

Abstract
Acute myeloid leukemia (AML) is a hematopoietic disorder in which there are too many immature blood-forming cells accumulating in the bone marrow and interfering with the production of normal blood cells. It has long been recognized that AML is a clinically heterogeneous disease characterized by a multitude of chromosomal abnormalities and gene mutations, which translate to marked differences in responses and survival following chemotherapy. The cytogenetic and molecular genetic aberrations associated with AML are not mutually exclusive and often coexist in the leukemic cells. AML is a disease of the elderly, with a mean age of diagnosis of 70 years. Adverse cytogenetic abnormalities increase with age, and within each cytogenetic group, prognosis with standard treatment worsens with age. In the past 20 years, there has been little improvement in chemotherapeutic regimens and hence the overall survival for patients with AML. A huge unmet need exists for efficacious targeted therapies for elderly patients that are less toxic than available chemotherapy regimens. The multitude of chromosomal and genetic abnormalities makes the treatment of AML a challenging prospect. A detailed understanding of the molecular changes associated with the chromosomal and genetic abnormalities in AML is likely to provide a rationale for therapy design and biomarker development. This review summarizes the variety of cytogenetic and genetic changes observed in AML and gives an overview of the clinical status of new drugs in development.
Keywords: acute myeloid leukemia, genetic abnormalities, new drugs
Introduction
Acute myeloid leukemia (AML) is a clonal hematopoietic disorder resulting from genetic alterations in normal hematopoietic stem cells. These alterations disrupt normal differentiation and/or cause excessive proliferation of abnormal immature leukemic cells known as blasts. As the disease progresses, blast cells accumulate in the bone marrow, blood, and organs and interfere with the production of normal blood cells. This leads to fatal infection, bleeding, or organ infiltration in the absence of treatment within 1 year of diagnosis.1-3 AML is characterized by more than 20% blasts in bone marrow. AML can arise de novo or secondarily either due to the progression of other diseases or due to treatment with cytotoxic agents (referred to as therapy-related AML). Up to 10% to 15% of patients with AML develop the disorder after treatment with cytotoxic chemotherapy (usually for a solid cancer). There are 2 main types of therapy-related AML. The “classic” alkylating-agent type has a latency period of 5 to 7 years and is often associated with abnormalities of chromosomes 5 and/or 7.4 Exposure to agents, such as etoposide and teniposide, that inhibit the DNA repair enzyme topoisomerase II is associated with secondary AML with a shorter latency period, usually 1 to 3 years, with rearrangements at chromosome 11q23.5 Drugs, such as chloramphenicol, phenylbutazone, chloroquine, and methoxypsoralen, can induce marrow damage that may later evolve into AML. Secondary AML may also occur because of progression of myelodysplastic syndrome (MDS) or chronic bone marrow stem cell disorders, such as polycythemia vera, chronic myeloid leukemia, primary thrombocytosis, or paroxysmal nocturnal hemoglobinuria.6,7 Secondary AML has a particularly poor prognosis and is not considered to be curable, with the exception of secondary acute promyelocytic leukemia (APL).8 This is largely due to the high percentage of secondary AML associated with multidrug resistance (MDR) mechanisms: up to 70% of secondary AML patients show overexpression of P-glycoprotein (Pgp) or other MDR mechanisms.9
The genetic changes in leukemic blasts make them ineffective at generating mature red blood cells, neutrophils, monocytes, and platelets. In addition, these AML blasts also inhibit normal blasts from differentiating into mature progeny. Inhibition does not result from “crowding out” of normal blasts; rather, inhibition may be mediated by various chemokines produced by AML blasts.10 AML progresses rapidly and is typically fatal within weeks or months if left untreated. The most common cause of death in AML is bone marrow failure, and the principal sign of marrow failure is infection. Potential fatal organ infiltration, most commonly involving the lung and the brain, becomes more likely as the disease progresses.
AML is the most common acute leukemia affecting adults, and its incidence increases with age.1 Although the majority of patients under age 60 years achieve complete remission (CR) with traditional anthracycline- and cytarabine-based induction regimens, the long-term survival rates continue to be poor at approximately 30% to 40%.11-13 The prognosis is even poorer for those with high-risk AML, such as those who are older, those who had preceding MDS or myeloproliferative disorders, or those with secondary AML from environmental exposures or prior chemotherapy. In such cases, CR is achieved in less than 40% of cases, with survival rates of less than 10%.13 While 60% to 80% of younger patients achieve CR with standard therapy, only about 20% to 30% of the overall patient population has long-term disease-free survival.3 Outcomes are worse for patients aged 60 years or over, with CR rates in the range of 40% to 55% and poor long-term survival rates.3 Along with age, remission rates and overall survival depend on a number of other factors, including cytogenetics, previous bone marrow disorders such as MDS, and comorbidities.3
Epidemiology and Etiology of AML
AML accounts for approximately 25% of all leukemias diagnosed in adults, and the median age at diagnosis is 67 years.13,14 In the United States, 43,050 new cases of leukemia were reported in the year 2010, of which 12,330 were new cases of AML. There were 21,840 patients who died in the year 2010 because of leukemia, of which 8,950 were attributed to AML.15 The incidence of AML in the <65 years’ age group is 1.8 cases per 100,000 patients, and the incidence in the >65 years’ age group is 17.9 cases per 100,000 patients.15 The incidence of AML is expected to increase in the future in line with the aging population, and along with its precursor myelodysplasia, AML prevalence appears to be increasing, particularly in the population older than 60 years of age, and represents the most common type of acute leukemia in adults. Table 1 shows the incidence and prevalence of AML in the United States and other developed countries.
