

Abstract
Metabotropic glutamate receptors (mGluRs) are G-protein-coupled receptors that modulate excitatory neurotransmission and synaptic plasticity. The group I mGluRs (mGluR1 and mGluR5) have long intracellular C-terminal domains, which interact with many proteins. Our previous studies identified calmodulin (CaM) as a strong regulator of mGluR5 trafficking and mGluR5-induced calcium signaling. Although it has been accepted that both mGluR1 and mGluR5 interact with CaM, we now show that CaM specifically binds mGluR5 and not mGluR1. We have identified a single critical residue in mGluR5 (L896) that is required for CaM binding. In mGluR1, mutation of the corresponding residue, V909, to leucine is sufficient to confer CaM binding to mGluR1. To investigate the functional effects of CaM binding, we examined the surface expression of mGluR1 and mGluR5 in hippocampal neurons. The mutation in mGluR1 (V909L) that confers CaM binding dramatically increases mGluR1 surface expression, whereas the analogous mutation in mGluR5 that disrupts CaM binding (L896V) decreases mGluR5 surface expression. In addition, the critical residue that alters CaM binding regulates mGluR internalization. Furthermore, we find that mGluR-mediated AMPA receptor endocytosis is enhanced by CaM binding to group I mGluRs. Finally, we show that calcium responses evoked by group I mGluRs are modulated by these mutations, which regulate CaM binding. Our findings elucidate a critical mechanism that specifically affects mGluR5 trafficking and signaling, and distinguishes mGluR1 and mGluR5 regulation.
Introduction
Metabotropic glutamate receptors (mGluRs) are G-protein-coupled receptors (GPCRs) that are widely expressed throughout the brain and are involved in synaptic transmission, development, and plasticity (Hermans and Challiss, 2001; Kim et al., 2008). The mGluRs (mGluR1–8) are subdivided into three groups based on their sequence similarity and pharmacological properties (Hermans and Challiss, 2001). The group I mGluRs (mGluR1 and mGluR5) are Gq-coupled receptors that activate phospholipase Cβ (PLCβ), leading to the release of intracellular calcium stores (Conn and Pin, 1997). In heterologous cells, group I mGluRs can regulate both intracellular Ca2+ release and cAMP production (Pin and Duvoisin, 1995). The activation of this dual signal transduction is determined by critical residues within the second and third intracellular loops (Francesconi and Duvoisin, 1998).
Group I mGluRs modulate a variety of ion channels and receptors (Hermans and Challiss, 2001; Snyder et al., 2001; Kelly et al., 2009; Johnston and Delaney, 2010; Lüscher and Huber, 2010). Group I mGluR activation results in the loss of synaptic AMPA and NMDA receptors (Snyder et al., 2001), induces long-term depression (LTD) (Bashir et al., 1993; Bolshakov and Siegelbaum, 1994), and promotes AMPA receptor endocytosis (Chowdhury et al., 2006; Park et al., 2008; Waung et al., 2008). This mGluR-LTD requires de novo protein synthesis and the activation of the phosphoinositide 3-kinase-Akt-mTOR (mammalian target of rapamycin) signaling pathway (Huber et al., 2000; Hou and Klann, 2004). In addition, group I mGluRs stimulate cap-dependent translation, by specifically regulating eIF4F (eukaryotic translation initiation factor 4F) activity (Banko et al., 2006).
Although mGluR1 and mGluR5 are both Gq-coupled mGluRs that stimulate intracellular Ca2+ release, they have distinct signaling properties. For example, mGluR5 induces Ca2+ oscillations, whereas mGluR1 activation results in a single Ca2+ transient (Kawabata et al., 1996). In addition, mGluR1 and mGluR5 are differentially expressed throughout the brain (Akbar et al., 1996; Ferraguti and Shigemoto, 2006). The intriguing differences in the distribution and regulation of group I mGluRs suggest they have distinct effects on neuronal function.
Group I mGluRs interact with many proteins, including Homer, Tamalin, Siah-1A, caveolin-1, and calmodulin (CaM) (Minakami et al., 1997; Tu et al., 1998; Ishikawa et al., 1999; Kitano et al., 2002; Enz, 2007; Francesconi et al., 2009). PKC phosphorylation of serine 901 (S901) in the mGluR5 C-terminal domain inhibits CaM binding, and the balance between PKC phosphorylation and CaM binding is a key determinant of mGluR5 trafficking and mGluR5-stimulated calcium signaling (Lee et al., 2008). Data support a model in which activation of mGluR5 induces PKC activity, and PKC phosphorylates mGluR5 S901, causing dissociation of CaM, and ultimately leads to mGluR5 internalization.
Here, we show that mGluR1 does not bind to CaM, despite significant sequence homology with mGluR5. Interestingly, a single residue substitution between mGluR1 and mGluR5 mediates CaM binding to group I mGluRs. This residue also profoundly affects group I mGluR surface expression, receptor signaling, and 3,5-dihydroxyphenylglycine (DHPG)-mediated AMPA receptor endocytosis. These findings demonstrate that CaM is a critical determinant of mGluR5 function, and reveal a molecular basis for underlying functional differences between the highly homologous group I mGluRs.
Materials and Methods
Constructs.
Myc-tagged mGluR1a was obtained as a gift from Dr. Paul J. Kammermeier (University of Rochester Medical Center, Rochester, NY) (Kammermeier and Yun, 2005). All mGluR1 constructs in this study are the mGluR1a isoform. A Myc epitope was inserted into the N terminus of mGluR5 (between amino acids 22 and 23) to make Myc-tagged mGluR5 (Lee et al., 2008). Glutathione S-transferase (GST) fusion protein constructs containing the C terminus of mGluR1 (amino acids 842–959) or mGluR5 (amino acids 828–944) were subcloned into pGEX vectors (GE Healthcare Bio-Sciences) as described previously (Kim et al., 2005). Additional mutations (described in Fig. 2A) were introduced into Myc-mGluR1, GST-mGluR1-Cprox (C-terminal proximal region), Myc-mGluR5, or GST-mGluR5-Cprox using the QuikChange site-directed mutagenesis system (Stratagene) following the manufacturer's instructions. All mutations were confirmed by sequence analysis. His-tagged CaM2 was used for coimmunoprecipitation with group I mGluRs.
Figure 2.
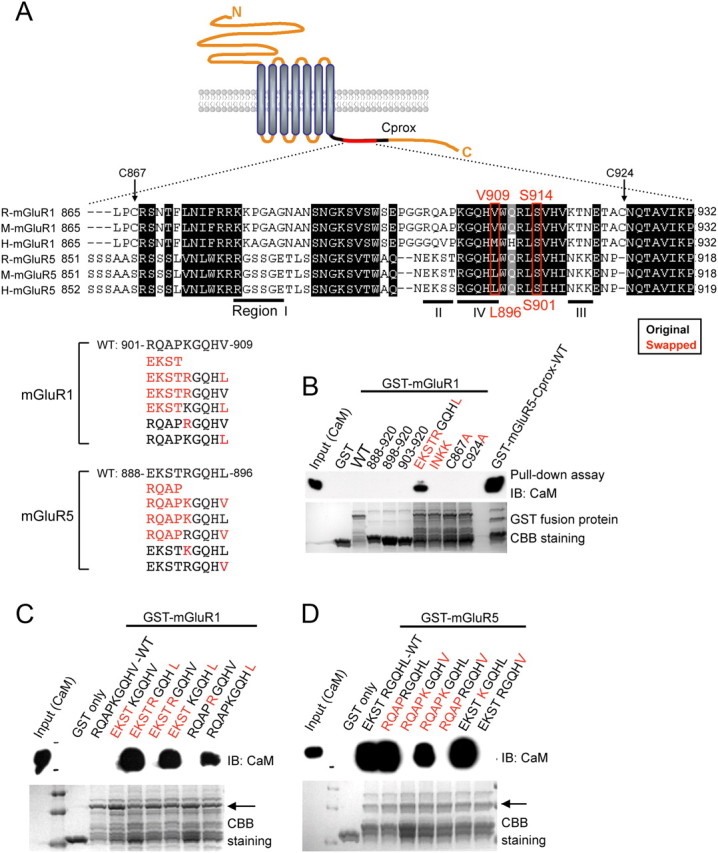
Defining critical determinants for CaM binding to group I mGluRs. A, Schematic showing a small region of the cytoplasmic C terminus of either mGluR1 or mGluR5 from rat, mouse, and human. The Cprox region is indicated as a dark line, and the aligned region is shown as a red line. The amino acids surrounding S901 of rat mGluR5 (S914 of mGluR1) were aligned using clustal W. In the alignment, dark boxes indicate the regions of high homology between mGluR1 and mGluR5. The mutant constructs of either rat mGluR1 or rat mGluR5 are shown, with the native sequences in black and the swapped residues in red. To determine the critical domain for CaM binding, we specifically focused on three regions (dark lines, regions I, II, and III) that have divergent sequences between mGluR1 and mGluR5. Region IV contains the critical CaM-binding residue, although this region shows high homology between mGluR1 and mGluR5. B–D, Recombinant CaM was pulled down with the indicated GST-mGluR1-Cprox (B, C) or GST-mGluR5-Cprox (D) proteins, either WT or containing amino acid substitutions, in the presence of 2 mm Ca2+, and subjected to Western blotting with CaM antibody. Coomassie Brilliant Blue staining was used for the detection of total amounts of GST fusion proteins.
