

Abstract
FDH (10-formyltetrahydrofolate dehydrogenase, the product of the ALDH1L1 gene), a major folate-metabolizing enzyme in the cytosol, is involved in the regulation of cellular proliferation. We have previously demonstrated that FDH is strongly and ubiquitously down-regulated in malignant human tumors and cancer cell lines. Here, we report that promoter methylation is a major mechanism controlling FDH levels in human cancers. A computational analysis has identified an extensive CpG island in the ALDH1L1 promoter region. It contains 96 CpG pairs and covers the region between −525 and +918 bp of the ALDH1L1 gene including the promoter, the entire exon 1, and a part of intron 1 immediately downstream of the exon. Bisulfite sequencing analysis revealed extensive methylation of the island (76%-95% of CpGs) in cancer cell lines. In agreement with these findings, treatment of FDH-deficient A549 cells with the methyltransferase inhibitor 5-aza-2′-deoxycytidine restored FDH expression. Analysis of the samples from patients with lung adenocarcinomas demonstrated methylation of the ALDH1L1 CpG island in tumor samples and a total lack of methylation in respective normal tissues. The same phenomenon was observed in liver tissues: the CpG island was methylation free in DNA extracted from normal hepatocytes but was extensively methylated in a hepatocellular carcinoma. Levels of ALDH1L1 mRNA and protein correlated with the methylation status of the island, with tumor samples demonstrating down-regulation of expression or even complete silencing of the gene. Our studies have also revealed that exon 1 significantly increases transcriptional activity of ALDH1L1 promoter in a luciferase reporter assay. Interestingly, the exon is extensively methylated in samples with a strongly down-regulated or silenced ALDH1L1 gene.
Keywords: ALDH1L1, CpG island methylation, folate metabolism, lung adenocarcinoma
Introduction
Tumors demonstrate significantly altered protein expression patterns compared to normal tissues, and some of the unique properties of cancer cells are acquired through a stepwise accumulation of heritable changes in expression of proto-oncogenes and tumor suppressor genes.1,2 Many other proteins, which are not tumor suppressors or oncogenes, are also up- or down-regulated in cancers.3 This provides a selective advantage for uncontrolled proliferation, one of the hallmarks of cancer.4 ALDH1L1 is one of the genes in which expression is strongly down-regulated in human cancers.5 The product of this gene, 10-formyltetrahydrofolate dehydrogenase (FDH), is an abundant cytosolic enzyme involved in folate pathways.6 Folate coenzymes are essential for cellular metabolism because they function as carriers of 1-carbon groups, participating in reactions of de novo nucleotide biosynthesis and amino acid biogenesis.7 Of the latter reactions, the remethylation of homocysteine to methionine is especially important because it provides the substrate for biosynthesis of S-adenosylmethionine, the universal methyl group donor in methylation reactions in the cell.8 FDH catalyzes the NADP+-dependent oxidative deformylation of 10-formyltetrahydrofolate to produce CO2 and tetrahydrofolate.6 This reaction removes 1-carbon groups from the reduced folate pool and restores the pool of tetrahydrofolate, the only form of the coenzyme in folate pathways capable of accepting such groups.
The FDH reaction limits the flow of folate-bound carbon groups towards biosynthetic processes.6 As such, this reaction controls the contribution of folate metabolism to cellular proliferation. The fact that FDH is present at a very high level in several tissues (it comprises about 1% of the total cytosolic protein in hepatocytes9) underscores the importance of this pathway for cellular function. The observation that FDH is a strong marker of astrocytes in the rat brain suggests a function for the enzyme in the nervous system.10 Of note, FDH expression is tissue specific, with some tissues lacking detectable levels of the protein.5 The ubiquitous lack of the enzyme was also revealed in different types of cancers compared to corresponding normal tissues.5,11 Furthermore, cancer cell lines also do not express the enzyme at detectable levels, and ectopic expression of the enzyme induces strong cytotoxicity and apoptosis.5,12 These findings indicate that disabling the FDH-dependent pathway is advantageous to rapidly proliferating cells.
While gain, loss, and mutation of genes have long been known to contribute to tumorigenesis, it has been increasingly recognized that epigenetic mechanisms play an important role in this process through the regulation of gene transcription.13,14 The main epigenetic modification of the human genome is methylation of cytosine residues in CpG dinucleotides.13,14 CpG dinucleotides are frequently clustered in small regions (0.5 to several kb) located near 5′ ends of genes, called “CpG islands”.13,15 Despite increased CpG density relative to the rest of the genome, CpG islands tend to remain remarkably unmethylated in normal cells.13,15 However, hypermethylation within CpG islands of tumor suppressor gene promoters is among the earliest and most common alterations in human malignancies.13-15 There is now a growing list of genes that display CpG island hypermethylation that results in transcriptional silencing associated with cancer.16 Thus, promoter methylation has emerged as a fundamental molecular lesion associated with the loss of tumor suppressor gene expression early in tumorigenesis.17
In humans, the ALDH1L1 gene is organized into 23 exons spanning about 77 kb on the long arm of chromosome 3 (3q21.2). The first exon of the gene is entirely nontranslated, and a long intron (about 22,000 bp) separates exons 1 and 2 (Fig. 1A). Computational analysis of the ALDH1L1 has identified a CpG island at the 5′ end of the gene, adjacent to the transcription start site (Fig. 1). In the present study, we have evaluated the methylation patterns within this region in cancer cell lines, normal human tissues, and human lung adenocarcinomas. We have further compared the ALDH1L1 CpG methylation between normal and tumor lung tissues from the same patients and correlated it with FDH expression levels.
Figure 1.