Table 1.
Number of Incidence and Prevalence Cases of Acute Myeloid Leukemia (AML) in the Major Pharmaceuticals Markets in 2010
| Markets | Incidence of new AML in 2010 | Prevalence of AML in 2010 |
|---|---|---|
| US | 12,330 | 25,180 |
| Europe | 12,923 | 22,790 |
| Japan | 3,173 | 5,820 |
| 28,426 | 53,790 |
Note: Incident cases are the new cases diagnosed within a particular time frame; prevalent cases are all cases present at a particular time. Prevalence is thus a function of incident cases and duration of disease.
Development of AML has been correlated with exposure to a variety of environmental agents, most likely due to links between exposure history and cytogenetic abnormalities. Radiation, benzene inhalation, alcohol use, smoking, dyes, and herbicide and pesticide exposure have all been implicated as potential risk factors for the development of AML.16,17 Survivors of the atomic bombs in Japan had an increased incidence of myeloid leukemias that peaked approximately 5 to 7 years following exposure.18 Therapeutic radiation also increases AML risk, particularly if given with alkylating agents such as cyclophosphamide, melphalan, and nitrogen mustard.
Diagnosis and Classification of AML
Demonstration of the accumulation of blasts resulting from the block in differentiation, characteristic of AML, is the essential requirement of diagnosis.19 The early signs of AML include fever, weakness and fatigue, loss of weight and appetite, and aches and pains in the bones or joints. Other signs of AML include tiny red spots in the skin, easy bruising and bleeding, frequent minor infections, and poor healing of minor cuts. The 2 systems commonly used in the classification of AML are the French-American-British (FAB) system and the World Health Organization (WHO) system. The FAB system is based on morphology and cytochemistry and recognizes 8 subtypes of AML, as shown in Table 2.20 In 1999, the WHO classification was introduced to include newer prognostic factors, such as molecular markers and chromosome translocations, and lowered the blast minimum criterion to 20%, thus including many cases classified as high-grade MDS in the FAB system.21 The WHO classification system identifies 4 AML subgroups: 1) AML with recurrent genetic abnormalities, 2) AML with multilineage dysplasia, 3) therapy-related AML and MDS, and 4) those that do not fall into any of these groups. This system created a minimum of 17 subclasses of AML, allowing physicians to identify subgroups of patients who might benefit from specific treatment strategies. Recently, a revised classification has been published as part of the fourth edition of the WHO monograph series.22 The aim of the revision was to incorporate new scientific and clinical information to refine diagnostic criteria for previously described neoplasms and to introduce newly recognized disease entities.
Table 2.
French-American-British (FAB) Classification of Acute Myeloid Leukemia (AML)
| FAB subtype | Morphological classification | % of all AML cases |
|---|---|---|
| AML-M0 | Undifferentiated acute myeloblastic leukemia | 5 |
| AML-M1 | Acute myeloblastic leukemia with minimal maturation | 15 |
| AML-M2 | Acute myeloblastic leukemia with maturation | 25 |
| AML-M3 | Acute promyelocytic leukemia | 10 |
| AML-M4 | Acute myelomonocytic leukemia | 20 |
| AML-M4 eos | Acute myelomonocytic leukemia with eosinophilia | 5 |
| AML-M5 | Acute monocytic leukemia | 10 |
| AML-M6 | Acute erythroid leukemia | 5 |
| AML-M7 | Acute megakaryoblastic leukemia | 5 |
Cytogenetic Abnormalities in AML
AML is characterized by a high degree of heterogeneity with respect to chromosome abnormalities, gene mutations, and changes in expression of multiple genes and microRNAs. Cytogenetic abnormalities can be detected in approximately 50% to 60% of newly diagnosed AML patients.23 The majority of AML cases are associated with nonrandom chromosomal translocations that often result in gene arrangements. Cytogenetics is the most important prognostic factor for predicting remission rate, relapse, and overall survival.23 Several chromosomal abnormalities such as monosomies or deletions of part or all of chromosomes 5 or 7 (–5/–7 AML) and trisomy 8 are common in AML.24 The chromosomal abnormalities also include the long arm of chromosome 11 (11q); balanced translocations between chromosomes 15 and 17 (t(15;17)); chromosomes 8 and 21 (t(8;21)); others such as (q22;q22), (q31;q22), and t(9;11); and inversion such as inv(16).25 Table 3 shows the most frequent chromosomal aberrations and their corresponding fusion genes in AML. The translocation in t(15;17) is always associated with APL and leads to the expression of PML-RARα oncofusion gene in hematopoietic myeloid cells.26 Generally, patients with APL t(15;17) phenotype represent a unique group characterized by distinct biological features and good prognosis, particularly when all-trans retinoic acid (ATRA) is used as part of remission induction.
