artificial neural networks with six input and six hidden units with sigmoidal transfer function. The recognition is based on the distribution of BLAST scores precomputed for the known domain groups in a database versus database comparison. Applications to the prediction of function are discussed.
Sequence similarity searching is a crucial step in analyzing newly determined protein sequences. Whereas similarity searching by programs such as BLAST (Pearson and Lipman 1988; Altschul et al. 1990, 1997) or FASTA (Wilbur and Lipman 1983; Lipman and Pearson 1985) allows the inference of homology and/or function in many cases, identification of multidomain proteins is often problematic because their similarities point to various unrelated protein families. The current best solution to this problem is the use of pattern databases that store the common sequence patterns of domain groups in the form of consensus representations (for review, see Attwood 2000). Various pattern representation methods are in use, including regular expressions (Bairoch et al. 1996), position-dependent frequency matrices (Gribskov et al. 1987), and Hidden Markov Models (HMMs) (Sonnhammer et al. 1998). All of these representations are based on multiple sequence alignments. Even though these consensus pattern representations — such as used in PROSITE (Hofmann et al. 1999), PRINTS (Attwood et al. 2000), PFAM (Bateman et al. 2000), PRODOM (Corpet et al. 2000), BLOCKS (Henikoff et al. 2000), PROTFAM (Mewes et al. 2000), INTERPRO (Apweiler et al. 2000), and others (for review, see Attwood 2000) — can be optimized so as to reach a very high prediction performance. It is well known that construction of multiple alignments as well as updating them with the stream of new domain sequences requires a substantial human overhead, which is partly due to the high computational complexity of the problem. BLAST or FASTA searches on domain sequence databases (Corpet et al. 2000; Murvai et al. 2000a) offer a good alternative, however, the evaluation of the output requires human judgement and/or iterative search strategies, such as those used by PSI-BLAST (Altschul et al. 1997). One of the underlying problems is that the known structural and functional domain groups are quite variable in terms of size, sequence-length, as well as similarity between the members, and especially, short and variable domain sequences are sometimes quite hard to detect (Atwood 2000).
Artificial neural networks (ANNs) have been used very successfully in biological sequence analysis for purposes as diverse as protein secondary structure prediction, recognition of signal peptide cleavage sites, gene recognition, etc. (for review, see Baldi and Brunak 1998). Representation of sequence data in a form suitable for nonrecursive ANNs can be quite difficult because of the varying length of the sequences. A common solution to this problem is to use a window sliding over the protein sequence (Jagla and Schuchhardt 2000). On the other hand, a sequence window encompassing, for example, 19 amino acids can be mapped to a 19 × 20 = 380 dimensional vector. Training an ANN recognizer for so many input parameters would require an enormous data set for training (Jagla and Schuchhardt 2000). Adaptive encoding techniques can be used to find a smaller number of relevant parameters in the course of the training process. However, recognition of a short pattern, such as a signal peptide cleavage site, still required 79 input parameters (Jagla and Schuchhardt 2000). Recognition of substantially longer protein domains may thus require a prohibitively large number of parameters. In addition, individual training strategies may be necessary for the >2000 known domain types, which may lead to formidable updating problems.
In a search for an automated and computationaly feasible ANN solution, we found that sequences can be encoded with as few as six parameters, provided that suitably encoded BLAST search outputs, rather than domain sequences, are used to train the ANNs. The underlying idea is as follows: Assume we want to decide whether a domain belongs to a given domain group. A query sequence is then first compared with a reference database of protein domain sequences by use of the BLAST program, and then two domain similarity parameters are calculated between the query and each of the domain/types found in the output. Another four values are then calculated that characterize the probability of the given domain belonging to the domain group, given the knowledge of the above two parameters and their distribution within the given domain group. These six parameters are then used to train a feed-forward ANN, both with positive and negative instances of the given domain group. We found that training is very fast and the resulting predictive performance is very good in comparison with other methods, which are typically also more time consuming.
RESULTS
The six parameters used for representing the similarity of a sequence to a domain group are schematically shown inFigure 1. NSD is the number of significant BLAST similarities (P <0.8) between the sequence and members of a given domain group, whereas AVS is the average BLAST score of these similarities. In addition, the empirical distributions of both the NSD and the AVS values were computed both for the group members and for the significantly similar (P <0.8) non-member sequences resulting in the functions P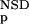 (·), P
(·), P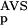 (·), P
(·), P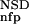 (·), and P
(·), and P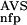 (·), respectively (Fig. 1). The latter four functions had been determined from a database versus database comparison carried out with the SBASE-A 7.0 domain sequence collection. Each domain sequence was then characterized with the six parameters, NSD, AVS, P
(·), respectively (Fig. 1). The latter four functions had been determined from a database versus database comparison carried out with the SBASE-A 7.0 domain sequence collection. Each domain sequence was then characterized with the six parameters, NSD, AVS, P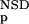 (NSD), P
(NSD), P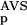 (AVS), P
(AVS), P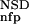 (NSD), and P
(NSD), and P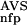 (AVS).
(AVS).
Figure 1.