Antibodies.
Anti-CaM, anti-mGluR1, and anti-mGluR5 antibodies were purchased from Millipore. Mouse anti-Myc (9E10), rabbit anti-Myc, mouse anti-FLAG, and mouse anti-α-tubulin were purchased from Sigma. Anti-CaM antibody was purchased from Millipore and anti-GST antibody was purchased from GeneTex. Alexa 488-, 568-, and 647-conjugated secondary antibodies were obtained from Invitrogen.
Cells.
HEK293 cells were maintained in DMEM containing 10% fetal bovine serum, 1% l-glutamine, and 0.1% gentamicin. Primary hippocampal neurons were prepared from embryonic day 18 Sprague Dawley rats as described previously (Roche and Huganir, 1995), and grown in Neurobasal medium supplemented with glutamine and B-27 supplement (Invitrogen). The use and care of animals used in this study followed the guidelines of the National Institutes of Health Animal Research Advisory Committee.
Fusion protein production and GST pull-down assay.
GST-tagged fusion proteins for wild-type (WT) and mutant C termini of mGluR1 or mGluR5 were purified as recommended by the manufacturer (GE Healthcare Bio-Sciences). In brief, DNA constructs were transformed into BL21 bacterial cells. Isopropyl 1-thio-d-galactopyranoside (1 mm) was added to 50 ml cultures in the mid log phase of growth (A550 = 0.5–1.0) to induce fusion protein expression. After 4 h of induction, cells were lysed in 5 ml of B-PER bacterial protein extraction reagent (Pierce). GST fusion proteins were purified according to the manufacturer's instructions (GE Healthcare Bio-Sciences). For pull-down assays, GST fusion proteins were incubated with recombinant CaM (1 μg, Millipore) in a total volume of 500 μl of binding buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 2 mm CaCl2, 0.5% Nonidet P-40, protease inhibitors, and phosphatase inhibitors) for 2 h at 4°C. The beads were washed four times with binding buffer, and the bound proteins were eluted by adding SDS-PAGE sample buffer and boiling for 5 min. Samples resolved by SDS-PAGE were transferred to a polyvinyl difluoride (PVDF) membrane (Millipore) and visualized by Western blotting. The binding of CaM to GST fusion proteins was analyzed by immunoblotting with anti-CaM antibody. The loaded GST fusion proteins were confirmed by immunoblotting with anti-GST antibody or by Coomassie Brilliant Blue staining.
Immunoblotting and immunoprecipitation.
Transiently transfected HEK293 cells were lysed with 1% Triton X-100 in Tris-buffered saline (TBS). Lysates were incubated with antibodies at 4°C overnight, and then with protein A/G agarose beads for 2 h. After washing beads three times with TBS containing 1% Triton X-100, the beads were incubated with SDS sample buffer at 37°C for 10 min. The eluted proteins were resolved on SDS-polyacrylamide gel, transferred to PVDF membrane, and subjected to Western blotting with the indicated primary and secondary antibodies.
Cell surface biotinylation assay.
HEK293 cells expressing Myc-mGluR1 (wild-type and V909L) or Myc-mGluR5 (wild-type and L896V) were rinsed three times with PBS containing 1 mm MgCl2 and 0.1 mm CaCl2 (PBS++) and then incubated with 1 mg/ml EZ-Link Sulfo-NHS-SS-Biotin (Pierce) in PBS++ for 20 min at 4°C. Cells were rinsed three times with cold 50 mm glycine in PBS++ to quench unreacted biotin and then lysed in TBS containing 1% Triton X-100 and protease inhibitor mixture (Roche) for 1 h at 4°C. Cell lysates were centrifuged at 13,000 × g for 10 min. The supernatant was incubated with streptavidin agarose beads (Pierce) for 2 h at 4°C and washed four times with TBS containing 1% Triton X-100. Precipitates were eluted and analyzed by immunoblotting with rabbit Myc antibody.
Immunofluorescence assay of receptor trafficking in neurons.
A fluorescence-based antibody uptake internalization assay was performed as described previously (Lavezzari et al., 2004),with some modifications. Cultured hippocampal neurons were transfected with Myc-mGluR1 (wild-type or V909L) or mGluR5 (wild-type or L896V). After 2 d, Myc antibody was added to the media to label surface-expressed mGluR1 and mGluR5 for 30 min at room temperature. Neurons were washed twice with PBS and then treated with the group I mGluR agonist DHPG (100 μm) for 30 min for mGluR5 and for 90 min for mGluR1 at 37°C. After washing with PBS, neurons were incubated with Alexa 568-conjugated (red) anti-mouse secondary antibody (Invitrogen) for 20 min at room temperature to label the surface receptors. After three washes in PBS, the cells were fixed with 4% paraformaldehyde/4% sucrose in PBS for 15 min, washed, and then permeabilized with 0.25% Triton X-100 for 5 min. After blocking with 10% normal goat serum, neurons were incubated with Alexa 488-conjugated (green) anti-mouse secondary antibody (Invitrogen) for 30 min to label internalized receptors. After washing with PBS, ProLong Antifade reagent (Invitrogen) was applied to neurons before mounting. Images were analyzed with a 63× objective on an LSM 510 confocal microscope (Zeiss). A series of optical sections were collected at intervals of 0.5 μm, and the figures show the maximum projections. For quantitative analysis, images from three or four dendrites per neuron (at least 15 neurons per experiment) were collected, and the amount of internalization was measured with MetaMorph 6.0 software (Universal Imaging Corp.). The values represent means ± SEM of three independent experiments. Significance was determined by using a Student's unpaired t test.
For immunostaining surface expression, the transfected neurons were incubated with saturated Myc antibody (9B11; Cell Signaling Technology) for 20 min at room temperature to label surface-expressed protein, followed by incubation with saturated Alexa 568-conjugated (red) anti-mouse IgG2a-specific secondary antibody (Invitrogen) for 20 min. Following extensive washing, the cells were fixed, permeabilized, incubated with 10% normal goat serum, and labeled with Myc antibody (9E10; Sigma) for 30 min at room temperature for staining intracellular receptor fractions. After washing with PBS, the cells were incubated with Alexa 488-conjugated (green) anti-mouse IgG1-specific secondary antibody for 30 min and mounted with a ProLong Antifade kit (Invitrogen). Images were collected and analyzed, as for the internalization assays. Red fluorescence intensity (surface expression) was divided by green fluorescence intensity (intracellular fraction) to calculate the intensity ratio.
AMPA receptor trafficking experiments.
The AMPA receptor endocytosis assay was performed as described previously (Park et al., 2008), with some modifications. Cultured hippocampal neurons were cotransfected with Myc-mGluR1 (wild-type or V909L) or mGluR5 (wild-type or L896V) in pIRES2-EGFP (internal ribosomal entry site 2-enhanced green fluorescent protein) and FLAG-GluR1. After 5 d, surface FLAG-GluR1 containing AMPA receptors was labeled with FLAG antibody in conditioned media at room temperature for 20 min. Cells were incubated at room temperature for 10 min with antagonists [10 μm 2-methyl-6-(phenylethynyl)-pyridine (MPEP) for cells transfected with Myc-mGluR1 and 50 μm 7-(hydroxyimino) cyclopropa[b]chromen-1a-carboxylate ethyl ester (CPCCOEt) for cells transfected with Myc-mGluR5] to inhibit the endogenous group I mGluR. After 5 min DHPG (50 μm) application, the neurons were incubated in conditioned media at 37°C for 15 min. To visualize the surface FLAG-GluR1, live neurons were incubated with an excess of Alexa 568-conjugated (red) anti-mouse IgG1 secondary antibody (Invitrogen) at 10°C or room temperature. Each antagonist was included in the incubation solution until fixation. After three washes in PBS, neurons were fixed and permeabilized as described above. After blocking with 10% normal goat serum, neurons were incubated with Alexa 647-conjugated (green) anti-mouse IgG1 secondary antibody (Invitrogen) for 30 min to label internalized FLAG-GluR1. For staining of EGFP, GFP antibody (Invitrogen) was added and followed by incubation with Alexa 488-conjugated (white) anti-rabbit-specific secondary antibody (Invitrogen). Image collection and analysis were the same as described above.
Calcium assays.
HeLa cells were transfected with Myc-mGluR1 [wild-type, V909L, or EKSTKGQHL (EKST+L) or Myc-mGluR5 (wild-type or L896V)]. Cells grown on coverglasses were preloaded with Fluo-4 AM for 30 min and then placed on the perfusion chamber under the confocal microscope. The cells were stimulated by perfusion of the different concentrations of glutamate in normal Tyrode's solution (in mm: 134 NaCl, 5.4 KCl, 2.5 CaCl2, 1.2 MgCl2, 14 d-glucose, 10.5 HEPES). Ca2+ responses were measured with excitation at 488 nm every 3 s. Concentration–response relationships were analyzed by nonlinear regression using Prism 4.0 software (GraphPad Software). For analyzing Ca2+ oscillation responses evoked by mGluR5 activation, intracellular Ca2+ levels were expressed as relative total fluorescence [ΔF/F0: ratio of fluorescence difference, stimulated-basal (FS-F0), to basal (F0) value as a function of time]. All data are represented as means ± SEM.