Table 3.
Acute Myeloid Leukemia (AML)–Associated Oncofusion Proteins
| Translocations | Oncofusion protein | Frequency of occurrence(% of AML) |
|---|---|---|
| t(8;21) | AML1-ETO | 10% |
| t(15;17) | PML-RARα | 10% |
| inv(16) | CBF□-MYH11 | 5% |
| der(11q23) | MLL-fusions | 4% |
| t(9;22) | BCR-ABL1 | 2% |
| t(6;9) | DEK-CAN | <1% |
| t(1;22) | OTT-MAL | <1% |
| t(8;16) | MOZ-CBP | <1% |
| t(7;11) | NUP98-HOXA9 | <1% |
| t(12;22) | MN1-TEL | <1% |
| inv(3) | RPN1-EVI1 | <1% |
| t(16;21) | FUS-ERG | <1% |
Many of the gene rearrangements involve a locus encoding a transcriptional activator, leading to expression of a fusion protein that retains the DNA-binding motifs of the wild-type protein. Moreover, in many instances, the fusion partner is a transcriptional protein that is capable of interacting with a corepressor complex.27 A commonly accepted paradigm is that through aberrant recruitment of a corepressor to a locus of active transcription, the fusion protein alters expression of target genes necessary for myeloid development, thus laying the groundwork for leukemic transformation.28 Potential targeting of this interaction has become a major focus for the development of novel therapeutics. ATRA serves as a prototype: by altering corepressor interaction with the APL fusion protein, ATRA effectively induces remission and has become a mainstay of treatment of this previously fatal disease.8 However, to date, APL represents both the most curable and the best-studied subtype of AML, while molecular data on other fusion proteins are limited or absent. Still, the work on PML-RARα has inspired the molecular analysis of many other AML-associated oncofusion proteins, especially AML1-ETO, CBFβ-MYH11, and MLL fusions.
Oncofusion Proteins Associated with AML
A total of 749 chromosomal aberrations have been catalogued in AML.29 The frequencies of the 4 most common translocations are between 3% and 10%, while for others, the prevalence is significantly smaller. The most frequent oncofusion proteins, PML-RARα, AML1-ETO, CBFβ-MYH11, and MLL fusions, are described below.
t(15;17), PML-RARα
The t(15;17) translocation is found in approximately 95% of APLs, a specific subtype of AML. The translocation results in the expression of the PML-RARα oncofusion gene in hematopoietic myeloid cells.8 The PML-RARα oncofusion protein acts as a transcriptional repressor that interferes with gene expression programs involved in differentiation, apoptosis, and self-renewal.8
t(8;21), AML1-ETO
Approximately 10% of AML cases carry the t(8;21) translocation, which involves the AML1 (RUNX1) and ETO genes, and express the resulting AML1-ETO fusion protein. AML1 is a DNA-binding transcription factor crucial for hematopoietic differentiation,30,31 while ETO is a protein harboring transcriptional repressor activities.32 The fusion protein AML1-ETO is suggested to function as a transcriptional repressor that blocks AML1-dependent transactivation in various promoter reporter assays, suggesting it may function as a dominant-negative regulator of wild-type AML1.33,34
inv(16), CBFβ-MYH11
inv(16) is found in approximately 8% of AML cases. inv(16) fuses the first 165 amino acids of core binding factor β (CBFβ) to the C-terminal coiled-coil region of a smooth muscle myosin heavy chain (MYH11). CBFβ-MYH11 fusion protein is suggested to cooperate with AML1 to repress transcription.35,36
11q23, MLL Rearrangements
Mixed lineage leukemia (MLL) is implicated in at least 10% of acute leukemias of various types. In general, the prognosis is poor for patients harboring MLL translocations.37 In these patients, the MLL protein fuses to 1 of >50 identified partner genes, resulting in an MLL fusion protein that acts as a potent oncogene.38 The amino-terminal portion of MLL serves as a targeting unit to direct MLL oncoprotein complexes to their target loci through DNA binding, whereas the fusion partner portion serves as an effecter unit that causes sustained transactivation.