Variables used to characterize domain similarities. Average BLAST similarity score (AVS) versus number of significant BLAST similarities (NSD) plot for Trypsin domains (red circles) from SBASE 7.0 (Murvai et al. 2000a). Each red circle represents a domain sequence. The AVS and NSD values are computed from the BLAST similarities between the sequence and other members of the group. Blue triangles represent domain sequences that are not Trypsin domains but still produce a significant BLAST similarity score with some of the Trypsin domains. The database versus database comparison was carried out with BLAST (Altschul et al. 1990), by use of a significance threshold of 0.8 and a minimum score of 32. (Inset) Schematic representation of the empirical distributions (see text for details). Values of P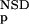 (NSD), and P
(NSD), and P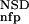 (NSD), are simply read out from the precomputed empirical distributions. A similar procedure is followed for the AVS scores. The resulting six values are the input parameters of the ANNs.
(NSD), are simply read out from the precomputed empirical distributions. A similar procedure is followed for the AVS scores. The resulting six values are the input parameters of the ANNs.
For the training of ANNs, we selected a few known domain types that represent various critical properties of the domain groups (Fig. 2). The Kringle domains form a tight group (Fig. 2A), that is, most members are significantly similar to all other members of the group, and this domain type can be identified more or less precisely with simple regular expression-type patterns such as the PROSITE signatures. The ANK domain-type (Fig. 2B) is more problematic. Its members are far less similar to each other than those of the Kringle group and cannot be described with a simple regular expression. The EGF-like domain-type is a large group (Fig. 2C) that does not separate well from the non-member neighbors, and this makes the recognition of the corresponding proteins harder. Finally, FN3, the fibronectin type III repeat is an especially variable group (Fig. 2D), with only three fully conserved amino acids residues within a segment of ∼100 amino acids, therefore, the traditional domain-recognition methods perform quite poorly on it.
Figure 2.

NSD versus AVS plots in the following domain groups (PROSITE or PFAM reference given in parenthesis): EFG-like [PROSITE:PDOC00021]; ABC Transporter [PROSITE:PS00211]; Kringle [PROSITE:PDOC00020]; Sushi (SCR) repeat [PFAM:PF00084]; Fibronectin III domain [PFAM:PF00041]; ANK repeat [PFAM:PF00023].
To evaluate the importance of each parameter, we carried out a set of training experiments using the EGF-like domain group as an example (Table 1). If only the NSD and AVS values were used, the performance was poor, as shown by a relatively low Matthews coefficient. Increasing the number of input parameters to four by including either the values of P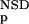 (NSD) and P
(NSD) and P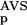 (AVS), or the values P
(AVS), or the values P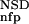 (NSD), and P
(NSD), and P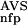 (AVS) substantially increased the prediction accuracy. The probability estimates P
(AVS) substantially increased the prediction accuracy. The probability estimates P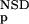 (NSD), P
(NSD), P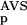 (AVS) were somewhat more efficient in this respect than the P
(AVS) were somewhat more efficient in this respect than the P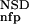 (NSD), and P
(NSD), and P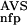 (AVS) parameters (Table 1, rows 3,4).
(AVS) parameters (Table 1, rows 3,4).
Table 1.
Optimization of Neural Network Architecture (Input Parameters vs. Performance on the EGF-Like Domain Type1)
| Training set | Test set | Total | ||||||||||||||||||||||||||||||||
| Parameter | No | tp | fp | fn | tn | C | tp | fp | fn | tn | C | tp | fp | fn | tn | C | ||||||||||||||||||
| 1 | NSD, AVS | 2 | 242 | 23 | 49 | 311 | 0.77 | 125 | 3 | 20 | 137 | 0.85 | 360 | 14 | 76 | 460 | 0.81 | |||||||||||||||||
| 2 | NSD, AVS, P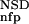 (NSD), P (NSD), P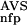 (AVS) (AVS) |
4 | 284 | 4 | 7 | 330 | 0.97 | 142 | 3 | 3 | 137 | 0.96 | 426 | 8 | 10 | 466 | 0.96 | |||||||||||||||||
| 3 | NSD, AVS, P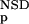 (NSD),P (NSD),P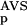 (AVS) (AVS) |
2 | 291 | 2 | 0 | 332 | 0.99 | 145 | 3 | 0 | 137 | 0.98 | 434 | 4 | 2 | 470 | 0.99 | |||||||||||||||||
| 4 | NSD, AVS, P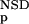 (NSD), P (NSD), P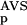 (AVS), P (AVS), P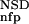 (NSD), P (NSD), P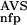 (AVS) (AVS) |
4 | 284 | 4 | 7 | 330 | 0.97 | 142 | 3 | 3 | 137 | 0.96 | 426 | 8 | 10 | 466 | 0.96 | |||||||||||||||||
tp, True positives; fp, false positives; tn, true negatives; fn, false negatives.
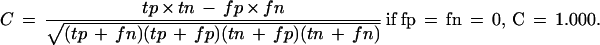 |
The sequence queries were presented in two alternative forms as follows: (1) by use of an entire protein sequence as the query; and (2) by use of only a selected potential similarity region of the query sequence. In the second case, the query was first compared with the domain database, and the potential similarity regions were identified by use of a cumulative similarity score versus sequence plot as described (Murvai et al. 1999). Contiguous regions longer than 11 residues were excised. On the average, a protein sequence contained 15–20 such similarity regions (even though very large proteins can have up to 100 such regions). These regions were then individually compared with the database. Table 2 compares the two strategies for various domain types; region selection performed better in most cases.