Results
Group I mGluRs differ in their binding to CaM
CaM binding to mGluR5 acts as a critical regulator for mGluR5 trafficking (Lee et al., 2008). Although never demonstrated, it has been accepted that mGluR1, another group I mGluR, also interacts with CaM (Ishikawa et al., 1999). To directly compare CaM binding, we conducted in vitro GST pull-down assays using recombinant CaM protein and GST-mGluR1-Cprox (residues 842–959), GST-mGluR5-Cprox (residues 828–944), and GST-mGluR7A CT (intracellular C-terminal region). In Figure 1A, we observed robust binding of CaM to mGluR5 and mGluR7, but not mGluR1. Next, we evaluated the ability of Myc-mGluR1 or Myc-mGluR5 to coimmunoprecipitate His-CaM2 from transfected HEK293 cells. Consistent with our in vitro results, we found that Myc-mGluR5 immunoprecipitated CaM, whereas Myc-mGluR1 did not (Fig. 1B). In addition, we immunoprecipitated endogenous mGluR1 or mGluR5 from rat whole-brain lysates, and again found that CaM associates with mGluR5, but not mGluR1 (Fig. 1C). Together, these results clearly demonstrate that CaM interacts specifically with mGluR5, but not mGluR1.
Figure 1.
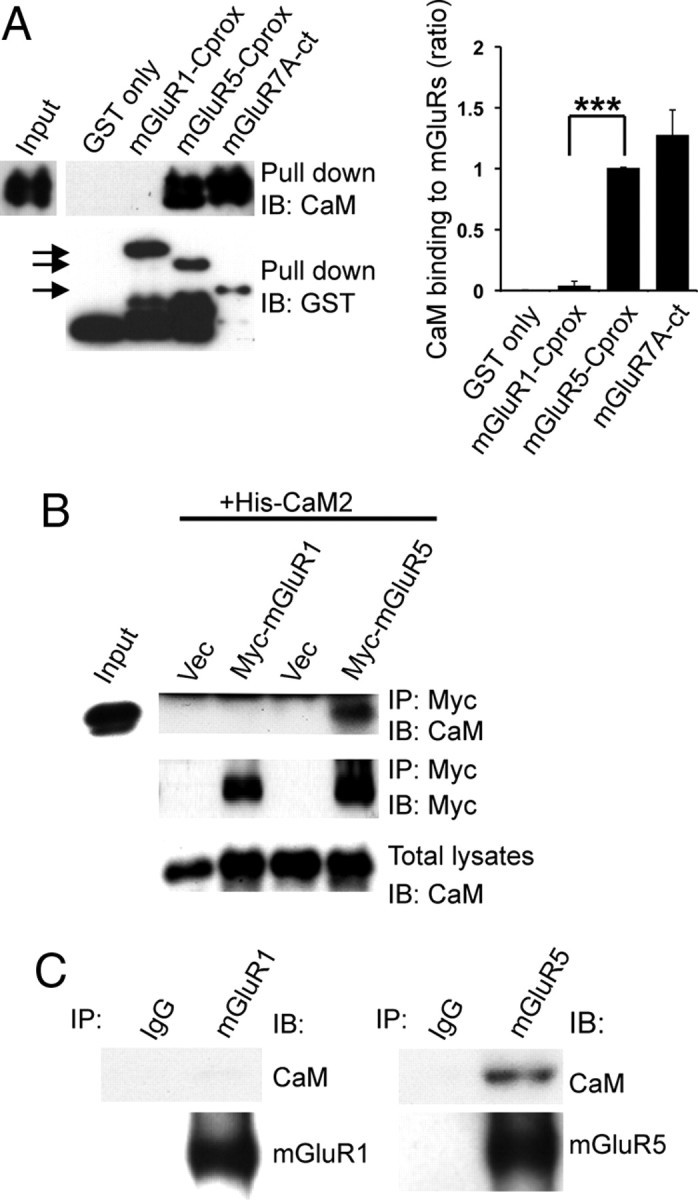
Differential binding of CaM to group I mGluRs. A, Recombinant CaM was subjected to a pull-down assay using GST fusion proteins containing mGluR1-Cprox (amino acids 842–959), mGluR5-Cprox (amino acids 828–944), or mGluR7A-ct (amino acids 851–915). Bound proteins were analyzed by SDS-PAGE and Western blotting using anti-CaM or anti-GST antibodies. The arrows indicate each GST fusion protein. The graph shows the ratio of CaM binding to GST-mGluRs normalized to GST-mGluR5-Cprox binding. Data represent means ± SEM (n = 4, ***p < 0.001). B, HEK293 cells were cotransfected with His-tagged CaM2 and Myc-tagged mGluR1 or mGluR5. Lysates were immunoprecipitated with anti-Myc antibody and analyzed by Western blotting using the indicated antibodies. C, Total rat brain lysates were immunoprecipitated with IgG (negative control), anti-mGluR1, or anti-mGluR5 antibodies. Immunoprecipitates were resolved by SDS-PAGE and immunoblotted with the indicated antibodies.
Mapping of a critical region for CaM binding
There is high sequence homology between mGluR1 and mGluR5 (Fig. 2A). To identify the CaM binding region on mGluR5, we made a variety of mutant constructs, exchanging single residues and stretches of amino acids between mGluR1 and mGluR5 (Fig. 2). As previously reported, S901 of mGluR5 is critical for CaM binding (Lee et al., 2008). Alignment of the region surrounding mGluR5 S901 (mGluR1 residues 865–932 and mGluR5 amino acids 851–918) revealed amino acid divergence and led us to make substitutions between mGluR1 and mGluR5 of both stretches of amino acids (depicted as regions I, II, III, and IV) and individual residues (red letters). We performed in vitro binding assays using WT and mutant forms of GST-mGluR1-Cprox or GST-mGluR5-Cprox. Swapping residues in regions I–III had no effect on CaM binding to either mGluR1 or mGluR5, suggesting that the residues in these regions are not essential determinants of CaM binding (data not shown).
In contrast, exchange of residues in region IV had a profound effect on CaM binding to mGluR1 and mGluR5. Inserting a segment of mGluR5 (888-EKSTRGQHL-896) into the analogous region of mGluR1 (901-RQAPKGQHV-909) (Fig. 2A) results in mutant mGluR1 binding to CaM (Fig. 2B). Within this critical stretch of amino acids, the only mGluR5 residues that differ from mGluR1 are EKST, arginine 892 (R892), and leucine 896 (L896) (Fig. 2A). The exchange of EKST or R892 did not confer CaM binding on mGluR1 or disrupt mGluR5-CaM binding (Fig. 2C,D). Surprisingly, we found that the single point mutation, GST-mGluR1 V909L, resulted in CaM binding, which could be further enhanced when combined with the mGluR1-EKSTRGQHL substitution (Fig. 2B,C). Conversely, GST-mGluR5-Cprox mutant fusion proteins with a single residue substitution of valine at L896 completely abolished CaM binding (Fig. 2D). These in vitro binding results indicate that a single residue (V909 in mGluR1 or L896 in mGluR5) is a critical determinant for CaM binding to group I mGluRs. In Figure 2B, smaller fragments (GST-mGluR1, amino acids 888–920, 898–920, or 903–920) and point mutants [GST-mGluR1, INKK (in region III), C867A, or C924A] were also examined, but did not show CaM binding.
We next analyzed full-length mGluR1 and mGluR5 for CaM binding. We transfected Myc-mGluR1 (WT, V909L, or EKST+L) or Myc-mGluR5 (WT, L896V, or S901D) into HEK293 cells, followed by immunoprecipitation with anti-mGluR1 or anti-mGluR5 antibody. Myc-mGluR1 V909L or mGluR1 EKST+L coimmunoprecipitated endogenous CaM, whereas mGluR1 WT did not (Fig. 3A). Although the mGluR1 EKST+L mutant (with 5 residues swapped) shows more robust CaM binding, the single residue substitution (V909L) was sufficient to confer CaM binding, consistent with the results of the in vitro binding assays. Similarly, we observed robust binding of endogenous CaM to mGluR5, but not to mGluR5 L896V (Fig. 3B). We also found that mGluR1 did not bind to CaM when valine 909 was mutated to methionine (data not shown), the analogous residue in human (Fig. 2A).
Figure 3.