Gene Mutations in AML
Approximately 40% to 50% of patients with AML have a normal karyotype and represent the largest subset of AML.39 All such cases of cytogenetically normal AML are currently categorized in the intermediate-risk group; yet, this group is quite heterogeneous, and not all patients in this subset have the same response to treatment. This is likely a result of the large variability in gene mutations and gene expression in this population. These alterations appear to fall into 2 broadly defined complementation groups. One group (class I) comprises mutations that activate signal transduction pathways and thereby increase the proliferation or survival, or both, of hematopoietic progenitor cells. The other complementation group (class II) comprises mutations that affect transcription factors or components of the cell cycle machinery and cause impaired differentiation.
Class I Mutations
Mutations in KIT, FLT3, and NRAS fall into the class I mutations.
KIT mutations
Although patients with AML and inv(16) and t(8;21) in general have a more favorable prognosis, there remains a significant failure rate, and the long-term disease-free survival rate is approximately 60%. Studies have shown that activating KIT mutations in approximately 30% to 40% of patients with inv(16) are associated with higher incidence of relapse and significantly lower survival. In those with t(8;21), the incidence of KIT mutations appears to be variable.40
FLT3 mutations
Fms-like tyrosine kinase 3 (FLT3) is a receptor tyrosine kinase that plays a key role in cell survival, proliferation, and differentiation of hematopoietic stem cells.41,42 It is frequently overexpressed in acute leukemias. FLT3 mutations occur in approximately 30% of AML patients and confer a poor prognosis. The 2 major types of mutations that occur are internal tandem duplication (ITD) mutations of the juxtamembrane region and point mutations in the tyrosine kinase domain (TKD), which frequently involve aspartic acid 835 of the kinase domain. Both mutations result in constitutive activation of the receptor’s tyrosine kinase activity in the absence of ligand.41 The incidence of FLT3 mutations also increases with age, but the FLT3 ITD mutations have less prognostic impact in patients >60 years of age possibly because other adverse prognostic factors are more prevalent.
RAS mutations
Mutations in NRAS and KRAS occur in approximately 10% and 5% of AML patients, respectively. IRASS mutations occur only rarely in conjunction with FLT3 mutations and do not appear to have a significant impact on AML survival.43
Class II Mutations
In addition, mutations in MLL, brain and acute leukemia gene (BAAL), Wilms tumor gene (WT-1), CCAAT/enhancer-binding protein α (CEBPα), and nucleoplasmin 1 (NPM1) have also been observed in AML patients.44-46 Recently, mutations in DNA methyltransferase gene DNMT3A have been identified in one third of patients with de novo AML with intermediate-risk cytogenetics.47 DNMT3A represents 1 of 3 human genes that encodes DNA methyltransferase that catalyzes the addition of methyl groups to cytosine within CpG dinucleotide, resulting in repression of nearby genes. Genomes with DNMT3A mutations commonly harbored additional mutations in FLT3, NPM1, and IDH1. The presence of any DNMT3A mutation, either alone or in combination with FLT3 ITD mutation, is associated with significantly shorter overall survival (OS).47
Prognostic Factors in AML
Prognostic factors can be divided into those associated with treatment-related death occurring before response can be assessed and those associated with resistance to treatment. The predictor of treatment-related death is the patient’s performance status. Therapy-related AML or AML arising after MDS is usually more resistant to treatment than de novo AML.48 However, age and cytogenetics are the most important prognostic factors for predicting remission rate, relapse, and OS in AML. Risk stratification based on cytogenetics divides patients into 3 main groups: patients with favorable, intermediate, and unfavorable cytogenetics depending on the presence or absence of specific chromosomal abnormalities (Table 4). Studies have shown that the 5-year survival rate was 55% for patients with favorable cytogenetics, 24% for patients with intermediate risk, and 5% for patients with poor-risk cytogenetics.24 Adverse cytogenetic abnormalities increase with age, and within each cytogenetic group, prognosis with standard treatment worsens with age.3 A recent study demonstrated that the percentage of patients with unfavorable cytogenetics has been shown to increase from 35% in patients below 56 years of age to 51% in patients over 75 years (Fig. 1).49
Table 4.
Acute Myeloid Leukemia (AML) Cytogenetic Risk Groups
| Karyotype | Frequency, % | Complete remission, % | Event-free survival, % |
|---|---|---|---|
| Favorable | |||
| t(8;21) | 5-10 | 90 | 60-70 |
| inv(16) | 5-10 | 90 | 60-70 |
| t(15;17) | 5-10 | 80-90 | 70 |
| Intermediate | |||
| Diploid, –Y | 40-50 | 70-80 | 30-40 |
| Unfavorable | |||
| −5/–7 | 20-30 | 50 | 5-10 |
| +8 | 10 | 60 | 10-20 |
| 11q23, 20q-, other | 10-20 | 60 | 10 |
Figure 1.