Table 2.
Comparison of Sequence Preprocessing Methods in Various Domain Groups
| Training set | Test set | Total | ||||||||||||||||||||||||
| Group | tp | fp | fn | tn | tp | fp | fn | tn | tp | fp | fn | tn | ||||||||||||||
| EGF-like domain | S | 281 | 13 | 10 | 34988 | 134 | 13 | 11 | 17488 | 415 | 22 | 21 | 52480 | |||||||||||||
| R | 291 | 5 | 0 | 329 | 145 | 4 | 0 | 136 | 436 | 7 | 0 | 467 | ||||||||||||||
| Fibronectin III Domain | S | 214 | 1 | 23 | 35054 | 100 | 12 | 13 | 17521 | 325 | 11 | 25 | 52577 | |||||||||||||
| R | 237 | 1 | 0 | 507 | 113 | 0 | 0 | 228 | 350 | 2 | 0 | 734 | ||||||||||||||
| Sushi domain (SCR repeat) | S | 105 | 0 | 1 | 35186 | 59 | 5 | 0 | 17582 | 165 | 0 | 0 | 52938 | |||||||||||||
| R | 106 | 0 | 0 | 77 | 59 | 0 | 0 | 32 | 165 | 0 | 0 | 109 | ||||||||||||||
| ANK repeat | S | 98 | 0 | 4 | 35190 | 55 | 52 | 2 | 17539 | 151 | 3 | 6 | 52778 | |||||||||||||
| R | 99 | 1 | 3 | 56 | 54 | 0 | 1 | 22 | 155 | 2 | 2 | 77 | ||||||||||||||
| ABC transporters | S | 530 | 0 | 0 | 34762 | 239 | 0 | 0 | 17407 | 769 | 0 | 0 | 52169 | |||||||||||||
| R | 530 | 3 | 0 | 90 | 239 | 2 | 0 | 43 | 769 | 1 | 0 | 137 | ||||||||||||||
| WD repeat | S | 184 | 3 | 6 | 35099 | 77 | 40 | 0 | 17529 | 261 | 3 | 6 | 52668 | |||||||||||||
| R | 186 | 2 | 4 | 168 | 76 | 0 | 1 | 92 | 262 | 2 | 5 | 260 | ||||||||||||||
S = whole sequence vs. R = regions.
On the basis of these preliminary experiments, we selected an architecture with six input parameters and one hidden layer of six elements (Fig. 3) and used pre-selected similarity regions (strategy b) for the training process. The performance of 40 such networks is shown in Table 3A. The number of false positives and false negatives is extremely low in most cases. In fact, these numbers compare quite favorably with those obtained with other systems in almost all cases. A representative comparison is shown in Table 4.
Figure 3.

The backpropagation neural network architecture used for domain recognition.
Table 3.
Performance of Neural Network Recognizers for Various Domain Groups
| A. Protein Domain Groups | Training set | Test set | Total | |||||||||||||||||||||
| tp | fp | fn | tn | tp | fp | fn | tn | tp | fp | fn | tn | |||||||||||||
| Immunoglobulin domain | 1721 | 24 | 15 | 2428 | 829 | 18 | 19 | 1155 | 2552 | 71 | 32 | 3554 | ||||||||||||
| Zinc finger, C2H2 type | 330 | 9 | 9 | 540 | 147 | 3 | 6 | 245 | 477 | 12 | 15 | 785 | ||||||||||||
| Protein kinase domain | 1120 | 0 | 0 | 264 | 522 | 0 | 0 | 119 | 1642 | 0 | 0 | 383 | ||||||||||||
| EF hand | 409 | 14 | 1 | 116 | 186 | 3 | 1 | 62 | 594 | 17 | 3 | 178 | ||||||||||||
| EGF-like domain | 291 | 2 | 0 | 332 | 145 | 3 | 0 | 137 | 436 | 3 | 0 | 471 | ||||||||||||
| WD repeat | 186 | 2 | 4 | 168 | 76 | 0 | 1 | 92 | 262 | 2 | 5 | 260 | ||||||||||||
| Fibronectin type III domain | 237 | 1 | 0 | 507 | 113 | 0 | 0 | 228 | 350 | 2 | 0 | 734 | ||||||||||||
| ABC transporters | 530 | 3 | 0 | 90 | 239 | 2 | 0 | 43 | 769 | 1 | 0 | 137 | ||||||||||||
| Globin | 595 | 2 | 2 | 200 | 269 | 1 | 0 | 92 | 866 | 3 | 0 | 292 | ||||||||||||
| Homeobox domain | 584 | 0 | 1 | 58 | 260 | 0 | 0 | 31 | 845 | 0 | 0 | 89 | ||||||||||||
| ANK repeat | 99 | 1 | 3 | 56 | 54 | 0 | 1 | 22 | 155 | 2 | 2 | 77 | ||||||||||||
| Sushi domain (SCR repeat) | 106 | 0 | 0 | 77 | 59 | 0 | 0 | 32 | 165 | 0 | 0 | 109 | ||||||||||||