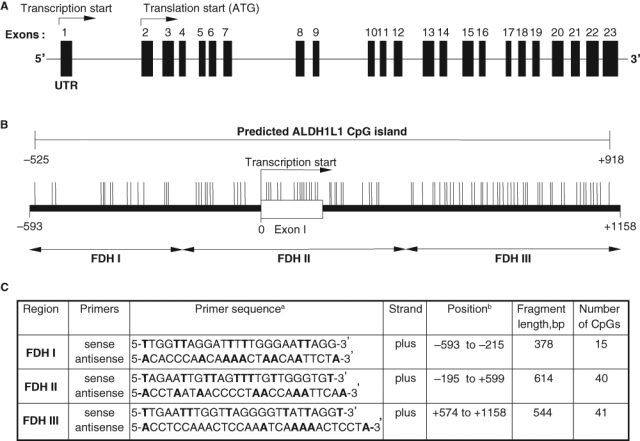
Schematic representation of human ALDH1L1 gene locus (A) and the CpG island in the promoter region (B). Double-headed arrows indicate location of PCR primers (C) for bisulfite genomic sequencing. CpG sites and their genomic positions within the CpG island (–525 to +918) are represented by vertical lines. Nucleotide positions are numbered relative to the transcriptional start site.
Results
The ALDH1L1 gene contains a CpG island near the transcription start site
Our previous studies have demonstrated that FDH is strongly and ubiquitously down-regulated in cancer cell lines and human tumors at both mRNA and protein levels.5 Such a phenomenon is often associated with increased methylation of genes near the promoter regions. Analysis of the ALDH1L1 gene (near the transcription start site) using CpG Island Searcher18 (www.cpgislands.com) revealed the presence of a CpG island totaling 96 CpG pairs and spanning a region of 1,443 bp (–525 to +918). This CpG island covers the region upstream of the transcription start, the first exon (which is nontranslatable), and part of the first intron adjacent to exon 1 (Fig. 1). Analysis of this 1,443-bp region using Proscan (www-bimas.cit.nih.gov/molbio/proscan), a promoter identification program,19 highlighted the portion of the sequence from –10 to –260 (relative to the transcriptional start site) as the putative promoter. The ALDH1L1 promoter lacks canonical TATA or CCAAT boxes often found within the first hundred nucleotides upstream of the transcription start. Instead, multiple SP1 transcription factor binding sites were predicted, which is a common feature of TATA-less promoters.20 Of note, the putative promoter contains a significant number of CpGs (21 dinucleotides) and is located in the middle of the CpG island.
Methylation status of the ALDH1L1 CpG island in cell lines.
We have examined the extent of the CpG island methylation in FDH-deficient A549, HepG2, and HCT116 cell lines using a bisulfite sequencing approach.21 In all examined cell lines, the ALDH1L1 CpG island was extensively methylated (Fig. 2): of the total of 96 CpGs, 74 in A549 cells (77%), 91 in HCT116 cells (95%), and 56 in HepG2 cells (58%) were methylated. In HepG2 cells, an additional 17 CpGs (17.5% of all CpGs in the region) were defined as partially methylated: in each case, only 50% of analyzed clones demonstrated methylation at a specific site (Table 1 of Suppl. Material). In HEK293 cells, which express detectable levels of FDH, the proximal promoter and the first exon remained totally nonmethylated, while some methylation (5 CpGs) was seen in the intron. Interestingly, in this cell line, partial methylation (50% of analyzed clones showed methylation at each specific site) was seen in the distal promoter (Fig. 2). It appears that this partial methylation does not affect the ability of HEK293 cells to express FDH.
Figure 2.
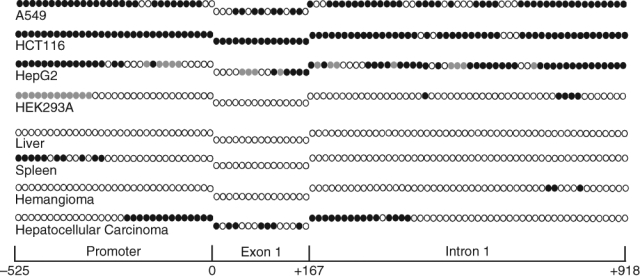
Methylation patterns of the ALDH1L1 CpG island in human tissues and cell lines. Individual CpG dinucleotides (positioned sequentially) are as follows: open circles = nonmethylated; closed circles = methylated; gray circles = methylated in one of the alleles (10 to 15 clones for each fragment were analyzed). Schematic at the bottom allows the assignment of CpGs to 1 of the 3 ALDH1L1 regions (promoter, exon 1, or intron 1).
Methylation status of the ALDH1L1 CpG island in normal human tissues
The fact that expression of ALDH1L1 in humans is highly tissue specific suggests tight gene regulation.5 In several tissues, FDH is present at very high levels, while there are tissues with undetectable levels of FDH mRNA.5 To study whether the difference in ALDH1L1 methylation contributes to its tissue-specific expression, we analyzed the methylation status of individual CpG dinucleotides in the human liver and spleen, organs with either very high or undetectable levels of FDH, respectively. Three different commercially available genomic DNA samples for each of the tissues were analyzed. These experiments revealed the lack of methylated CpGs in the ALDH1L1 island region in liver DNA (Fig. 2). In contrast to liver samples, we observed methylation of the CpG pairs in the distal part of the ALDH1L1 promoter in DNA samples from the spleen (Fig. 2). However, the proximal promoter as well as exon 1 and the downstream intronic sequence remain completely unmethylated.
Restoration of FDH expression in A549 cells
In many instances, genes silenced by hypermethylation can be reactivated by treatment with 5-aza-2′-deoxycytidine (5-aza-dC), a potent and specific inhibitor of DNA methyltransferases.22 In the cell, this agent is incorporated into DNA and upon binding becomes covalently linked to DNA methyltransferases, thus blocking their activity.22 This further results in significant loss of CpG methylation after replication. After treatment of A549, HepG2, or HCT116 cells with 1 µM 5-aza-dC for 96 hours, we observed the appearance of FDH mRNA as well as protein (Fig. 3). These results are in agreement with studies that demonstrated decreased DNA methylation and increased expression of certain proteins in 5-aza-dC–treated cells,23-28 and they indicate that ALDH1L1 promoter methylation is the mechanism silencing this gene in tumors.