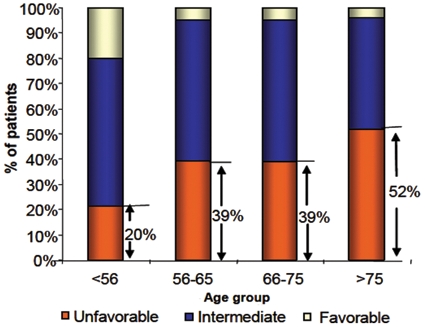
Cytogenetic risk group by age group. Adapted with permission from Appelbaum FR, Gundacker H, Head DR, et al. Age and acute myeloid leukemia. Blood. 2006;107:3481-5.
Treatment of AML
The primary objective of therapy for AML is to achieve and maintain CR. CR is defined as a marrow with less than 5% blasts, a neutrophil count greater than 1,000, and a platelet count greater than 100,000. CR is the only response that leads to a cure or at least an extension in survival. The probability of AML recurrence sharply declines to <10% after 3 years in CR.50 For the past 30 years, treatment of AML has consisted of the combination of an anthracycline, such as daunorubicin or idarubicin, and cytarabine.51 Treatment of AML is divided into 2 phases: 1) remission induction therapy (with possible postinduction) and 2) postremission therapy.52 Generally, AML treatment includes at least one course of intensive induction chemotherapy followed by an additional course of intensive consolidation therapy and then maintenance therapy.
Remission Induction Therapy
In induction therapy, the goal is to achieve a marked reduction in the number of malignant cells in order to establish normal hematopoiesis. A standard form of induction therapy consists of a standard dose of cytarabine (SDAraC, 100-200 mg/m2), administered by continuous infusion for 7 days and combined with an anthracycline administered intravenously for 3 days (referred to as 7 + 3 regimen). With standard induction regimens, remission is achieved in about 65% to 85% of younger patients but in less than 50% of patients over 60 years of age.2,53 This approach results in a long-term disease-free survival of approximately 30%, with treatment-related mortality of 5% to 10%. A number of studies have been conducted to improve the CR rate by use of alternative anthracyclines, incorporation of high-dose AraC (HDAraC), or addition of other agents such as etoposide, fludarabine, or cladribine. However, presently, there is no conclusive evidence to recommend one 7 + 3 induction regimen over another. However, these studies clearly support the conclusion that further intensification of the induction regimen is not associated with an increased CR rate.
In patients who fail to achieve CR following induction therapy, postinduction therapy is recommended. Postinduction therapy with standard-dose cytarabine is recommended in patients who have received standard-dose cytarabine induction and have significant residual blasts.52 In other cases, postinduction therapy may consist of hematopoietic stem cell transplantation if a suitable donor can be found.
Postremission Therapy
There are mainly 2 types of postremission therapy:
Consolidation therapy is usually administered at doses approaching those used during induction.
Maintenance therapy is usually defined as therapy less myelosuppressive than therapy used to produce remission. Typically, patients receive the same regimen used during induction at approximately monthly intervals for 4 to 12 months.
Consolidation Therapy
Although obtaining an initial remission is the first step in controlling the disease, it is important that patients continue with consolidation therapy to achieve a durable remission. Patients who do not receive consolidation therapy will relapse within 6 to 9 months.54,55 Consolidation therapy can consist of chemotherapy or hematopoietic stem cell transplantation (HSCT), and the choice of therapy is typically dependent on patient age, comorbidities, chance of recurrence based on cytogenetics, and whether a patient has a suitable donor for HSCT.3 The use of HSCT is less common in patients aged over 60 years because of increased risks of transplant-related morbidity and mortality. Consolidation therapy comprises treatment with additional courses of intensive chemotherapy after the patient has achieved CR, usually with higher doses of the same drugs used during the induction period. High-dose AraC (2-3 g/m2) is now standard consolidation therapy for patients aged <60 years of age. The median disease-free survival for patients who receive only the induction therapy is 4 to 8 months. However, 35% to 50% of adults aged <60 years who receive consolidation treatment survive 2 to 3 years.55 HSCT has a central role in the treatment of AML. However, because of the morbidity and mortality of the procedure, it tends to be used in patients who have a substantial risk of relapse.56 APL, a subtype of AML, is treated differently from other subtypes of AML; the vitamin A derivative ATRA (Vesanoid, Roche, Basel, Switzerland) can induce differentiation of leukemic promyelocytes, resulting in high remission rates.8 Older patients are generally treated with lower intensity therapies such as subcutaneous cytarabine or hydroxyl urea in an attempt to minimize treatment-related mortality.