| RNA recognition motif (=RRM, RBD, or RNP domain) | 215 | 1 | 2 | 50 | 99 | 1 | 1 | 26 | 315 | 2 | 2 | 76 | ||||||||||||
| Tetratricopeptide repeat | 98 | 0 | 0 | 152 | 45 | 0 | 0 | 74 | 143 | 0 | 0 | 226 | ||||||||||||
| Short chain dehydrogenase | 316 | 0 | 0 | 55 | 142 | 0 | 0 | 29 | 458 | 0 | 0 | 84 | ||||||||||||
| Trypsin | 296 | 0 | 1 | 215 | 146 | 0 | 0 | 97 | 442 | 0 | 1 | 312 | ||||||||||||
| Dead/H box helicase domain | 277 | 1 | 4 | 499 | 117 | 0 | 2 | 238 | 397 | 2 | 3 | 736 | ||||||||||||
| Kazal-type serine protease inhibitor domain | 211 | 0 | 0 | 84 | 99 | 0 | 0 | 38 | 310 | 0 | 0 | 122 | ||||||||||||
| Response regulator receiver domain | 242 | 0 | 0 | 20 | 112 | 0 | 0 | 11 | 354 | 0 | 0 | 31 | ||||||||||||
| Spectrin repeat | 21 | 0 | 1 | 293 | 21 | 0 | 1 | 124 | 43 | 0 | 1 | 417 | ||||||||||||
| 4FE-4S ferredoxins and rel. iron-sulf. clust.bind.domains | 183 | 0 | 0 | 51 | 86 | 0 | 0 | 24 | 269 | 0 | 0 | 75 | ||||||||||||
| RAS family | 203 | 0 | 0 | 32 | 96 | 0 | 0 | 14 | 299 | 0 | 0 | 46 | ||||||||||||
| SH3 domain | 185 | 0 | 0 | 27 | 94 | 0 | 0 | 6 | 279 | 0 | 0 | 33 | ||||||||||||
| APOA/APOE-repeat | 33 | 0 | 0 | 498 | 16 | 5 | 2 | 229 | 51 | 12 | 0 | 720 | ||||||||||||
| Zinc-binding dehydrogenases | 152 | 0 | 0 | 74 | 80 | 0 | 1 | 26 | 233 | 0 | 0 | 100 | ||||||||||||
| Cytochrome C | 142 | 1 | 6 | 73 | 71 | 0 | 1 | 33 | 213 | 1 | 7 | 106 | ||||||||||||
| ATPases associated with various cell. activites (AAA) | 122 | 0 | 0 | 102 | 69 | 0 | 0 | 38 | 191 | 0 | 0 | 140 | ||||||||||||
| Helix-loop-helix DNA-binding domain | 153 | 0 | 1 | 21 | 74 | 0 | 0 | 13 | 227 | 0 | 1 | 34 | ||||||||||||
| Alpha amylase | 135 | 0 | 0 | 118 | 69 | 0 | 0 | 51 | 204 | 0 | 0 | 169 | ||||||||||||
| HSP70 protein | 130 | 1 | 0 | 247 | 63 | 0 | 0 | 117 | 193 | 0 | 0 | 365 | ||||||||||||
| Ligand-binding domain of nuclear hormone receptors | 176 | 1 | 0 | 13 | 81 | 0 | 0 | 10 | 257 | 0 | 0 | 14 | ||||||||||||
| Protein-tyrosine-phosphatase domain | 110 | 0 | 0 | 42 | 56 | 0 | 0 | 20 | 166 | 0 | 0 | 62 | ||||||||||||
| Lectin in C-type domain | 129 | 0 | 0 | 23 | 59 | 0 | 0 | 15 | 188 | 0 | 0 | 38 | ||||||||||||
| Intermediate filament proteins | 127 | 2 | 0 | 192 | 61 | 2 | 0 | 86 | 188 | 2 | 0 | 280 | ||||||||||||
| Zinc-binding metalloprotease domain | 148 | 1 | 0 | 4 | 75 | 0 | 0 | 1 | 223 | 1 | 0 | 5 | ||||||||||||
| ACYL carrier protein domain | 102 | 0 | 0 | 8 | 54 | 0 | 0 | 2 | 156 | 0 | 0 | 10 | ||||||||||||
| PAS domain | 77 | 0 | 0 | 52 | 46 | 0 | 0 | 15 | 123 | 0 | 0 | 67 | ||||||||||||
| FOS/JUN DNA-binding domain | 108 | 0 | 0 | 69 | 64 | 0 | 0 | 23 | 172 | 0 | 0 | 92 | ||||||||||||
| Snake toxin | 102 | 0 | 1 | 31 | 51 | 0 | 0 | 12 | 153 | 0 | 1 | 43 | ||||||||||||
| B. Functional Groups | Training set | Test set | Total | |||||||||||||||||||||
| tp | fp | fn | tn | tp | fp | fn | tn | tp | fp | fn | tn | |||||||||||||
| Permeases | 289 | 22 | 12 | 593 | 139 | 7 | 4 | 270 | 428 | 29 | 16 | 863 | ||||||||||||
| Sensory transduction histidine kinases | 177 | 2 | 3 | 167 | 87 | 2 | 1 | 75 | 264 | 2 | 4 | 244 | ||||||||||||
| Glycosyltransferases I | 204 | 1 | 1 | 103 | 95 | 0 | 0 | 46 | 300 | 2 | 0 | 148 | ||||||||||||
| Thiol-disulfide isomerase and thioredoxins | 139 | 0 | 4 | 65 | 77 | 1 | 4 | 17 | 218 | 1 | 6 | 82 | ||||||||||||
| Serine/threonine protein kinases | 1107 | 1 | 10 | 264 | 515 | 1 | 2 | 121 | 1622 | 1 | 12 | 386 | ||||||||||||
Table 4.