Figure 3.
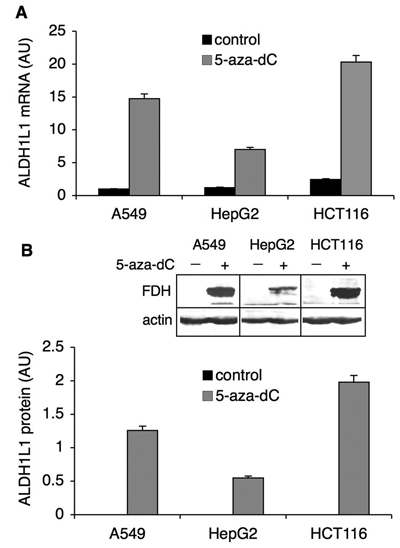
Reversal of ALDH1L1 silencing by 5-aza-dC. Levels of ALDH1L1 mRNA (A) and protein (B) before (control) and after the treatment with 5-aza-dC (1 µM). Levels of mRNA were measured by real-time PCR (normalized to β-actin mRNA and presented relative to the untreated control). Protein levels were quantified from Western blot assays (inset) and normalized to β-actin levels as a loading control. Average ± SD of 3 independent experiments is shown. AU = arbitrary units.
Identification of a core ALDH1L1 promoter
We examined the ALDH1L1 promoter using deletion analysis and luciferase reporter assays. Eleven different constructs were generated and tested for the ability to induce luciferase expression (Fig. 4A). The total of 5 ALDH1L1 promoter sequences of different length (from the region located upstream of the transcription start site) were analyzed (constructs II, IV, VI, VIII, and X) (Fig. 4A). These experiments revealed that the ALDH1L1 promoter itself is a weak one: no activation of luciferase expression above basal levels (obtained with the promoterless vector) was observed (Fig. 4C). We further tested the effect of exon 1 on the activity of the ALDH1L1 promoter. In these experiments, the entire exon 1 sequence was added to the same basic promoter constructs (Fig. 4A). Interestingly, inclusion of the exon into the constructs significantly enhanced the promoter activity, and this enhancement occurred with all tested constructs (Fig. 4C). In agreement with our computational analysis of the promoter, the examination of exon-containing constructs indicated that the core ALDH1L1 promoter locates within about 300 nucleotides upstream of the transcription start site.
Figure 4.
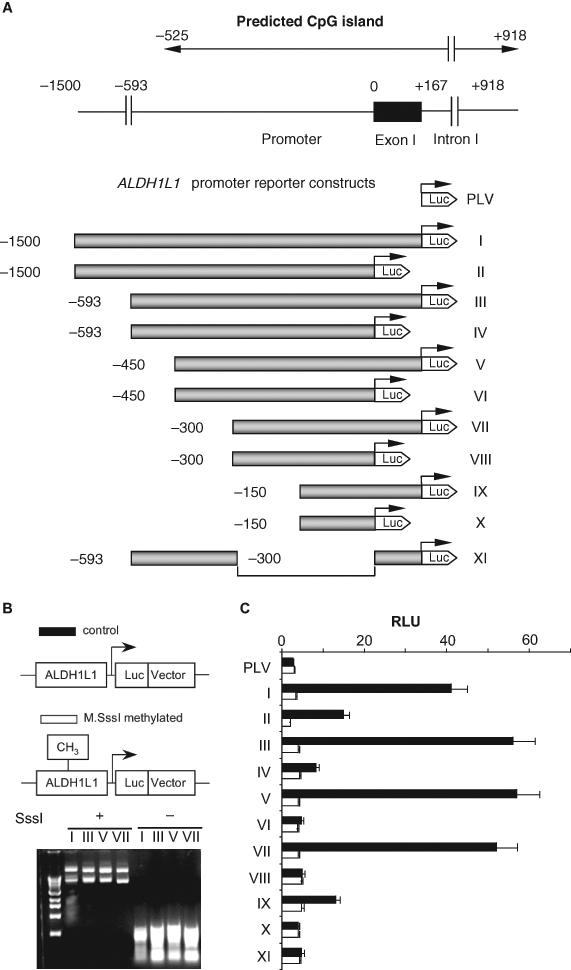
The effect of in vitro methylation on the activity of ALDH1L1 promoter (luciferase assays). (A) Schematic representation of constructs used in a luciferase assay (in each construct, the promoter is shown as a shaded bar, the luciferase gene is shown as a white arrowhead, and the transcription start site is indicated by arrows). Position of the ALDH1L1 CpG island (top diagram) is indicated. (B) Methylation analysis of reporter constructs. Top panel: schematic presentation of methylated and unmethylated plasmids. Bottom panel: agarose gel electrophoresis of methylated (SssI +) and nonmethylated (SssI –) reporter plasmids digested with methylation-sensitive restriction endonucleases, HpaII and HhaI (both enzymes do not cut methylated DNA). (C) Transcriptional activity of ALDH1L1 promoter constructs transiently transfected into A549 cells: closed bars = nonmethylated constructs; open bars = methylated constructs. Luciferase activity (±SD) of 3 independent experiments is shown. PLV = promoterless vector (pGL-basic plasmid).