Maintenance Therapy
Maintenance therapy, which is considered less myelosuppressive than the induction and consolidation forms of treatment, is used in patients who have previously obtained CR. It is a strategy to further reduce the number of residual leukemic cells and prevent a relapse. Its role in the routine management of AML patients is controversial and depends mainly on the intensity of induction and consolidation therapies.52
Treatment of Relapsed and Refractory Disease
Despite the substantial progress in the treatment of newly diagnosed AML, 20% to 40% of patients still do not achieve remission with standard induction chemotherapy, and 50% to 70% of first CR patients are expected to relapse over 3 years.57 The prognosis for patients with AML refractory to first-line treatment or in first or subsequent relapse is generally poor. The duration of first remission in relapsed patients is the most important prognostic factor correlating with the probability of second CR and survival.58 Patients who relapsed in less than 6 months have a significantly poor prognosis compared to patients who relapsed after a first CR lasting >6 months. Treatment strategies for relapse are dependent on patient age.52 For patients less than 60 years old who have experienced an early (<6 months) relapse after induction chemotherapy, the US National Comprehensive Cancer Network (NCCN) guidelines recommend participation in a clinical trial or HSCT.52 However, if patients have relapsed after a long (6 months or greater) remission, they can be retreated with a chemotherapy regimen or a development drug in the context of a clinical trial.52 The recommended option for patients aged 60 years or older is participation in a clinical trial.52
HSCT is the most commonly used treatment modality at relapse in patients aged below 60 years. In older patients, use of HSCT at relapse is rare, and single agents including azacitidine (Vidaza, Celgene, Summit, NJ), gemtuzumab ozogamicin (Mylotarg, Pfizer, New York City, NY), and hydroxyurea are most commonly used, although there is a lack of clear consensus over the optimum regimen.
Age Is a Major Determinant of Survival
Treatment recommendations for AML patients differ depending on whether patients are above or below 60 years old.52 Table 5 shows the treatment outcomes based on age criteria. Survival in AML depends on age, with significantly lower survival rates reported for older adults.3 Statistics from the Surveillance, Epidemiology and End Results (SEER) Program from 1996 to 2002 show 5-year survival rates of 34.4% for adults aged below 65 years and 4.3% for those aged 65 years or older.54 While selected older patients can benefit from standard therapies, this group of patients experiences greater treatment-related toxicity, lower remission rates, shorter disease-free survival, and shorter OS times.3 Older adults are less likely to achieve CR and to remain relapse free if they have achieved CR.3 In addition, these patients are more likely to experience treatment-related death, which is in the range of 15% to 30% in reported clinical trials.3 This is because patients over the age of 60 years are characterized by a higher prevalence of unfavorable cytogenetics and myelodysplasia, a greater incidence of MDR, and more frequent comorbidities that often make them unsuitable for intensive treatment.3
Table 5.
Outcomes in Acute Myeloid Leukemia (AML) by Age Criteria
| Age <60 y | Age >60 y | |
|---|---|---|
| Complete response, % | 70 | 45 |
| Disease-free survival, % | 45 | 20 |
| Early death, % | 10 | 25 |
| Overall survival, % | 30 | 10 |
| Median survival, mo | 24 | 10 |
Novel Agents in the Pipeline for AML
Identification of specific gene mutations, chromosomal translocations, and alterations in signaling pathways and gene transcription in AML has led to the development of a number of targeted agents. A number of therapeutic approaches are being investigated in the treatment of AML (Table 6). These include histone deacetylase inhibitors, DNA methyl transferase inhibitors, retinoid X receptor agonists, proteosome inhibitors, antiangiogenesis inhibitors, FLT3 inhibitors, farnesyl transferase inhibitors, mTOR inhibitors, poly ADP-ribose polymerase (PARP) inhibitors, MEK1/2 inhibitors, modulators of drug resistance, and immune-modulating agents.59 In addition, a number of traditional chemotherapeutics in new formulations are also being investigated. Table 7 lists the molecules that are being investigated in late-stage clinical trials for AML. Clinical trial results of key drugs in AML are summarized below.
Table 6.