Comparison of Various Prediction Methods on Selected Domain Groups
| Domain type (number in SWISS-PROT 38) | Number of errors2 | |||
|---|---|---|---|---|
| Ann (present method) | PROSITE | PRINTS | PFAM | |
| EGF (340) | 3 | 12 | 108 | 17 |
| Fibronectin type 3 (241) | 2 | n.a. | 27 | 7 |
| Trypsin (273) | 0 | 26 | 0 | 0 |
| ANK-repeat (119) | 0 | n.a.3 | n.a. | 9 |
| WD-repeat (247) | 7 | 25 | 15 | 8 |
| Cytochrome C (78) | 8 | 4 | 5 | 3 |
The total number of sequences that contain the given domain type in Swiss-Prot 38. The release numbers of PROSITE, PRINTS, and PFAM are those used by Swiss-Prot 38.
Errors for PROSITE, PRINTS and PFAM were determined from the Swiss-Prot annotation (e.g., a sequence annotated as having an EGF repeat but having no cross-reference to a given pattern database was considered not detected by the corresponding method). Thus, this number contains only false negatives. On the other hand, the errors of ANN contain both false positives and false negatives.
n.a. = Not available (i.e., the domain type is not included in the corresponding method).
The general applicability of the system was tested on an experimental data set of proteins annotated according to their biological functions. This data set was created by merging the COG database of orthologous protein groups and a functionally well-annotated subset of SWISS-PROT and PIR databases into a nonredundant set of 81 079 sequences (Methods). This was necessary as a preliminary analysis (not shown) revealed that many of the COG groups are too small for training ANNs, and in addition, BLAST found too few significantly similar neighbors for many of the larger groups. From the resulting data set we chose a number of groups, listed in Table 3B, and identified the members on the basis of their annotations. Then, the P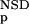 (·), P
(·), P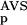 (·), P
(·), P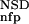 (·), and P
(·), and P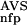 (·) functions were determined from a database versus database comparison, and the neural networks trained for the selected groups as described in Methods. The predictive performance of the ANNs trained for these functional groups is only slightly inferior to those obtained with the domain groups (Table 3B). In some cases, there is a conspicuously high number of false predictions, for example, of 428 permease sequences, 29 false positives (6.8%) and 16 false negatives (3.7%) were found. We mention, however, that much of the false predictions seem to be due to erroneous functional annotations in the databases. Of the 35 false positives and 38 false negatives listed in Table 3B, 21 and 13 of them, respectively, coincide with conflicting annotations between various sequence databases such as SWISS-PROT, PIR, and COG. However, it has to be pointed out that this comparison was carried out on an experimental data set, therefore, the results cannot be used to draw conclusions on the quality of the underlying databases. We also note that the selected sequence groups represent especially difficult cases (i.e., the separation of group members from non-group members is rather poor as compared with other sequence groups). It appears that for the present method, the case of functional clusters are not more difficult than that of domain clusters; on the other hand, the accuracy of functional annotations may become a limiting factor.
(·) functions were determined from a database versus database comparison, and the neural networks trained for the selected groups as described in Methods. The predictive performance of the ANNs trained for these functional groups is only slightly inferior to those obtained with the domain groups (Table 3B). In some cases, there is a conspicuously high number of false predictions, for example, of 428 permease sequences, 29 false positives (6.8%) and 16 false negatives (3.7%) were found. We mention, however, that much of the false predictions seem to be due to erroneous functional annotations in the databases. Of the 35 false positives and 38 false negatives listed in Table 3B, 21 and 13 of them, respectively, coincide with conflicting annotations between various sequence databases such as SWISS-PROT, PIR, and COG. However, it has to be pointed out that this comparison was carried out on an experimental data set, therefore, the results cannot be used to draw conclusions on the quality of the underlying databases. We also note that the selected sequence groups represent especially difficult cases (i.e., the separation of group members from non-group members is rather poor as compared with other sequence groups). It appears that for the present method, the case of functional clusters are not more difficult than that of domain clusters; on the other hand, the accuracy of functional annotations may become a limiting factor.