Inhibition of the transcriptional activity of the ALDH1L1 promoter by in vitro methylation
We further studied the effect of in vitro methylation of the ALDH1L1 promoter/luciferase constructs on the protein expression in A549 cells. To confirm the completeness of in vitro methylation, plasmid constructs were subjected to the treatment with methylation-sensitive restriction endonucleases, HpaII and HhaI. The lack of cleaved fragments indicated that methylation was complete (Fig. 4B). In these experiments, a strong decrease in luciferase activity was observed upon methylation as compared to the activity in control cells transfected with nonmethylated plasmid (Fig. 4C). In fact, methylation completely disabled the promoter’s ability to direct the reporter gene expression. Interestingly, these experiments revealed that methylation of the constructs, which include exon 1, abolished the previously observed enhancing effect on transcription. This finding strongly indicates that methylation within exon 1 is involved in the regulation of ALDH1L1 expression.
ALDH1L1 CpG island methylation correlates with the levels of FDH in lung adenocarcinomas
We studied whether ALDH1L1 CpG island methylation plays a role in protein silencing in human tumors. Our initial experiments with samples from liver tissues demonstrated strong methylation of the ALDH1L1 promoter region in hepatocellular carcinoma (Fig. 2). In support of the role of methylation in ALDH1L1 silencing, the promoter is completely nonmethylated in the normal liver (Fig. 2). Interestingly, analysis of the ALDH1L1 promoter in a benign liver tumor hemangioma also revealed the lack of methylation (Fig. 2). To further evaluate the role of ALDH1L1 methylation in gene silencing, we examined samples from lung adenocarcinomas and samples from normal lung tissue of the same individuals. A total of 10 matched pairs were analyzed (Fig. 5). All samples from normal lung tissues revealed a complete lack of methylation at any of the 96 CpG dinucleotides in the ALDH1L1 CpG island. Accordingly, these tissues contained ALDH1L1 mRNA and protein (Fig. 5). In contrast, a variable degree of methylation was observed in all tumor samples. One sample (#1262) showed partial methylation in one CpG in the intron region (in 5 of 11 analyzed clones, this site was methylated) (Table 3 of Suppl. Material) (Fig. 5). This weak methylation did not result in a decrease in ALDH1L1 mRNA or protein levels (Fig. 5). Similarly, another sample with only partial methylation of 4 CpGs (#3549) did not show a decrease in the mRNA or protein levels (Fig. 5). Conversely, 8 samples with more extensive methylation of the island demonstrated a strong decrease in mRNA and protein levels, with 4 samples appearing to have completely silenced ALDH1L1 (Fig 5). Of note, these 4 samples (#1258, #1245, #1261, and #3905) were those with most extensive methylation within exon 1 (Fig. 5). Paired t test analysis of normal tissues and adenocarcinomas demonstrated that differences in CpG island methylation and ALDH1L1 protein levels were highly statistically significant (P = 0.0001) (Fig. 5).
Figure 5.
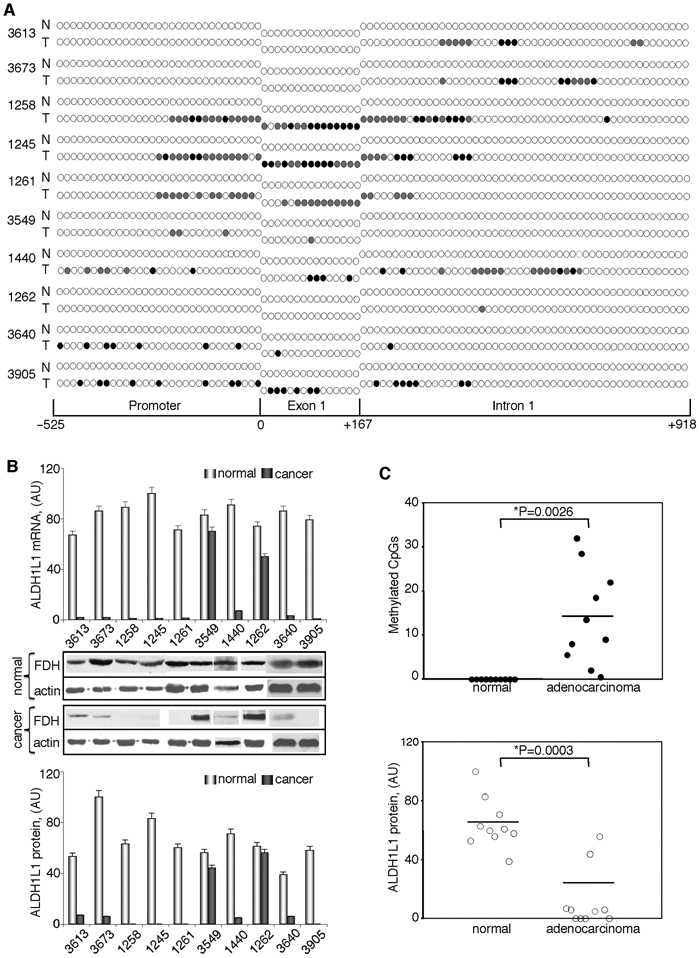
ALDH1L1 is silenced in human lung adenocarcinomas via methylation within its CpG island. CpG island methylation patterns (A) and alterations in mRNA and protein levels (B) of ALDH1L1 in patients with lung adenocarcinomas. Open circle = nonmethylated CpG; closed circle = methylated CpG; gray circles = CpG methylated in one of the alleles (8 to 14 clones for each fragment were analyzed). Schematic depicts position of CpGs within ALDH1L1 promoter, exon 1, or intron 1. Levels of mRNA were measured by real-time PCR and are presented in arbitrary units (top bar graph). Average ± SD of 3 independent experiments is shown. Protein levels were assessed by Western blot assays; levels of β-actin are shown as loading controls (middle panel). Bottom bar graph: quantification of the Western blots normalized by actin levels. (C) Paired t test analysis of the CpG island methylation (top panel) and ALDH1L1 protein levels (bottom panel) in matching normal and adenocarcinoma tissue samples. Statistically significant differences are indicated by an asterisk. Y-axis in the upper panel shows the number of methylated CpGs (partially methylated sites were assigned the value of 0.5; in all cases, partial methylation was 50% if an even number of clones was analyzed and close to this value if an odd number of clones was analyzed) (Table 3 of Suppl. Material).