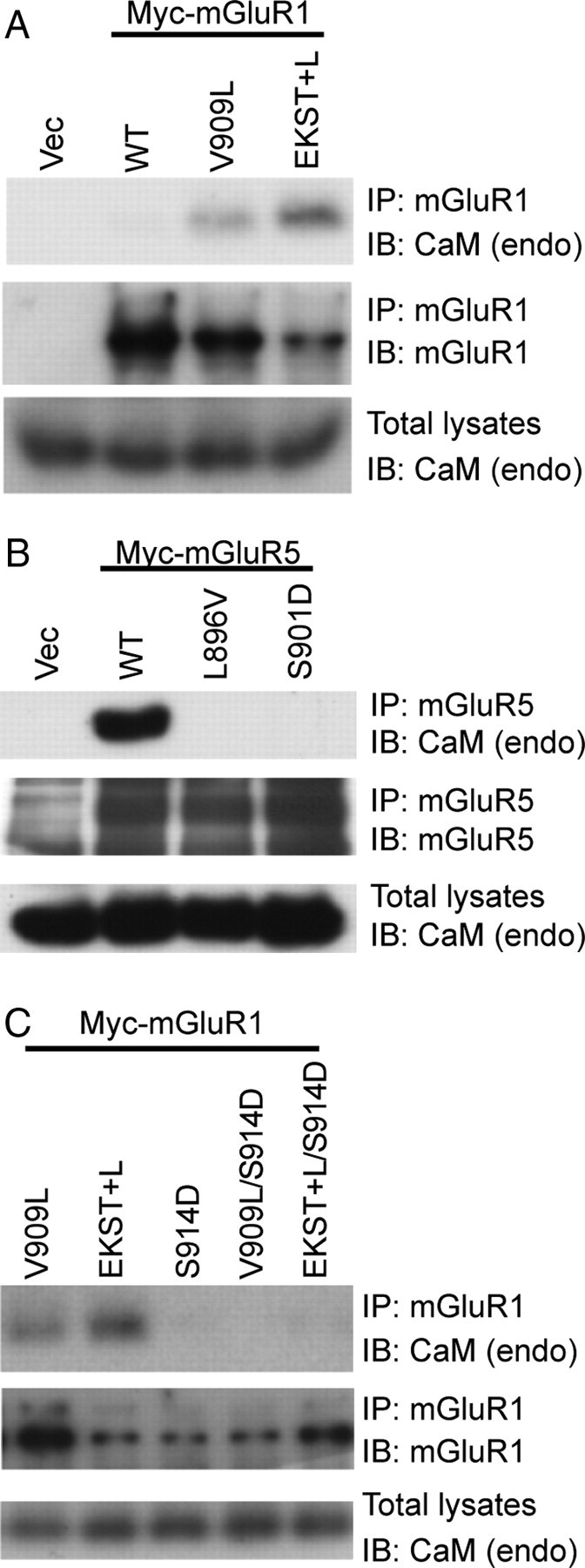
Substitution of a single residue in mGluR1 (V909L) or mGluR5 (L896V) regulates CaM binding. HEK293 cells were transfected with WT or mutant Myc-mGluR1 (A, C) or Myc-mGluR5 (B) constructs. Myc-mGluRs were immunoprecipitated from lysates with anti-mGluR1 (A, C) or anti-mGluR5 (B) antibodies, resolved by SDS-PAGE, and immunoblotted with antibodies as indicated. Total cell lysates were probed with CaM antibody to detect endogenous CaM (endo). C, Myc-mGluR1 S914D, V909L/S914D, and EKST+L/S914D were transfected, and tested for CaM binding.
As reported previously, mGluR5 S901D disrupted CaM binding (Lee et al., 2008). We also examined mGluR1 S914D containing a point mutation in the serine (S914) analogous to mGluR5 S901 that regulates CaM binding. Using coimmunoprecipitation assays from cell lysates expressing Myc-mGluR1 (S914D, V909L/S914D, or EKST+L/S914D), we found that mGluR1 S914D disrupted CaM binding of mGluR1 V909L and EKST+L (Fig. 3C), similar to mGluR5 S901D. These results demonstrate that mGluR1, unlike mGluR5, does not interact with CaM, and define a critical single residue substitution that reverses the CaM-binding properties of the two group I mGluRs.
It is surprising that a single residue substitution regulates the differential CaM binding of group I mGluRs, especially because valine and leucine are structurally similar, with non-polar side chains. Analysis of the mGluR1 sequence using a CaM target database (http://calcium.uhnres.utoronto.ca/ctdb/flash.htm), a web-based database for proteins containing CaM-binding sites (Yap et al., 2000), revealed that mGluR1 has a putative CaM-binding motif in the second intracellular loop, where mGluR1 and mGluR5 share a high level of sequence identity. We generated GST-fusion proteins containing this loop region of mGluR1 and mGluR5, and performed in vitro binding assays with CaM. However, we did not detect any CaM binding (data not shown). Therefore, our data are consistent with the C-terminal region of mGluR5 being the dominant region for CaM binding.
CaM binding to group I mGluRs increases constitutive surface expression
CaM binding to mGluR5 has been shown to stabilize receptor surface expression (Lee et al., 2008). Mutation of the PKC phosphorylation site on mGluR5, S901D, dramatically reduces CaM binding and causes a decrease in mGluR5 surface expression (Lee et al., 2008). To investigate whether a substitution of the critical residue in mGluR1 (V909L) or mGluR5 (L896V) affects receptor surface expression, we performed cell surface biotinylation assays, using heterologous cells expressing Myc-mGluR1 (WT or V909L) or Myc-mGluR5 (WT or L896V). The constitutive surface expression of Myc-mGluR1 V909L was significantly increased compared with WT (Fig. 4A,C), whereas the surface expression of Myc-mGluR5 L896V was markedly reduced (Fig. 4B,C).
Figure 4.

CaM binding to group I mGluRs increases constitutive surface expression. A, B, Surface expression of mGluR1 and mGluR5 was analyzed using a cell-surface biotinylation assay in heterologous cells. HEK293 cells were transfected with the indicated Myc-mGluR1 (A) or Myc-mGluR5 (B) constructs. Biotinylated surface proteins were isolated with streptavidin-agarose beads and subjected to Western blotting using anti-Myc antibody. C, The amount of surface-expressed receptors was normalized to total mGluR expression from total lysates. Quantification is depicted as a histogram, including results from three independent experiments. Data represent means ± SEM (for mGluR1, *p < 0.05; and for mGluR5, **p < 0.01). D–G, Constitutive surface expression of Myc-mGluR1, Myc-mGluR1 V909L, Myc-mGluR5, or Myc-mGluR5 L896V in primary cultured neurons. Hippocampal neurons were transiently transfected with Myc-mGluR1 (WT or V909L) (D) or Myc-mGluR5 (WT or L896V) (F). The surface receptors (Sur, red) and intracellular receptors (Intra, green) were visualized as described in Materials and Methods. The panels on the right show higher-magnification images of boxed regions (D, F). Each scale bar for the whole-cell image and the magnified image indicates 20 and 5 μm, respectively. E, G, Quantification of the fluorescence microscopy is presented as histograms for Myc-mGluR1 (E) and for Myc-mGluR5 (G). The data were quantified by measuring ratios of surface pool/intracellular fraction using MetaMorph software. Data represent means ± SEM (E, n = 3, **p < 0.01; and G, n = 4, *p < 0.05).
In addition, we characterized the steady-state surface expression of the WT and mutant group I mGluRs expressed in neurons using fluorescence-based antibody staining. Cultured primary hippocampal neurons were transfected with Myc-mGluR1 (WT or V909L) or Myc-mGluR5 (WT or L896V), followed by immunostaining for surface-expressed (red) or intracellular (green) receptors. The ratio of surface to intracellular receptors was analyzed. We found that mGluR1 V909L, which binds CaM, showed a dramatic increase in surface expression compared with WT (Fig. 4D,E). Conversely, surface expression of mGluR5 L896V, which is unable to bind CaM, was decreased (Fig. 4F,G). Therefore, both biotinylation assays and fluorescence microscopy assays reveal that CaM binding enhances the cell surface stability of group I mGluRs.
Agonist-induced internalization of mGluR1 or mGluR5 is regulated by CaM binding in neurons
We next addressed the role of CaM binding on agonist-stimulated internalization of group I mGluRs. It is known that endocytosis of mGluR5 S901A, which constitutively binds CaM regardless of phosphorylation, is decreased following stimulation with the potent group I mGluR agonist DHPG (Lee et al., 2008). Cultured hippocampal neurons expressing Myc-mGluR1, Myc-mGluR1 V909L, Myc-mGluR5, or Myc-mGluR5 L896V were subjected to agonist-induced internalization assays. The internalization of mGluR1 V909L was significantly reduced compared with that of mGluR1 WT (Fig. 5A,B), consistent with CaM binding decreasing mGluR1 V909L endocytosis. Conversely, mGluR5 L896V showed increased internalization compared with WT (Fig. 5C,D), demonstrating that CaM binding is a critical factor for mGluR5 trafficking.
Figure 5.