Therapeutic Strategies Being Investigated in the Treatment of Acute Myeloid Leukemia (AML)
| Therapeutic approach | Examples |
|---|---|
| Epigenetic regulation | Histone deacetylase inhibitors: vorinostat, panobinostat, belinostat DNA methyl transferase inhibitors: Vidaza, Dacogen |
| Differentiation-inducing therapeutics | Retinoid X receptor agonists Arsenic trioxide |
| Angiogenesis inhibition | Inhibition of angiogenesis: Velcade Thalomid, Revlimid |
| Inhibition of signaling pathways | Tyrosine kinase inhibitors: midostaurin, lestaurtinib, sorafenib, KW-2449, AC220 Cell cycle inhibitors: ON 01910.Na Farnesyl transferase inhibitors: Zarnestra, Sarasar mTOR inhibitors: Afinitor, PI-103, temsirolimus, GSK21110183 PARP inhibitors: ABT-888 MEK1/2 inhibitors: AZD6244, AS703026, PD98059, GSK1120212 Bcl-2 inhibitors: oblimersen, obatoclax, ABT-263 XIAP inhibitor: AEG-35156 Aminopeptidase inhibitors (tosedostat) |
| Modulation of drug resistance | Valspodar, zosuquidar |
| Modified traditional chemotherapeutics | Nucleoside analogs: clofarabine, sapacitabine, elacytarabine Alkylating drugs: irofulven, Temodar, Onrigin Topoisomerase inhibitors: Hycamtin |
| Immune therapy | Antibodies: Mylotarg, lintuzumab, Avastin, T-cell targeted therapy |
Table 7.
List of Molecules in the Late-Stage Pipeline for Acute Myeloid Leukemia (AML)
| Molecule | Class | Mechanism of action | Company | Phase (US) |
|---|---|---|---|---|
| Amonafide (AS1413) | Cytotoxic | Topoisomerase II inhibitor | Antisoma | III |
| Clofarabine | Cytotoxic | Purine nucleoside | Genzyme | III |
| Decitabine | Cytotoxic | DNA hypomethylating agent | Eisai/Johnson & Johnson | III |
| Midostaurin (PKC412) | Targeted | Flt-3 tyrosine kinase inhibitor | Novartis | III |
| PR1 peptide antigen vaccine | Immunotherapy | Peptide-based therapeutic vaccine | The Vaccine Company | III |
| Daunorubicin | Cytotoxic | Topoisomerase inhibitor | Gilead | III |
| Tipifarnib | Targeted | Farnesyl transferase inhibitor | Janssen | III |
| Theralux | Photodynamic | Radical formation stimulant | Kiadis | III |
| Lestaurtinib (CEP-701) | Targeted | TKI including Flt-3, TrkA, and JAK2 | Cephalon | III |
| Belinostat (PXD-101) | Cytotoxic | HDAC inhibitor | Spectrum Pharmaceuticals | III |
| ON 01910.Na | Cytotoxic | Cell cycle inhibitor | Onconova Therapeutics | II |
Flt-3 Inhibitors
Despite an exciting rationale for the use of FLT3 tyrosine kinase inhibitors (TKIs) in AML, the clinical results have so far been modest. Several FLT3 inhibitors are currently being developed such as PKC412 (midostaurine), lestaurtinib, sorafenib, AC-220, CEP-701, and sunitinib. Clinical trials of FLT3 inhibitors as monotherapy have resulted in frequent responses in peripheral blasts but less frequent significant responses in bone marrow blasts. The responses also tend to be short lived, lasting anywhere from weeks to months. These results using FLT3 inhibitors as single agents in AML have been, perhaps not surprisingly, disappointing. Full-blown clinical AML likely represents a multitude of leukemogenic mutations, only one of which, and perhaps a late one at that, is the FLT3-activating mutation. Trials of these agents in combination with chemotherapy are ongoing and show very encouraging responses, but clinical responses appear to correlate with in vitro sensitivity of the blasts and the achievement of adequate levels of FLT3 inhibition in vivo. The pharmacodynamics studies associated with these trials are thus very important.60,61 Whether these responses ultimately improve long-term outcome of patients and whether they may be particularly beneficial for patients with FLT3 mutations compared to those with FLT3 wild-type (WT) are being investigated.
Midostaurin
Midostaurin was originally developed as a protein kinase C inhibitor. It was also found to be a potent inhibitor of FLT3 phosphorylation and cell proliferation. NCT00651261 is a phase III trial looking at midostaurin added to daunorubicin + cytarabine in newly diagnosed AML. Novartis (Basel, Switzerland) is the first company to get US Food and Drug Administration (FDA) approval to study an Flt-3 inhibitor in the front line. The protocol is to give daunorubicin and cytarabine with or without midostaurin, followed by high-dose cytarabine and midostaurin. The 514-patient trial was scheduled to be complete in March 2009 but is still accruing patients.
Lestaurtinib (CEP-701)
A phase II study of the Flt-3 inhibitor lestaurtinib (CEP-701) as first-line treatment for older AML patients demonstrated clinical improvement in 60% with mutations and in 23% with wild-type FLT3. Lestaurtinib also had biological and clinical activity in relapsed/refractory AML.62 The pivotal CEP-701 trial in relapsed/refractory AML is flawed because Cephalon (Frazer, PA) did not collect samples in the control arm and in patients who initially responded to the drug but then relapsed. Thus, it is not going to be possible to know whether different outcomes are due to differences in mutations in each arm.