The training ANN recognizers for all 116 difficult domain groups took less than 3 h of CPU time on a single processor SUN Ultra-Enterprise 450 server (300 MHz), excluding the time of database versus database comparison with BLAST. The time required to analyze an average protein sequence (300 amino acids) with all of the ANN recognizers is typically below 1 min per sequence, and at least 95% of this time is spent with the BLAST search. (The estimates refer to a nonoptimized program code).
DISCUSSION
Even though ANNs have a relatively long history in biological sequence analysis, recognition of domain types is a new application area that at first may appear problematic because of diversity of data. Simple prediction methods can be used for many of the domain types, whereas others, especially the large and heterogeneous groups, are usually problematic. From the 1515 domain groups in the SBASE-A domain collection, 116 groups are difficult in the sense that categorization could not be achieved by simple statistical methods (Murvai et al. 2000b). Our primary goal was to design ANN recognizers for these difficult cases.
The ANN architecture used here works best on larger groups, the false-positive and false-negative rates were lower than 2.8% (EF-hand domain) and 3.2% (Cytochromse C), respectively, and in many cases there were no false positives and false negatives. The recognition performance deteriorates for groups with <50 members (the APOA/APOE is a typical example). The recognition accuracy of the present ANN method compares quite favorably with other methods (Table 4). It has to be noted that ANNs presented here can be trained in a quasi-automated manner, whereas the conventional methods of domain recognition require multiple alignment as well as substantial human intervention. The human intervention necessary during ANN training was needed to check the input sequence data. With properly selected positive and negative sets, the training is fully automated.
We found that training speed and prediction accuracy substantially improved by selecting the most similar subset of the negative group for the training. On the other hand, the training process is quite sensitive to errors in the database; even one unidentified positive domain erroneously assigned to the negative group (or vice versa) can lead to a conspicuously low recognition accuracy (i.e., >30% false positives or false negatives). In this respect, ANNs provide a sensitive quality test for the domain groups. Of the 116 difficult domain groups of SBASE-A 7.0 domain collection, ANN training revealed a total of 129 potentially mistaken annotations in 67 groups (not shown).
We surmise that the apparent success of this approach can be attributed primarily to the efficiency of the data-encoding process. First of all, not the sequences themselves, but only BLAST outputs are compared. This process can be best pictured as evaluating the query in a similarity space rather than within the sequence space. Second, a protein domain sequence database is used for comparison so that the search outputs can be directly evaluated in terms of domain similarities. In this way, the preprocessed data incorporates a vast amount of biological knowledge.
The idea underlying the entire process is quite simple: The similarity between a query and a domain group depends on two main parameters, the NSD and the AVS (Hegyi and Pongor 1993; Murvai et al. 1999). The higher these values, the higher the similarity. On the other hand, the final similarity will also depend on two intuitive criteria, the tightness of the group (the similarity of the group members to each other) and the separation of the group members from other sequences (how many non-member sequences have significant similarities with the members of the group) (Murvai et al. 2000b). Clearly, the same NSD, AVS value pair may have one meaning when a group is tight and separated from the neighborhood (like the Kringle domains in Fig. 3), and another meaning if the group is scattered and overlapping with the neighborhood (like the EGF-like domains in Fig. 3). The cumulative frequency distributions Pp and Pnfp were designed to capture these properties, and, in fact, including these parameters into the training substantially improves the predictive power. Pp seemed to be more effective in improving predictive power than Pnfp.
The main difficulty for domain sequence recognition is the data explosion that makes sequence pattern databases very difficult to update. Sequence pattern recognizers that are based on multiple sequence alignment can be very efficiently optimized so as to reach very low error rates, but they may need careful reoptimization as novel or atypical domain sequences appear. In contrast, the current artificial neural network procedure can be quite simply updated by adding new sequences to the training set, and, unlike with multiple alignment, little human intervention is necessary. For this reason, we hope that this approach will be useful in different fields of genome sequence analysis.
METHODS
Sequence Data
Sequences of multidomain proteins were taken from SWISS-PROT (Bairoch and Apweiler 2000) and PIR (Barker et al. 2000) databases and were merged into a nonredundant database containing 52 938 sequences. The domain sequences (79 478 individual domains classified in 1515 domain groups) were taken from SBASE-A 7.0 (Murvai et al. 2000a). The protein sequences characterized by function were taken from an experimental data set on the basis of COG (Clusters of Orthologous Groups) database that contains 28 141 sequences from 21 complete genomes (Tatusov et al. 2000). First, a nonredundant data set was created from the COG database and from those sequences of the SWISS-PROT and PIR databases that contained functional annotations in their feature tables. A few sufficiently large (>200 members) groups were chosen at random as examples for ANN analysis (Table 3B) and their members identified on the basis of their annotations as given in the various databases. The sequence annotations were also checked by visual inspection. The rest of the sequence groups were not checked in detail as they served only as negative examples in the ANN training
Data Encoding
The encoding of data is based on a prior analysis of the domain groups on the bais of a database versus database comparison carried out with the BLAST program (Altschul et al. 1997) on the domain sequences of SBASE-A 7.0 by use of ungapped alignments. Similarities with P values below 0.8 were taken into consideration; for brevity, these are termed significant similarities. The value of 0.8 was chosen empirically, although we have found that in the range above 0.7 the results are not sensitive to the actual value of this parameter. Each domain sequence was then characterized with two variables, the number of significant similarities to members of its own group (NSD), and the average BLAST similarity score of the significant similarities computed for the same members (AVS). In addition, the distribution of the values of these two variables were also computed for each domain group (Fig. 1). Namely, the empirical distributions of both the NSD and AVS values were computed both for the group-members (red), and for the significantly similar non-member sequences (blue), resulting in the functions P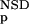 (·), P
(·), P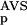 (·), P
(·), P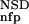 (·), and P
(·), and P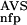 (·), respectively.