Discussion
While cancer cells display dramatically different protein expression patterns compared to normal cells,2 there are not too many examples of the lack of an abundant metabolic enzyme such as FDH. Most of the proteins in folate pathways, in contrast to FDH, demonstrate increased expression in tumors. The well-known examples are the folate receptor, which is highly expressed in cancer cells, and DHFR, in which elevation in response to antifolate treatment is one of the main mechanisms of the resistance to these drugs.29,30 Interestingly, one of the folate enzymes, glycine N-methyltransferase, similarly to FDH is strongly down-regulated in cancers and exerts inhibitory effects on tumorigenesis; it was suggested to be a putative tumor susceptibility gene.31 Likewise, the evidence from our laboratory suggests that FDH is a putative tumor suppressor. In addition to its strong down-regulation in cancers (with complete silencing in many cases),5,11,32 FDH induces distinct apoptotic cascades with c-Jun N-terminal kinases and p53 as downstream mediators.33-35 Canonical tumor suppressor genes, however, have been viewed as those deleted or mutated in cancer,36,37 which is not the case for ALDH1L1. Numerous studies have also demonstrated epigenetic alterations of tumor suppressor genes in human cancers, which often result in repression of the transcription.38 It has been proposed to classify tumor suppressor genes that are unaltered at the genetic level but have diminished expression as class II, in contrast to class I genes that are mutated or deleted in cancers.39 Since class II tumor suppressors are often silenced through promoter methylation, methylated genes are potential candidates as tumor suppressors.40,41 In this regard, our study has demonstrated that methylation of ALDH1L1 CpG island is a major mechanism regulating expression of the gene in tumors.
Analysis of the human genome has indicated that about 72% of genes contain CpG islands within their promoters.42 However, cytosines of CpG pairs located in the islands typically stay nonmethylated in normal cells.38 Since methylation of CpG islands is a mechanism to repress gene expression, the abundance of the islands suggests that many promoters can be potentially regulated in a methylation-dependent manner, a type of epigenetic regulation especially common in tumorigenesis. It should be noted that genomes of malignant cells are generally hypomethylated compared to normal cells, and the actual number of methylated CpGs is still relatively low compared to nonmethylated islands (e.g., in lung cancers, only about 0.5% to 3% of all CpG islands are methylated43,44). Thus, apparently only a limited number of genes are silenced in cancers through this mechanism, and it would be expected that these are the genes for which loss provides selective advantage to malignant cells. In support of this notion, hypermethylation of genes for many tumor suppressors was detected in malignant cells including RB, p16, RASSF1A, GSTP1, DAPK, and TIMP3.38,44,45 Epigenetic regulation of some genes, however, could be associated with a later stage of tumor development rather than be a causative factor in the malignant transformation.44
Typically, CpG islands are found at the 5′ ends of genes near the transcription start site often covering the promoter region, but such islands have also been identified far downstream of the transcription start site.46,47 In several cases, it has been demonstrated that the methylation of islands remotely located from the transcription start site also results in gene silencing,48-50 and both the methylation within exons as well as within introns has been shown to regulate gene expression.51-53 In this regard, methylation of exon 1 of genes would be expected to be a common trend because CpG islands are often extended into the first exon and intron of a gene.44 Likewise, it has been suggested that the maintenance of the region extending several hundred bases downstream of the transcription start site methylation free is important for efficient transcription.54
It is hard to predict how many CpGs should be methylated to silence a gene. In some cases, methylation of 1 or 2 CpG sites is sufficient to strongly repress the activity of a promoter, and it has been shown that methylation of as little as 7% of CpGs resulted in gene quiescence in experiments with episomes.46 Our study indicates that methylation of a few CpGs within the ALDH1L1 CpG island is sufficient to significantly down-regulate this gene expression. More extensive methylation seen in cancer cell lines and several patient samples resulted in complete gene silencing. The ALDH1L1 CpG island covers not only the promoter but also the entire exon 1 and the adjacent intronic region. Analysis of the 5′ end of the human ALDH1L1 gene for the presence of consensus transcription factor binding sites revealed that the sequence of about 600 bp immediately upstream of the transcription start has a high density of such sites, especially in the region from 0 to –250 bp. In contrast, exon 1 and the adjacent intronic region have a very low density of such sites. In agreement with this analysis, our studies have indicated that the core promoter lies within about 300 nucleotides upstream of the transcription start site. In our experiments, the promoter itself revealed very weak activation of luciferase transcription, perhaps suggesting a requirement for enhancer elements. Interestingly, the addition of exon 1 to the reporter vector considerably enhanced luciferase expression that indicates an important role of the exon in the regulation of ALDH1L1 gene transcription. In contrast, the intronic sequence did not produce similar effects (data not shown). While methylation within all these elements was determined, we did not differentiate the role of the promoter versus exon 1 or the intronic methylation in ALDH1L1 silencing.
It is unclear at present whether silencing of the ALDH1L1 gene is a prerequisite for malignant transformation or if it is a later event in tumor development. If the former is true, FDH perhaps could be considered as a bona fide tumor suppressor. In support of such a possibility, strong methylation of ALDH1L1 promoter was seen in lung adenocarcinomas even at an early stage (samples #3640 and #3905 are stage 1 adenocarcinomas) (Fig. 5), while a nonmalignant tumor, hemangioma, did not reveal any methylated sites. Another question is whether the extent of ALDH1L1 methylation and associated down-regulation of its expression correlate with tumor grade. In regard to this question, a study in the United Kingdom demonstrated that FDH was down-regulated in low-grade bladder tumors and was silenced in high-grade tumors (personal communication from Dr. Margaret Knowles). While the promoter methylation was not evaluated in that study, we suggest that the observed phenomenon is a result of stronger methylation in more advanced tumors. Overall, in the present work, we have demonstrated that FDH is down-regulated in human tumors through methylation of the CpG island. Although this study provides evidence of methylation-dependent down-regulation of ALDH1L1 in lung adenocarcinomas and a hepatocellular carcinoma, we hypothesize that perhaps some other cancer types will reveal a similar correlation.