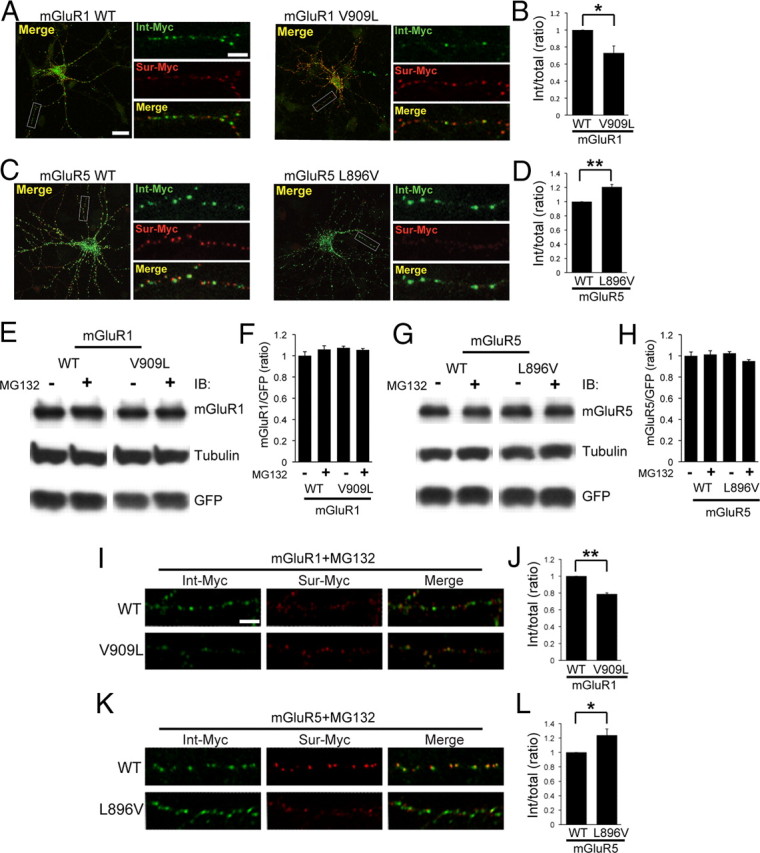
Agonist-induced internalization of mGluR1 or mGluR5 is regulated by CaM binding in neurons. A, C, Cultured hippocampal neurons expressing Myc-mGluR1 (WT or V909L) (A) or Myc-mGluR5 (WT or L896V) (C) were incubated with Myc antibody for 30 min at room temperature, followed by stimulation with 100 μm DHPG at 37°C. The surface pool (Sur-Myc, red) and internalized pool of receptors (Int-Myc, green) were visualized as described in Materials and Methods. The region in the white box is shown at higher magnification below. Each scale bar for the whole-cell image and the magnified image indicates 20 and 5 μm, respectively. B, D, Quantification is presented as a histogram. The values indicate the ratio of internalized receptor compared with total. The data represent means ± SEM (B, n = 3, *p < 0.05; and D, n = 4, **p < 0.01). E–H, The total expression level of mGluR1 or mGluR5 was not affected by CaM binding. HEK cells were transfected with Myc-mGluR1-pIRES2-EGFP (WT or V909L) (E) or Myc-mGluR5-pIRES2-EGFP (WT or L896V) (G). Two days after transfection, cells were treated with 10 μm MG132 at 37°C for 3 h, lysed, and then subjected to Western blotting with indicated antibodies. F, H, The total expression level of Myc-mGluRs was normalized to each GFP expression level. Quantification is depicted as a histogram. Data represent means ± SEM (n = 3; the statistical analysis showed no significance). I, K, Cultured hippocampal neurons expressing Myc-mGluR1 (WT or V909L) (I) or Myc-mGluR5 (WT or L896V) (K) were pretreated with 10 μm MG132 for the cells at 37°C for 1 h. The internalization assays were conducted as above, and MG132 was included in all remaining steps. The surface pool (Sur-Myc, red) and internalized pool of receptors (Int-Myc, green) were visualized. Scale bar, 5 μm. J, L, Quantification is presented as a histogram. The values indicate the ratio of internalized receptor compared with total. The data represent means ± SEM (J, n = 3, **p < 0.01; and L, n = 4, *p < 0.05).
We also evaluated the total protein levels of mGluR1, mGluR5, or their mutants in the presence of MG132 [N-benzyloxycarbonyl (Z)-Leu-Leu-leucinal], an inhibitor of proteasomal degradation by Western blotting. We found that total protein levels were not changed between WT and mutant (Fig. 5E–H). In addition, MG132 treatment did not alter the total expression level of receptors. Furthermore, we performed the internalization assays in the presence of MG132, and found that mGluR1 V909L still showed decreased internalization compared with WT (Fig. 5I,J), and mGluR5 L896V showed increased endocytosis (Fig. 5K,L). These results showed that CaM binding is a critical factor for group I mGluR trafficking and does not depend on changes in protein degradation.
Mutations in mGluR1 or mGluR5 that affect CaM binding also modulate DHPG-mediated AMPA receptor endocytosis in neurons
mGluR5 surface density is correlated with receptor activity (Lee et al., 2008), and activation of group I mGluRs can induce LTD (Bashir et al., 1993; Bolshakov and Siegelbaum, 1994), during which, AMPA receptors are internalized (Snyder et al., 2001; Xiao et al., 2001). To examine the correlation between the surface density and the activity of receptor of the group I mGluRs (WT or CaM-binding mutants), we performed a group I mGluR-mediated AMPA receptor endocytosis assay. FLAG-GluR1 was cotransfected with Myc-mGluR1 (WT or V909L) or Myc-mGluR5 (WT or L896V) in a pIRES2-EGFP vector into cultured neuronal cells. To analyze the endocytosis of FLAG-GluR1, surface receptors were labeled with anti-FLAG antibody on live neurons. To evaluate the specific role of each group I mGluR in DHPG-mediated AMPA receptor endocytosis, we pretreated with an antagonist to block the other endogenous group I mGluR receptor (MPEP for mGluR1-transfected cells and CPCCOEt for mGluR5-transfected cells). CPCCOEt is an mGluR1-selective noncompetitive antagonist, whereas MPEP is a highly selective noncompetitive antagonist for mGluR5. After staining internalized (green) and surface (red) GluR1, the ratios of internalized to total receptors (internalized plus surface) were measured. The CaM binding mGluR1 V909L markedly increased the internalization of FLAG-GluR1 compared with mGluR1 WT (Fig. 6A,B). In contrast, mGluR5 L896V showed decreased AMPA receptor endocytosis compared with WT (Fig. 6C,D). The decrease in AMPA receptor endocytosis observed with mGluR5 L896V was modest. This was likely due in part to the high expression of endogenous mGluR5 in these cultures. Together, these findings suggest that CaM binding to group I mGluRs and the subsequent increase in receptor surface expression also results in increased group I mGluR-mediated intracellular signaling.
Figure 6.

CaM binding mutations in mGluR1 and mGluR5 modulate DHPG-dependent AMPA receptor endocytosis in neurons. A, C, Hippocampal neurons expressing FLAG-GluR1 and Myc-mGluR1 (A) or Myc-mGluR5 (C) were stimulated with 50 μm DHPG for 5 min, followed by an additional 15 min incubation at 37°C in conditioned media with a group I mGluR antagonist. For mGluR1-transfected cells, 10 μm MPEP, an mGluR5 antagonist, was used (A), and for cells transfected with mGluR5, 50 μm CPCCOEt, an mGluR1 antagonist, was used (C). Cells were stained for internalized (Int, green) or surface (Sur, red) FLAG-GluR1, and GFP (from pIRES2-EGFP vector, white, right). The bottom shows higher-magnification images of the individual processes boxed in the top. Each scale bar for the whole-cell image and the magnified image indicates 20 and 5 μm, respectively. B, D, Quantification is presented as a histogram. The values indicate the normalized value of the FLAG-GluR1 internalized fraction compared with the total FLAG-GluR1. The data represent means ± SEM (n = 4, *p < 0.05, **p < 0.01).
Glutamate-induced calcium responses are modulated by CaM binding to group I mGluRs
Many GPCR agonists stimulate intracellular Ca2+ release. In many cases, the Ca2+ signal is concentration dependent, affecting the proportions of cells in which [Ca2+]i responses can be detected, as well as increases in [Ca2+]i in individual cells. In addition, the density of GPCRs on the plasma membrane also strongly regulates Ca2+ responses (Steinhoff et al., 2000; Nash et al., 2002). We therefore measured the concentration–response relationship of group I mGluRs (WT vs mutants). As shown in Figure 7, glutamate-stimulated Ca2+ responses were recorded and the proportion of cells that responded increased in a concentration-dependent manner. The mGluR1 mutations (V909L or EKST+L), which increase surface expression of mGluR1, caused a leftward shift in the concentration–response curves compared with that of mGluR1 WT (EC50 6.44 ± 0.80 μm for WT; 4.34 ± 0.47 μm for V909V; 2.59 ± 0.812 μm for EKST+L) (Fig. 7A). In contrast, mGluR5 L896V caused a significant rightward shift in the concentration–response curve compared with that of mGlulR5 WT (EC50 0.50 ± 0.04 μm for WT; 0.96 ± 0.09 μm for L896V) (Fig. 7B).
Figure 7.

Glutamate-induced Ca2+ responses are modulated by CaM binding to group I mGluRs. A, B, Concentration–response relationships were measured with HeLa cells expressing Myc-mGluR1 (WT, V909L, or EKST+L) (A) or Myc-mGluR5 (WT or L896V) (B). The cells were loaded with Fluo-4 AM, a Ca2+ indicator, and time-lapse images of cells under perfusion with glutamate were collected. Glutamate (0.1, 0.3, 1, 3, 10, 30, and 100 μm) was applied for 3 min, washed out for 1.5 min, and then an additional dose of glutamate was applied. As the concentration of glutamate increases, the proportion of cells with detectable increases in [Ca2+]i above basal values increases, dependent on glutamate concentration. The graphs show the normalized populations of cells with Ca2+ responses for mGluR1 (A) and mGluR5 (B). The data represent means ± SEM (A, n = 5, *p < 0.05; and B, n = 5, *p < 0.05). C, D, Representative traces of Ca2+ oscillations evoked by mGluR5 WT (C) or mGluR5 L896V (D) activation with 3 μm glutamate.