(·), respectively.
Encoding of the sequences for the training of and the recognition by an ANN corresponding to a given group was then carried out as follows. A sequence was compared with the reference database (SBASE-A 7.0) by use of BLAST, and the NSD and AVS were computed for the given group from the list of hits with P <0.8 (this is exactly the same procedure that was applied in the preprocessing phase). Then the values of P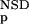 (NSD), P
(NSD), P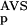 (AVS), P
(AVS), P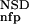 (NSD), and P
(NSD), and P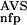 (AVS) were determined by looking up the precomputed values of these functions. The sxi values obtained in this way served then as input parameters, their magnitude was normalized to the interval [−1, +1].
(AVS) were determined by looking up the precomputed values of these functions. The sxi values obtained in this way served then as input parameters, their magnitude was normalized to the interval [−1, +1].
Training and Performance Evaluation
Several network architectures were trained and tested. The architecture was varied both in terms of the number of hidden layers, the number of hidden neurons within the hidden layers, and the number of inputs, that is, many different alternative domain representations were tested. The Matrix-Backpropagation (MBP) package of D. Anguita (1994), which implements a batch-back propagation algorithm with an adaptive learning rate and momentum (Vogl et al. 1993) was used. MBP is an efficient implementation of the back-propagation algorithm that includes a per-epoch adaptive technique for gradient descent (Anguita et al. 1994). It was consistently observed that increasing the number of input vector elements improved the performance when this number was small, but the performance approached saturation as more parameters were included. Using Occam's Razor, we present the results for the smallest nets that achieved the near-best performance. The training and test sets were kept strictly separated and the number of false positives and false negatives, as well as the Matthews coefficient (Matthews 1975) — a version of the Pearson correlation coefficient — were used to quantitatively follow the improvement of the predictive performance.
For a given domain-type, we selected first those protein sequences that contained at least one copy of the domain in question (positive set). The group was subdivided by random sampling into a training set (66.6 %) and a test set (33.3 %), the latter set being withheld from the training. For negative instances, we used a selected subset from the rest of the database; those sequences that showed the highest similarity to the given domain group, in fact, did not contain any copy of the given domain (close neighbors). The sequences that were selected by the actual implementation were those that had either their NSD or their AVS value (or both) higher than 60% of the lowest corresponding values within the given domain group. This filtering yielded negative sets one to four times the size of the positive sets (see Table 2).
A mixture of experts approach was then used to further improve the generalization capability of the networks: For each domain-group, five ANNs were trained, differing from each other in their initial sets of weights. A query sequence was then analyzed with all five neural networks and the final decision was based on a majority vote, that is, a domain type was assigned if it was detected by at least three of the ANNs. The robustness of the method is shown by the fact that 99.7% of the positive and 94.9% of the negative decisions listed in Table 3A were reached by a 5 : 0 vote.
Acknowledgments
This work was supported in part by EMBnet, the European Molecular Biology Network, in the framework of EU grant no. ERBBIO4-CT96–0030. J.M. is the recipient of an International Centre for Genetic Engineering and Biotechnology (ICGEB) fellowship. The SBASE domain sequence library is maintained collaboratively by ICGEB and the Agricultural Biotechnology Center, Gödöllö, Hungary. The advice of Drs. Eugene V. Koonin and David Landsman, National Center for Biotechnology Information, National Institutes of Health, is gratefully acknowledged. The authors thank Masa Cemazar for comments on the manuscript and the members of the Artificial Intelligence Group of Szeged University for useful discussions.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL pongor@icgeb.trieste.it; FAX 39-040-226555.
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.168701.