Materials and Methods
Cell culture and human tissue samples
Cell lines were obtained from the American Tissue Culture Collection (Manassas, VA) and maintained according to the manufacturer’s protocols. Cell media and reagents were purchased from Invitrogen (Carlsbad, CA) unless indicated otherwise. Human tumor samples from patients with stages I to IV adenocarcinomas (stage of the disease was determined by a certified pathologist) and matched nonmalignant tissues were obtained from the MUSC Hollings Cancer Center Tissue Biorepository (Charleston, SC). Frozen samples were stored at –80°C until analyzed. Genomic DNA from the spleen, liver, and hemangioma as well as hepatocellular carcinoma was obtained from BioChain Institute Inc. (Hayward, CA).
Treatment with 5-aza-2′-deoxycytidine
A549 cells were seeded at low density (about 2 × 106 cells) in 100-mm cell culture plates. Twenty-four hours later, 5-aza-2′-deoxycytidine (5-aza-dC) was added (1 µM final concentration). Expression levels of ALDH1L1 were analyzed after 4 days of treatment with 5-aza-dC. Medium was removed, and cells were lysed directly in the plates with RIPA buffer (for protein analysis) or in the commercial lysis buffer from the RNAeasy kit (for mRNA analysis) (Qiagen, Venlo, the Netherlands).
Bisulfite genomic sequencing
Genomic DNAs from 25 mg of human tissue or 107 cultured cells were isolated using the PureLink Genomic DNA Mini Kit (Invitrogen). Total extracted or commercially available DNA was used for bisulfite-mediated conversion of unmethylated cytosines to uracils using an EpiTect Bisulfite Kit (Qiagen) according to the manufacturer’s directions. After treatment, ALDH1L1 gene fragments were amplified by PCR using specific primer pairs complementary to the genomic sequences lacking CpG dinucleotides (Fig. 1C). Amplified DNA was purified with the Geneclean Kit (MP Biomedicals LLC, Solon, OH) after agarose gel electrophoresis. The purified fragments were cloned into a PCR 2.1 vector (Invitrogen) according to the manufacturer’s protocol. For each fragment, 8 to 14 (cancer tissues) or 4 to 8 (normal tissues) clones were sequenced at the Clemson University Genomics Institute DNA Sequencing Facility (Clemson, SC) using M13 reverse primer. Each clone represents the methylation profile of an individual DNA strand. Completeness of the conversion of unmethylated cytosines to uracils was confirmed by evaluation of non-CpG cytosine conversion. DNA methylation patterns were analyzed using BiQ Analyzer software (biq-analyzer.bioinf.mpi-sb.mpg.de).55
Reporter constructs
Sequence comprised of ALDH1L1 promoter plus the entire exon 1 (total of 1,667 bp) was amplified by PCR and cloned into pGL3-Basic vector (Promega, Fitchburg, WI) upstream of the firefly luciferase cDNA. Truncations of the promoter were carried out by site-directed mutagenesis using a QuickChange kit (Stratagene, La Jolla, CA). All constructs were confirmed by sequencing.
In vitro methylation assays
Reporter vectors (10 µg) were methylated in vitro using 30 U of SssI methyltransferase (M.SssI, New England Biolabs, Ipswich, MA) and 1 µM S-adenosylmethionine according to the manufacturer’s directions. Control reactions contained the same components with the exception of S-adenosylmethionine. Methylation of plasmids was confirmed by digestion with a mixture of methylation-sensitive endonucleases, HpaII and HhaI.
Luciferase reporter assay
Reporter constructs were transfected in A549 cells using Amaxa nucleofector (Lonza, Basel, Switzerland) according to the manufacturer’s manual. Luminescence was measured 48 hours posttransfection using a Bright-Glo Luciferase assay system (Promega). The pGL3-Basic plasmid (promoterless) (Promega) was used in each experiment to determine the basal levels of luciferase expression. Each construct was tested in 3 independent transfection experiments. A dual luciferase reporter assay system (Promega) was used to normalize experiments for transfection efficiency (the pRL-TK plasmid expressing Renilla luciferase was cotransfected with each reporter vector at a ratio of 1:10).
Real-time PCR
Total RNA was isolated from 2 × 106 cells or 25 mg of tissue samples using an RNAeasy protect Kit (Qiagen). One to 2 µg of total RNA was used in a reverse-transcription reaction to generate cDNA as earlier described.56 Real-time PCR was performed using 100 ng of the total cDNA, RT2Sybr-Green/Rox qPCR Master mix, and gene-specific real-time PCR primers (SABiosciences, Frederick, MD) with the ABI 7300 Q-PCR system (Applied Biosystems, Foster City, CA) as described by the manufacturer. Primers were designed to generate a specific ALDH1L1 PCR product of 108 bp. Levels of ALDH1L1 mRNA were normalized by the levels of actin as the internal standard. All primer pairs have been tested for dimerization and the amplification of only one product. The fold change in mRNA expression was calculated using 2−ΔΔCt.57
Western blot analysis
Protein extracts from approximately 106 cultured cells or 25 mg of frozen tissue were subjected to SDS-PAGE followed by immunoblotting with specific antibodies as described.5 Actin was used as a loading control. Intensity of corresponding protein bands was quantified using Quantity One Software (Bio-Rad Laboratories, Hercules, CA).
Supplementary Material
Footnotes
Supplementary material for this article is available on the Genes & Cancer website at http://ganc.sagepub.com/supplemental.