Activation of mGluR5 evokes Ca2+ oscillations, and the frequency of Ca2+ oscillations is correlated with mGluR5 density on the cell membrane (Kawabata et al., 1996; Nash et al., 2002; Lee et al., 2008). Therefore, reducing mGluR5 density on the plasma membrane by mutation of L896V will likely decrease mGluR5-induced Ca2+ oscillation frequency. To test this hypothesis, we analyzed [Ca2+]i responses recorded from HeLa cells expressing mGluR5 (WT or L896V). Glutamate (3 μm) triggered Ca2+ oscillations in mGluR5 WT or L896V (Fig. 7C,D), and the frequency of Ca2+ oscillations evoked by glutamate was significantly attenuated with mGluR5 L896V compared with WT (mean ± SEM, 20.7 ± 2.0 mHz for WT and 13.0 ± 1.0 mHz for L896V, for the number of analyzed cells, n = 26 for mGluR5 WT and n = 14 for mGluR5 L896V, p < 0.05).
Discussion
The group I mGluRs (mGluR1 and mGluR5) are postsynaptic Gq-coupled GPCRs that share high sequence homology, a similar pharmacological profile, and common downstream signal transduction pathways (Conn and Pin, 1997). Activation of either mGluR1 or mGluR5 can induce LTD (Volk et al., 2006). In addition, they share many interacting proteins, including Siah-1A, Homer, Tamalin, PP1γ1 (one catalytic isoform of protein phosphatase 1C), and Norbin (Tu et al., 1998; Ishikawa et al., 1999; Kitano et al., 2002; Croci et al., 2003; Wang et al., 2009).
Despite the well documented similarities between mGluR1 and mGluR5, several studies have suggested that these receptors have some distinct signaling properties and protein–protein interactions. In this study, we show that CaM binding is specific for mGluR5. Furthermore, our in vitro binding and coimmunoprecipitation assays demonstrate that a single residue (V909 for mGluR1; L896 for mGluR5) is a critical regulator for CaM binding and that exchanging this residue between mGluR1 and mGluR5 alters the CaM-binding properties of the group I mGluRs. CaM binding regulates constitutive surface expression and endocytosis of both group I mGluRs. The surface expression of CaM binding-competent mGluR1 (V909L) was significantly increased compared with that of WT, whereas CaM binding-deficient mGluR5 (L896V) showed a decrease in surface expression. Moreover, when group I mGluRs are activated with DHPG, the CaM-binding forms of mGluR1/5 induced more internalization of AMPA receptors, consistent with increased surface expression of the mGluRs leading to enhanced intracellular signaling. Since CaM is critical for mGluR5 trafficking (Lee et al., 2008), it is possible that some of the observed differences in group I mGluR signaling may be a result of differential CaM binding.
A previous study has shown that the group I mGluR-binding protein, Siah-1, competes with CaM for binding to both mGluR5 and GST-mGluR1 (R1–3) (Ishikawa et al., 1999). In contrast, our current study shows that mGluR1, unlike mGluR5, does not associate with CaM. However, the authors did not show the mGluR1-CaM interaction data. In addition, they reported that a greater amount of CaM was required for the competition assay with Siah-1 for mGluR1 binding (Ishikawa et al., 1999), suggesting that the interaction was significantly weaker than the one observed between mGluR5 and CaM. We now show that a single residue (V909 for mGluR1 and L896 for mGluR5) determines the CaM-binding properties of mGluR5 and mGluR1.
It is known that the CaM binding consensus sequence is relatively loose (Rhoads and Friedberg, 1997). In general, CaM recruitment signaling (CRS) motifs consist of hydrophobic and basic residues, and a 15–30 aa stretch that tends to form an α-helix. According to structural analyses of CaM–target complexes, CaM binding occurs through bulky hydrophobic residues, Phe, Trp, Ile, Leu, or Val (Rhoads and Friedberg, 1997; Yap et al., 2000). Based on the distance between key hydrophobic residues, the CRS motifs are categorized into four classes: 1–14, 1–10, IQ, and other (Rhoads and Friedberg, 1997; Yap et al., 2000). Based on sequence alignments, mGluR5 is categorized into the unclassified group, meaning that CaM-binding motifs in mGluR5 are more divergent than those of other CaM-binding proteins. Although both valine and leucine are hydrophobic, we find that this substitution profoundly affects CaM binding of group I mGluRs; a structural explanation for this difference awaits further studies.
Minakami et al. (1997), using in vitro binding and gel-shift assays, previously described two CaM-binding sites in the mGluR5 C-terminal region, site I (amino acids 841–869 in rat mGluR5) and site II (amino acids 889–917). However, a more recent study, describing the Norbin–mGluR5 interaction and the competition between Norbin and CaM for binding to mGluR5, reported that CaM binding was only disrupted by a site II mutation and not a mutation in site I (Wang et al., 2009). Hence, we directly investigated the ability of mGluR5 site I (residues 841–869 and 841–885) to associate with CaM and found little, if any, detectable CaM binding in these regions (data not shown). In contrast, the regions encompassing site II (residues 889–917 and 858–907) display robust binding to CaM (data not shown). The L896 residue that our data show is critical for mGluR5 binding to CaM, as well as the S901 PKC phosphorylation site (Lee et al., 2008), both reside within site II (Fig. 3B), and our data are therefore consistent with site II being a core CaM-binding domain.
We now show that increased surface expression of either mGluR1 or mGluR5 enhanced internalization of AMPA receptors. These results are consistent with CaM binding stabilizing receptors on the cell surface, which results in enhanced downstream signaling. GPCR interacting proteins can regulate receptor surface expression and activity. One example is the serotonin 1B receptor (5-HT1BR), which interacts with p11 (also know as S100A10, calpactin-I light chain, or annexin II light chain), a unique member of the S100 EF-hand protein family (Donato, 1999; Svenningsson et al., 2006). Coexpression of p11 and 5-HT1BR resulted in increased surface expression of 5-HT1BR in COS-7 cells compared with cells expressing only 5-HT1BR. In addition, COS-7 cells cotransfected with p11 and 5-HT1BR displayed enhanced 5-HT1BR signaling efficacy, compared with cells expressing only 5-HT1BR. These data are consistent with increased surface-expressed 5-HT1BR resulting in more signaling activity, although there is a possibility that p11 can enhance the activity of 5-HT1BR independent of the enhancement of 5-HT1BR surface expression.
PKC phosphorylates mGluR5 on serine 901 and disrupts CaM binding to mGluR5. The mutation of S901 to alanine allows mGluR5 to bind CaM regardless of PKC phosphorylation, resulting in increased surface mGluR5 following agonist treatment (Lee et al., 2008). In Ca2+ oscillation assays, in which agonist treatment stimulated mGluR5-induced Ca2+ oscillations, mGluR5 S901A exhibited an increase in Ca2+ oscillation frequency and more prolonged Ca2+ oscillations compared with WT mGluR5 (Lee et al., 2008), suggesting that more surface-expressed receptor can induce more prolonged intracellular signaling. Here, we demonstrated that CaM binding-competent forms of mGluR1 show increased Ca2+ signaling as evidenced by a leftward shift in the concentration–response curves. In addition mGluR5 L896V, a CaM binding-deficient form, displays a dramatic decrease in Ca2+ oscillation frequency. These data support an important role for CaM binding in regulating receptor trafficking and the function of group I mGluRs.
Several recent studies have reported a link between caveolin-1 and mGluR trafficking and signaling. Specifically, caveolin-1 overexpression reduces the internalization of mGluR1, whereas knockdown of caveolin-1 increases the internalization of mGluR1 (Francesconi et al., 2009). In addition, mutations in caveolin-1-binding motifs present within the intracellular loops of mGluR1 reduced surface expression of mGluR1. More recently, the same group showed that caveolin-1 knock-out (KO) mice display impaired mGluR-LTD (Takayasu et al., 2010). Together, these results suggest that the group I mGluR interacting protein (caveolin-1) regulates receptor trafficking and mGluR-mediated LTD, although interestingly, surface expression of group I mGluR was not significantly altered in hippocampal slices of caveolin-1 KO mice (Takayasu et al., 2010). A possible role for caveolin-1 in regulating CaM-dependent trafficking of group I mGluRs would be an important new line of investigation.
CaM also interacts with ionotropic glutamate receptors. Notably, the NMDA receptor NR1 subunit directly binds to CaM, modulating channel function and synaptic plasticity (Ehlers et al., 1996; Wyszynski et al., 1997; Wechsler and Teichberg, 1998; Zhang et al., 1998; Krupp et al., 1999). Specifically, CaM binding reduces the open probability of NMDA receptors. In addition, CaM competes with other interacting proteins, spectrin and α-actinin, for binding to NMDA receptors, thereby modulating channel function (Wyszynski et al., 1997; Wechsler and Teichberg, 1998; Krupp et al., 1999). CaM also acts as a signaling modulator by interacting with a variety of GPCRs: D2 dopamine receptor, 5-HT2A receptor, opioid receptor, V2 vasopressin receptor, and angiotensin II (AT1A) receptor (Thomas et al., 1999; Wang et al., 1999; Bofill-Cardona et al., 2000; Nickols et al., 2004; Turner and Raymond, 2005). In addition, other mGluRs (mGluR4, 5, 7, and 8) are known to interact with CaM (O'Connor et al., 1999; El Far et al., 2001). Furthermore, CaM antagonists also prevent inhibition of excitatory neurotransmission via presynaptic mGluRs (O'Connor et al., 1999).