REFERENCES
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anguita D, Parodi G, Zunino R. An efficient implementation of BP on RISC-based workstations. Neurocomputing. 1994;6:57–65. [Google Scholar]
- Apweiler R, Attwood TK, Bairoch A, Bateman A, Birney E, Bucher P, Codani JJ, Corpet F, Croning MDR, Durbin R, et al. InterPro – An integrated documentation resource for protein families, domains and functional sites. Bioinformatics. 2000;16:1145–1150. doi: 10.1093/bioinformatics/16.12.1145. [DOI] [PubMed] [Google Scholar]
- Attwood TK. The quest to deduce protein function from sequence: The role of pattern databases. Int J Biochem Cell Biol. 2000;32:139–155. doi: 10.1016/s1357-2725(99)00106-5. [DOI] [PubMed] [Google Scholar]
- Attwood TK, Croning MD, Flower DR, Lewis AP, Mabey JE, Scordis P, Selley JN, Wright W. PRINTS-S: The database formerly known as PRINTS. Nucleic Acids Res. 2000;28:225–227. doi: 10.1093/nar/28.1.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwood TK. The role of pattern databases in sequence analysis. Briefings in Bioinformatics. 2000;1:45–59. doi: 10.1093/bib/1.1.45. [DOI] [PubMed] [Google Scholar]
- Bairoch A, Apweiler R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000;28:45–48. doi: 10.1093/nar/28.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch A, Bucher P, Hofmann K. The PROSITE database, its status in 1995. Nucleic Acids Res. 1996;24:189–196. doi: 10.1093/nar/24.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldi P, Brunak S. Bioinformatics: The machine learning approach. Cambridge, MA: MIT Press; 1998. [Google Scholar]
- Barker WC, Garavelli JS, Huang H, McGarvey PB, Orcutt BC, Srinivasarao GY, Xiao C, Yeh LS, Ledley RS, Janda JF, et al. The protein information resource (PIR) Nucleic Acids Res. 2000;28:41–44. doi: 10.1093/nar/28.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A, Birney E, Durbin R, Eddy SR, Howe KL, Sonnhammer EL. The Pfam protein families database. Nucleic Acids Res. 2000;28:263–266. doi: 10.1093/nar/28.1.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corpet F, Servant F, Gouzy J, Kahn D. ProDom and ProDom-CG: Tools for protein domain analysis and whole genome comparisons. Nucleic Acids Res. 2000;28:267–269. doi: 10.1093/nar/28.1.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribskov M, McLachlan AD, Eisenberg D. Profile analysis: Detection of distantly related proteins. Proc Natl Acad Sci. 1987;84:4355–4358. doi: 10.1073/pnas.84.13.4355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegyi H, Pongor S. Predicting potential domain homologies from FASTA search results. Comput Appl Biosci. 1993;9:371–372. doi: 10.1093/bioinformatics/9.3.371. [DOI] [PubMed] [Google Scholar]
- Henikoff JG, Greene EA, Pietrokovski S, Henikoff S. Increased coverage of protein families with the blocks database servers. Nucleic Acids Res. 2000;28:228–230. doi: 10.1093/nar/28.1.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann K, Bucher P, Falquet L, Bairoch A. The PROSITE database, its status in 1999. Nucleic Acids Res. 1999;27:215–219. doi: 10.1093/nar/27.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagla B, Schuchhardt J. Adaptive encoding neural networks for the recognition of human signal peptide cleavage sites. Bioinformatics. 2000;16:245–250. doi: 10.1093/bioinformatics/16.3.245. [DOI] [PubMed] [Google Scholar]
- Lipman DJ, Pearson WR. Rapid and sensitive protein similarity searches. Science. 1985;227:1435–1441. doi: 10.1126/science.2983426. [DOI] [PubMed] [Google Scholar]
- Matthews BW. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim Biophys Acta. 1975;405:442–451. doi: 10.1016/0005-2795(75)90109-9. [DOI] [PubMed] [Google Scholar]
- Mewes HW, Frishman D, Gruber C, Geier B, Haase D, Kaps A, Lemcke K, Mannhaupt G, Pfeiffer F, Schuller C, et al. MIPS: A database for genomes and protein sequences. Nucleic Acids Res. 2000;28:37–40. doi: 10.1093/nar/28.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murvai J, Vlahovicek K, Barta E, Parthasarathy S, Hegyi H, Pfeiffer F, Pongor S. The domain-server: Direct prediction of protein domain-homologies from BLAST search. Bioinformatics. 1999;15:343–344. doi: 10.1093/bioinformatics/15.4.343. [DOI] [PubMed] [Google Scholar]
- Murvai J, Vlahovicek K, Barta E, Cataletto B, Pongor S. The SBASE protein domain library, release 7.0: A collection of annotated protein sequence segments. Nucleic Acids Res. 2000a;28:260–262. doi: 10.1093/nar/28.1.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murvai J, Vlahovicek K, Pongor S. A simple probabilistic scoring method for protein domain identification. Bioinformatics. 2000b;16:1155–1156. doi: 10.1093/bioinformatics/16.12.1155. [DOI] [PubMed] [Google Scholar]
- Pearson WR, Lipman DJ. Improved tools for biological sequence comparison. Proc Natl Acad Sci. 1988;85:2444–2448. doi: 10.1073/pnas.85.8.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnhammer EL, Eddy SR, Birney E, Bateman A, Durbin R. Pfam: Multiple sequence alignments and HMM-profiles of protein domains. Nucleic Acids Res. 1998;26:320–322. doi: 10.1093/nar/26.1.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogl TP, Mangis JK, Rigler AK, Zink WT, Alkon DL. Accelerating the convergence of the back-propagation method. Biol Cybernetics. 1993;59:257–263. [Google Scholar]
- Wilbur WJ, Lipman DJ. Rapid similarity searches of nucleic acid and protein data banks. Proc Natl Acad Sci. 1983;80:726–730. doi: 10.1073/pnas.80.3.726. [DOI] [PMC free article] [PubMed] [Google Scholar]