The author(s) declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
This work was supported in part by the National Institutes of Health [grant number CA095030].
References
- 1. Hirsch-Ginsberg CF, Stass SA, Freireich EJ. Principles of molecular oncology. In: Hirsch-Ginsberg CF, Stass SA, editors. Molecular basis of oncology. Cambridge, MA: Blackwell Science Inc.; 1995. p. 1-20 [Google Scholar]
- 2. Hanash S, Taguchi A. The grand challenge to decipher the cancer proteome. Nat Rev Cancer. 2010;10:652-60 [DOI] [PubMed] [Google Scholar]
- 3. Furuta E, Okuda H, Kobayashi A, Watabe K. Metabolic genes in cancer: their roles in tumor progression and clinical implications. Biochim Biophys Acta. 2010;1805:141-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57-70 [DOI] [PubMed] [Google Scholar]
- 5. Krupenko SA, Oleinik NV. 10-formyltetrahydrofolate dehydrogenase, one of the major folate enzymes, is down-regulated in tumor tissues and possesses suppressor effects on cancer cells. Cell Growth Differ. 2002;13:227-36 [PubMed] [Google Scholar]
- 6. Krupenko SA. FDH: an aldehyde dehydrogenase fusion enzyme in folate metabolism. Chem Biol Interact. 2009;178:84-93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wagner C. Biochemical role of folate in cellular metabolism. In: Bailey LB, editor. Folate in health and disease. New York: Marcel Dekker Inc.; 1995. p. 23-42 [Google Scholar]
- 8. Glynn SA, Albanes D. Folate and cancer: a review of the literature. Nutr Cancer. 1994;22:101-19 [DOI] [PubMed] [Google Scholar]
- 9. Kisliuk RL. Folate biochemistry in relation to antifolate selectivity. In: Jackman AL, editor. Antifolate drugs in cancer therapy. Totowa, NJ: Humana Press; 1999. p. 13-36 [Google Scholar]
- 10. Cahoy JD, Emery B, Kaushal A, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264-78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rodriguez FJ, Giannini C, Asmann YW, et al. Gene expression profiling of NF-1-associated and sporadic pilocytic astrocytoma identifies aldehyde dehydrogenase 1 family member L1 (ALDH1L1) as an underexpressed candidate biomarker in aggressive subtypes. J Neuropathol Exp Neurol. 2008;67:1194-204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Oleinik NV, Krupenko SA. Ectopic expression of 10-formyltetrahydrofolate dehydrogenase in a549 cells induces g(1) cell cycle arrest and apoptosis. Mol Cancer Res. 2003;1:577-88 [PubMed] [Google Scholar]
- 13. Baylin SB, Herman JG. DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trends Genet. 2000;16:168-74 [DOI] [PubMed] [Google Scholar]
- 14. Robertson KD. DNA methylation, methyltransferases, and cancer. Oncogene. 2001;20:3139-55 [DOI] [PubMed] [Google Scholar]
- 15. Esteller M, Herman JG. Cancer as an epigenetic disease: DNA methylation and chromatin alterations in human tumours. J Pathol. 2002;196:1-7 [DOI] [PubMed] [Google Scholar]
- 16. Esteller M. Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet. 2007;8:286-98 [DOI] [PubMed] [Google Scholar]
- 17. Jones PA. DNA methylation and cancer. Oncogene. 2002;21:5358-60 [DOI] [PubMed] [Google Scholar]
- 18. Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci U S A. 2002;99:3740-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Prestridge DS. Predicting Pol II promoter sequences using transcription factor binding sites. J Mol Biol. 1995;249:923-32 [DOI] [PubMed] [Google Scholar]
- 20. McKnight S, Tjian R. Transcriptional selectivity of viral genes in mammalian cells. Cell. 1986;46:795-805 [DOI] [PubMed] [Google Scholar]
- 21. Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A. 1996;93:9821-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Christman JK. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene. 2002;21:5483-95 [DOI] [PubMed] [Google Scholar]
- 23. Boivin AJ, Momparler LF, Hurtubise A, Momparler RL. Antineoplastic action of 5-aza-2′-deoxycytidine and phenylbutyrate on human lung carcinoma cells. Anticancer Drugs. 2002;13:869-74 [DOI] [PubMed] [Google Scholar]
- 24. Li M, Yin Y, Hua H, et al. The reciprocal regulation of gamma-synuclein and IGF-I receptor expression creates a circuit that modulates IGF-I signaling. J Biol Chem. 2010;285:30480-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Torres L, Avila MA, Carretero MV, et al. Liver-specific methionine adenosyltransferase MAT1A gene expression is associated with a specific pattern of promoter methylation and histone acetylation: implications for MAT1A silencing during transformation. FASEB J. 2000;14:95-102 [DOI] [PubMed] [Google Scholar]
- 26. Schagdarsurengin U, Wilkens L, Steinemann D, et al. Frequent epigenetic inactivation of the RASSF1A gene in hepatocellular carcinoma. Oncogene. 2003;22:1866-71 [DOI] [PubMed] [Google Scholar]
- 27. Bender CM, Pao MM, Jones PA. Inhibition of DNA methylation by 5-aza-2′-deoxycytidine suppresses the growth of human tumor cell lines. Cancer Res. 1998;58:95-101 [PubMed] [Google Scholar]
- 28. Mund C, Hackanson B, Stresemann C, Lubbert M, Lyko F. Characterization of DNA demethylation effects induced by 5-Aza-2′-deoxycytidine in patients with myelodysplastic syndrome. Cancer Res. 2005;65:7086-90 [DOI] [PubMed] [Google Scholar]
- 29. Jackman AL, Theti DS, Gibbs DD. Antifolates targeted specifically to the folate receptor. Adv Drug Deliv Rev. 2004;56:1111-25 [DOI] [PubMed] [Google Scholar]