Receptor trafficking is an important mechanism regulating GPCR desensitization and modulation (Ferguson, 2001; Gainetdinov et al., 2004). For several GPCRs, CaM is an important regulator of trafficking. For example, pharmacological inhibition of CaM prevents agonist-stimulated endocytosis of the 5-HT1A receptor (Della Rocca et al., 1999). In addition, the endocytosis of some mGluRs (mGluR5 and mGluR7) is affected by CaM binding. CaM binding to mGluR7 decreases its interaction with the synaptic PDZ (PSD-95/Dlg/ZO-1) protein PICK1 (PKC-interacting protein 1), resulting in an increase in mGluR7 endocytosis (Suh et al., 2008). Conversely, mGluR5 endocytosis is inhibited by CaM binding, and the receptor is stabilized on the cell surface (Lee et al., 2008).
Our findings now demonstrate that CaM binding differentially regulates mGluR1 and mGluR5, the two homologous members of the group I mGluR family. In particular, CaM binding regulates mGluR5 signaling and trafficking. Therefore, CaM acts as a specific and important modulator of group I mGluR function that confers distinct effects on mGluR5 trafficking and intracellular signaling.
Footnotes
The National Institute of Neurological Disorders and Stroke (NINDS) Intramural Research Program supported this work. We thank Dr. Carolyn Smith (NINDS Light Imaging Facility) for assistance with confocal microscopy, James Nagle and Debbie Kauffman (NINDS Sequencing Facility) for DNA sequence analysis, and Dr. Elizabeth Webber for critical reading of this manuscript.
References
- Akbar MT, Rattray M, Powell JF, Meldrum BS. Altered expression of group I metabotropic glutamate receptors in the hippocampus of amygdala-kindled rats. Brain Res Mol Brain Res. 1996;43:105–116. doi: 10.1016/s0169-328x(96)00162-3. [DOI] [PubMed] [Google Scholar]
- Banko JL, Hou L, Poulin F, Sonenberg N, Klann E. Regulation of eukaryotic initiation factor 4E by converging signaling pathways during metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2006;26:2167–2173. doi: 10.1523/JNEUROSCI.5196-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashir ZI, Jane DE, Sunter DC, Watkins JC, Collingridge GL. Metabotropic glutamate receptors contribute to the induction of long-term depression in the CA1 region of the hippocampus. Eur J Pharmacol. 1993;239:265–266. doi: 10.1016/0014-2999(93)91009-c. [DOI] [PubMed] [Google Scholar]
- Bofill-Cardona E, Kudlacek O, Yang Q, Ahorn H, Freissmuth M, Nanoff C. Binding of calmodulin to the D2-dopamine receptor reduces receptor signaling by arresting the G protein activation switch. J Biol Chem. 2000;275:32672–32680. doi: 10.1074/jbc.M002780200. [DOI] [PubMed] [Google Scholar]
- Bolshakov VY, Siegelbaum SA. Postsynaptic induction and presynaptic expression of hippocampal long-term depression. Science. 1994;264:1148–1152. doi: 10.1126/science.7909958. [DOI] [PubMed] [Google Scholar]
- Chowdhury S, Shepherd JD, Okuno H, Lyford G, Petralia RS, Plath N, Kuhl D, Huganir RL, Worley PF. Arc/Arg3.1 interacts with the endocytic machinery to regulate AMPA receptor trafficking. Neuron. 2006;52:445–459. doi: 10.1016/j.neuron.2006.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- Croci C, Sticht H, Brandstätter JH, Enz R. Group I metabotropic glutamate receptors bind to protein phosphatase 1C. Mapping and modeling of interacting sequences. J Biol Chem. 2003;278:50682–50690. doi: 10.1074/jbc.M305764200. [DOI] [PubMed] [Google Scholar]
- Della Rocca GJ, Maudsley S, Daaka Y, Lefkowitz RJ, Luttrell LM. Pleiotropic coupling of G protein-coupled receptors to the mitogen-activated protein kinase cascade. Role of focal adhesions and receptor tyrosine kinases. J Biol Chem. 1999;274:13978–13984. doi: 10.1074/jbc.274.20.13978. [DOI] [PubMed] [Google Scholar]
- Donato R. Functional roles of S100 proteins, calcium-binding proteins of the EF-hand type. Biochim Biophys Acta. 1999;1450:191–231. doi: 10.1016/s0167-4889(99)00058-0. [DOI] [PubMed] [Google Scholar]
- Ehlers MD, Zhang S, Bernhadt JP, Huganir RL. Inactivation of NMDA receptors by direct interaction of calmodulin with the NR1 subunit. Cell. 1996;84:745–755. doi: 10.1016/s0092-8674(00)81052-1. [DOI] [PubMed] [Google Scholar]
- El Far O, Bofill-Cardona E, Airas JM, O'Connor V, Boehm S, Freissmuth M, Nanoff C, Betz H. Mapping of calmodulin and Gbetagamma binding domains within the C-terminal region of the metabotropic glutamate receptor 7A. J Biol Chem. 2001;276:30662–30669. doi: 10.1074/jbc.M102573200. [DOI] [PubMed] [Google Scholar]
- Enz R. The trick of the tail: protein-protein interactions of metabotropic glutamate receptors. Bioessays. 2007;29:60–73. doi: 10.1002/bies.20518. [DOI] [PubMed] [Google Scholar]
- Ferguson SS. Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharmacol Rev. 2001;53:1–24. [PubMed] [Google Scholar]
- Ferraguti F, Shigemoto R. Metabotropic glutamate receptors. Cell Tissue Res. 2006;326:483–504. doi: 10.1007/s00441-006-0266-5. [DOI] [PubMed] [Google Scholar]
- Francesconi A, Duvoisin RM. Role of the second and third intracellular loops of metabotropic glutamate receptors in mediating dual signal transduction activation. J Biol Chem. 1998;273:5615–5624. doi: 10.1074/jbc.273.10.5615. [DOI] [PubMed] [Google Scholar]
- Francesconi A, Kumari R, Zukin RS. Regulation of group I metabotropic glutamate receptor trafficking and signaling by the caveolar/lipid raft pathway. J Neurosci. 2009;29:3590–3602. doi: 10.1523/JNEUROSCI.5824-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainetdinov RR, Premont RT, Bohn LM, Lefkowitz RJ, Caron MG. Desensitization of G protein-coupled receptors and neuronal functions. Annu Rev Neurosci. 2004;27:107–144. doi: 10.1146/annurev.neuro.27.070203.144206. [DOI] [PubMed] [Google Scholar]
- Hermans E, Challiss RA. Structural, signalling and regulatory properties of the group I metabotropic glutamate receptors: prototypic family C G-protein-coupled receptors. Biochem J. 2001;359:465–484. doi: 10.1042/0264-6021:3590465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou L, Klann E. Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2004;24:6352–6361. doi: 10.1523/JNEUROSCI.0995-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Kayser MS, Bear MF. Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science. 2000;288:1254–1257. doi: 10.1126/science.288.5469.1254. [DOI] [PubMed] [Google Scholar]
- Ishikawa K, Nash SR, Nishimune A, Neki A, Kaneko S, Nakanishi S. Competitive interaction of seven in absentia homolog-1A and Ca2+/calmodulin with the cytoplasmic tail of group 1 metabotropic glutamate receptors. Genes Cells. 1999;4:381–390. doi: 10.1046/j.1365-2443.1999.00269.x. [DOI] [PubMed] [Google Scholar]
- Johnston J, Delaney KR. Synaptic activation of T-type Ca2+ channels via mGluR activation in the primary dendrite of mitral cells. J Neurophysiol. 2010;103:2557–2569. doi: 10.1152/jn.00796.2009. [DOI] [PubMed] [Google Scholar]
- Kammermeier PJ, Yun J. Activation of metabotropic glutamate receptor 1 dimers requires glutamate binding in both subunits. J Pharmacol Exp Ther. 2005;312:502–508. doi: 10.1124/jpet.104.073155. [DOI] [PubMed] [Google Scholar]
- Kawabata S, Tsutsumi R, Kohara A, Yamaguchi T, Nakanishi S, Okada M. Control of calcium oscillations by phosphorylation of metabotropic glutamate receptors. Nature. 1996;383:89–92. doi: 10.1038/383089a0. [DOI] [PubMed] [Google Scholar]
- Kelly L, Farrant M, Cull-Candy SG. Synaptic mGluR activation drives plasticity of calcium-permeable AMPA receptors. Nat Neurosci. 2009;12:593–601. doi: 10.1038/nn.2309. [DOI] [PubMed] [Google Scholar]
- Kim CH, Braud S, Isaac JT, Roche KW. Protein kinase C phosphorylation of the metabotropic glutamate receptor mGluR5 on Serine 839 regulates Ca2+ oscillations. J Biol Chem. 2005;280:25409–25415. doi: 10.1074/jbc.M502644200. [DOI] [PubMed] [Google Scholar]
- Kim CH, Lee J, Lee JY, Roche KW. Metabotropic glutamate receptors: phosphorylation and receptor signaling. J Neurosci Res. 2008;86:1–10. doi: 10.1002/jnr.21437. [DOI] [PubMed] [Google Scholar]