- 30. Chu E, Allegra CJ. Antifolates. In: Chabner BA, Longo DL, editors. Cancer chemotherapy and biotherapy: principles and practice. Philadelphia: Lippincott-Raven Publishers; 1996. p. 109-48 [Google Scholar]
- 31. Tseng TL, Shih YP, Huang YC, et al. Genotypic and phenotypic characterization of a putative tumor susceptibility gene, GNMT, in liver cancer. Cancer Res. 2003;63:647-54 [PubMed] [Google Scholar]
- 32. Leonard JF, Courcol M, Mariet C, et al. Proteomic characterization of the effects of clofibrate on protein expression in rat liver. Proteomics. 2006;6:1915-33 [DOI] [PubMed] [Google Scholar]
- 33. Oleinik NV, Krupenko NI, Priest DG, Krupenko SA. Cancer cells activate p53 in response to 10-formyltetrahydrofolate dehydrogenase expression. Biochem J. 2005;391:503-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Oleinik NV, Krupenko NI, Krupenko SA. Cooperation between JNK1 and JNK2 in activation of p53 apoptotic pathway. Oncogene. 2007;26:7222-30 [DOI] [PubMed] [Google Scholar]
- 35. Ghose S, Oleinik NV, Krupenko NI, Krupenko SA. 10-formyltetrahydrofolate dehydrogenase-induced c-Jun-NH2-kinase pathways diverge at the c-Jun-NH2-kinase substrate level in cells with different p53 status. Mol Cancer Res. 2009;7:99-107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Islam MQ, Islam K. A new functional classification of tumor-suppressing genes and its therapeutic implications. Bioessays. 2000;22:274-85 [DOI] [PubMed] [Google Scholar]
- 37. Haber D, Harlow E. Tumour-suppressor genes: evolving definitions in the genomic age. Nat Genet. 1997;16:320-2 [DOI] [PubMed] [Google Scholar]
- 38. Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148-59 [DOI] [PubMed] [Google Scholar]
- 39. Lee SW, Tomasetto C, Sager R. Positive selection of candidate tumor-suppressor genes by subtractive hybridization. Proc Natl Acad Sci U S A. 1991;88:2825-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang M, Martin KJ, Sheng S, Sager R. Expression genetics: a different approach to cancer diagnosis and prognosis. Trends Biotechnol. 1998;16:66-71 [DOI] [PubMed] [Google Scholar]
- 41. Kohno T, Yokota J. How many tumor suppressor genes are involved in human lung carcinogenesis? Carcinogenesis. 1999;20:1403-10 [DOI] [PubMed] [Google Scholar]
- 42. Saxonov S, Berg P, Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci U S A. 2006;103:1412-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shiraishi M, Sekiguchi A, Terry MJ, et al. A comprehensive catalog of CpG islands methylated in human lung adenocarcinomas for the identification of tumor suppressor genes. Oncogene. 2002;21:3804-13 [DOI] [PubMed] [Google Scholar]
- 44. Pfeifer GP, Rauch TA. DNA methylation patterns in lung carcinomas. Semin Cancer Biol. 2009;19:181-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4:143-53 [DOI] [PubMed] [Google Scholar]
- 46. Jones PA. The DNA methylation paradox. Trends Genet. 1999;15:34-7 [DOI] [PubMed] [Google Scholar]
- 47. Medvedeva YA, Fridman MV, Oparina NJ, et al. Intergenic, gene terminal, and intragenic CpG islands in the human genome. BMC Genomics. 2010;11:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li B, Goyal J, Dhar S, et al. CpG methylation as a basis for breast tumor-specific loss of NES1/kallikrein 10 expression. Cancer Res. 2001;61:8014-21 [PubMed] [Google Scholar]
- 49. Ferguson AT, Evron E, Umbricht CB, et al. High frequency of hypermethylation at the 14-3-3 sigma locus leads to gene silencing in breast cancer. Proc Natl Acad Sci U S A. 2000;97:6049-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pao MM, Tsutsumi M, Liang G, Uzvolgyi E, Gonzales FA, Jones PA. The endothelin receptor B (EDNRB) promoter displays heterogeneous, site specific methylation patterns in normal and tumor cells. Hum Mol Genet. 2001;10:903-10 [DOI] [PubMed] [Google Scholar]
- 51. Strathdee G, Davies BR, Vass JK, Siddiqui N, Brown R. Cell type-specific methylation of an intronic CpG island controls expression of the MCJ gene. Carcinogenesis. 2004;25:693-701 [DOI] [PubMed] [Google Scholar]
- 52. Kim SK, Jang HR, Kim JH, et al. CpG methylation in exon 1 of transcription factor 4 increases with age in normal gastric mucosa and is associated with gene silencing in intestinal-type gastric cancers. Carcinogenesis. 2008;29:1623-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lu A, Gupta A, Li C, et al. Molecular mechanisms for aberrant expression of the human breast cancer specific gene 1 in breast cancer cells: control of transcription by DNA methylation and intronic sequences. Oncogene. 2001;20:5173-85 [DOI] [PubMed] [Google Scholar]
- 54. Appanah R, Dickerson DR, Goyal P, Groudine M, Lorincz MC. An unmethylated 3′ promoter-proximal region is required for efficient transcription initiation. PLoS Genet. 2007;3:e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bock C, Reither S, Mikeska T, Paulsen M, Walter J, Lengauer T. BiQ Analyzer: visualization and quality control for DNA methylation data from bisulfite sequencing. Bioinformatics. 2005;21:4067-8 [DOI] [PubMed] [Google Scholar]
- 56. Krupenko NI, Dubard ME, Strickland KC, Moxley KM, Oleinik NV, Krupenko SA. ALDH1L2 is the mitochondrial homolog of 10-formyltetrahydrofolate dehydrogenase. J Biol Chem. 2010;285:23056-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402-8 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.