- Kitano J, Kimura K, Yamazaki Y, Soda T, Shigemoto R, Nakajima Y, Nakanishi S. Tamalin, a PDZ domain-containing protein, links a protein complex formation of group 1 metabotropic glutamate receptors and the guanine nucleotide exchange factor cytohesins. J Neurosci. 2002;22:1280–1289. doi: 10.1523/JNEUROSCI.22-04-01280.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupp JJ, Vissel B, Thomas CG, Heinemann SF, Westbrook GL. Interactions of calmodulin and alpha-actinin with the NR1 subunit modulate Ca2+-dependent inactivation of NMDA receptors. J Neurosci. 1999;19:1165–1178. doi: 10.1523/JNEUROSCI.19-04-01165.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavezzari G, McCallum J, Dewey CM, Roche KW. Subunit-specific regulation of NMDA receptor endocytosis. J Neurosci. 2004;24:6383–6391. doi: 10.1523/JNEUROSCI.1890-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Lee J, Choi KY, Hepp R, Lee JY, Lim MK, Chatani-Hinze M, Roche PA, Kim DG, Ahn YS, Kim CH, Roche KW. Calmodulin dynamically regulates the trafficking of the metabotropic glutamate receptor mGluR5. Proc Natl Acad Sci U S A. 2008;105:12575–12580. doi: 10.1073/pnas.0712033105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, Huber KM. Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron. 2010;65:445–459. doi: 10.1016/j.neuron.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minakami R, Jinnai N, Sugiyama H. Phosphorylation and calmodulin binding of the metabotropic glutamate receptor subtype 5 (mGluR5) are antagonistic in vitro. J Biol Chem. 1997;272:20291–20298. doi: 10.1074/jbc.272.32.20291. [DOI] [PubMed] [Google Scholar]
- Nash MS, Schell MJ, Atkinson PJ, Johnston NR, Nahorski SR, Challiss RA. Determinants of metabotropic glutamate receptor-5-mediated Ca2+ and inositol 1,4,5-trisphosphate oscillation frequency. Receptor density versus agonist concentration. J Biol Chem. 2002;277:35947–35960. doi: 10.1074/jbc.M205622200. [DOI] [PubMed] [Google Scholar]
- Nickols HH, Shah VN, Chazin WJ, Limbird LE. Calmodulin interacts with the V2 vasopressin receptor: elimination of binding to the C terminus also eliminates arginine vasopressin-stimulated elevation of intracellular calcium. J Biol Chem. 2004;279:46969–46980. doi: 10.1074/jbc.M407351200. [DOI] [PubMed] [Google Scholar]
- O'Connor V, El Far O, Bofill-Cardona E, Nanoff C, Freissmuth M, Karschin A, Airas JM, Betz H, Boehm S. Calmodulin dependence of presynaptic metabotropic glutamate receptor signaling. Science. 1999;286:1180–1184. doi: 10.1126/science.286.5442.1180. [DOI] [PubMed] [Google Scholar]
- Park S, Park JM, Kim S, Kim JA, Shepherd JD, Smith-Hicks CL, Chowdhury S, Kaufmann W, Kuhl D, Ryazanov AG, Huganir RL, Linden DJ, Worley PF. Elongation factor 2 and fragile X mental retardation protein control the dynamic translation of Arc/Arg3.1 essential for mGluR-LTD. Neuron. 2008;59:70–83. doi: 10.1016/j.neuron.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pin JP, Duvoisin R. The metabotropic glutamate receptors: structure and functions. Neuropharmacology. 1995;34:1–26. doi: 10.1016/0028-3908(94)00129-g. [DOI] [PubMed] [Google Scholar]
- Rhoads AR, Friedberg F. Sequence motifs for calmodulin recognition. FASEB J. 1997;11:331–340. doi: 10.1096/fasebj.11.5.9141499. [DOI] [PubMed] [Google Scholar]
- Roche KW, Huganir RL. Synaptic expression of the high-affinity kainate receptor subunit KA2 in hippocampal cultures. Neuroscience. 1995;69:383–393. doi: 10.1016/0306-4522(95)00253-f. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Philpot BD, Huber KM, Dong X, Fallon JR, Bear MF. Internalization of ionotropic glutamate receptors in response to mGluR activation. Nat Neurosci. 2001;4:1079–1085. doi: 10.1038/nn746. [DOI] [PubMed] [Google Scholar]
- Steinhoff M, Vergnolle N, Young SH, Tognetto M, Amadesi S, Ennes HS, Trevisani M, Hollenberg MD, Wallace JL, Caughey GH, Mitchell SE, Williams LM, Geppetti P, Mayer EA, Bunnett NW. Agonists of proteinase-activated receptor 2 induce inflammation by a neurogenic mechanism. Nat Med. 2000;6:151–158. doi: 10.1038/72247. [DOI] [PubMed] [Google Scholar]
- Suh YH, Pelkey KA, Lavezzari G, Roche PA, Huganir RL, McBain CJ, Roche KW. Corequirement of PICK1 binding and PKC phosphorylation for stable surface expression of the metabotropic glutamate receptor mGluR7. Neuron. 2008;58:736–748. doi: 10.1016/j.neuron.2008.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svenningsson P, Chergui K, Rachleff I, Flajolet M, Zhang X, El Yacoubi M, Vaugeois JM, Nomikos GG, Greengard P. Alterations in 5-HT1B receptor function by p11 in depression-like states. Science. 2006;311:77–80. doi: 10.1126/science.1117571. [DOI] [PubMed] [Google Scholar]
- Takayasu Y, Takeuchi K, Kumari R, Bennett MV, Zukin RS, Francesconi A. Caveolin-1 knockout mice exhibit impaired induction of mGluR-dependent long-term depression at CA3-CA1 synapses. Proc Natl Acad Sci U S A. 2010 doi: 10.1073/pnas.1015553107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas WG, Pipolo L, Qian H. Identification of a Ca2+/calmodulin-binding domain within the carboxyl-terminus of the angiotensin II (AT1A) receptor. FEBS Lett. 1999;455:367–371. doi: 10.1016/s0014-5793(99)00904-7. [DOI] [PubMed] [Google Scholar]
- Tu JC, Xiao B, Yuan JP, Lanahan AA, Leoffert K, Li M, Linden DJ, Worley PF. Homer binds a novel proline-rich motif and links group 1 metabotropic glutamate receptors with IP3 receptors. Neuron. 1998;21:717–726. doi: 10.1016/s0896-6273(00)80589-9. [DOI] [PubMed] [Google Scholar]
- Turner JH, Raymond JR. Interaction of calmodulin with the serotonin 5-hydroxytryptamine2A receptor. A putative regulator of G protein coupling and receptor phosphorylation by protein kinase C. J Biol Chem. 2005;280:30741–30750. doi: 10.1074/jbc.M501696200. [DOI] [PubMed] [Google Scholar]
- Volk LJ, Daly CA, Huber KM. Differential roles for group 1 mGluR subtypes in induction and expression of chemically induced hippocampal long-term depression. J Neurophysiol. 2006;95:2427–2438. doi: 10.1152/jn.00383.2005. [DOI] [PubMed] [Google Scholar]
- Wang D, Sadée W, Quillan JM. Calmodulin binding to G protein-coupling domain of opioid receptors. J Biol Chem. 1999;274:22081–22088. doi: 10.1074/jbc.274.31.22081. [DOI] [PubMed] [Google Scholar]
- Wang H, Westin L, Nong Y, Birnbaum S, Bendor J, Brismar H, Nestler E, Aperia A, Flajolet M, Greengard P. Norbin is an endogenous regulator of metabotropic glutamate receptor 5 signaling. Science. 2009;326:1554–1557. doi: 10.1126/science.1178496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waung MW, Pfeiffer BE, Nosyreva ED, Ronesi JA, Huber KM. Rapid translation of Arc/Arg3.1 selectively mediates mGluR-dependent LTD through persistent increases in AMPAR endocytosis rate. Neuron. 2008;59:84–97. doi: 10.1016/j.neuron.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wechsler A, Teichberg VI. Brain spectrin binding to the NMDA receptor is regulated by phosphorylation, calcium and calmodulin. EMBO J. 1998;17:3931–3939. doi: 10.1093/emboj/17.14.3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyszynski M, Lin J, Rao A, Nigh E, Beggs AH, Craig AM, Sheng M. Competitive binding of alpha-actinin and calmodulin to the NMDA receptor. Nature. 1997;385:439–442. doi: 10.1038/385439a0. [DOI] [PubMed] [Google Scholar]
- Xiao MY, Zhou Q, Nicoll RA. Metabotropic glutamate receptor activation causes a rapid redistribution of AMPA receptors. Neuropharmacology. 2001;41:664–671. doi: 10.1016/s0028-3908(01)00134-4. [DOI] [PubMed] [Google Scholar]
- Yap KL, Kim J, Truong K, Sherman M, Yuan T, Ikura M. Calmodulin target database. J Struct Funct Genomics. 2000;1:8–14. doi: 10.1023/a:1011320027914. [DOI] [PubMed] [Google Scholar]
- Zhang S, Ehlers MD, Bernhardt JP, Su CT, Huganir RL. Calmodulin mediates calcium-dependent inactivation of N-methyl-d-aspartate receptors. Neuron. 1998;21:443–453. doi: 10.1016/s0896-6273(00)80553-x. [DOI] [PubMed] [Google Scholar]