AC220
AC220 is a receptor tyrosine kinase inhibitor (TKI), demonstrated to have potent and specific in vitro and in vivo activity against the FLT3 tyrosine kinase. Ambit Biosciences (San Diego, CA) is running a phase II study of Flt-3 inhibitor, AC-220, in relapsed/refractory AML.63 Its claim is that the drug is more potent so it could be a 1-pill qd therapy for this setting. Other Flt-3 inhibitors have shown initial responses in refractory AML. All have produced short remissions.
Sorafenib
Sorafenib is a multikinase inhibitor that is approved for the treatment of metastatic renal cell and hepatocellular carcinoma. In a phase II study, 18 patients with newly diagnosed AML and mutated FLT3 were enrolled to receive sorafenib, idarubicin, and Ara-C. There were 94% of the patients who achieved a morphological CR/CRp and 6% who achieved PR. This regimen was found to be effective in reducing the mutant clones.64 However, a large prospective study is needed to confirm the results from the small observational studies. A randomized, placebo-controlled, double-blind, phase II trial concluded that 1) the addition of sorafenib to standard 7 + 3 chemotherapy did not prolong disease-free survival in patients older than 60 years of age with AML; 2) lower rates of response and higher rates of early death were found with sorafenib versus placebo; 3) there was no difference in OS; and 4) the study was not significantly powered to detect treatment difference in patients positive for FLT3 ITD. Study investigators concluded that sorafenib should not be given to older patients not selected for FLT3 ITD status. Efficacy of sorafenib in FLT3 ITD–positive patients needs further study.65
Old Drugs in New Formulations
CPX-351
CPX-351 is a liposomal formulation that encapsulates cytarabine and daunorubicin at a 5:1 molar ratio. A recently concluded multicenter, randomized, open-label phase IIB study showed that CPX-351 is safe, well tolerated, and associated with low early mortality in treatment-naive elderly patients with AML. Early signals of efficacy of CPX-351 were encouraging when compared with standard cytarabine/daunorubicin 7 + 3 regimen, particularly in patients considered to have high-risk factors. Numerical, but not statistically significant, increases in response rates (66.7% v. 51.2%; P = 0.0712) and OS were noted. The results showed that liposomal encapsulation of this chemotherapy doublet changed the safety profile by reducing nonhematological toxicities including hair loss, gastrointestinal toxicities, and hepatic toxicity while retaining hematopoietic cytotoxicity.66
Nucleoside Analogs
Clofarabine
Clofarabine is a new nucleoside analog and potent inhibitor of both ribonucleotide reductase and DNA polymerase. AML patients were enrolled in a phase II study to receive clofarabine plus low-dose Ara-C induction, followed by consolidation with clofarabine plus low-dose Ara-C alternating with decitabine. Clofarabine plus low-dose cytarabine achieved high response rates with a manageable toxicity profile and low induction mortality in patients age ≥60 years with previously untreated AML. Longer follow-up and comparisons with conventional therapy will help establish whether this combination also has a survival advantage.67 In October 2009, the FDA refused to approve clofarabine for use in previously untreated elderly AML without completion of an additional trial. Data from the CLASSIC I study (NCT00317642) of cytarabine ± clofarabine are expected to show a benefit in patients aged >55 years with AML in CR rate, progression-free survival, and OS.
Sapacitabine
Sapacitabine is an orally available nucleoside analog in phase II trials, in advanced MDS/AML, and in cutaneous T-cell lymphoma. In terms of efficacy, Cyclacel (Berkeley Heights, NJ) did not present any results to suggest that it is better than azacitidine or decitabine.
Inhibitors of Angiogenesis
Lenalidomide
Lenalidomide is now used in the treatment of various hematological malignancies; the anticancer effects are probably caused by several mechanisms. Preliminary data presented at the American Society of Hematology (ASH) annual meeting in 2009 showed that AML patients were responsive to lenalidomide in a nonspecific fashion, meaning that patients did not necessarily have deletion 5q (del(5q)) lesions. However, recent studies on SWOG S0605 in a single-arm phase II trial reported that single-agent therapy with lenalidomide demonstrated modest activity (response rate: 14%) in older patients (60 years of age or older) with AML and del(5q). Use of higher lenalidomide doses in induction therapy may help overcome effects of additional chromosomal abnormalities. NCT01016600, opening in January 2010, is an open-label trial looking at lenalidomide + azacitidine in relapsed/refractory younger AML or first-line older AML.68
DNA Methyl Transferase Inhibitors
Dacogen and Vidaza
The CR rates for the hypomethylating agents are lower than they are with low-dose cytarabine. In first-line AML, the CR rate for Vidaza (Celgene) is 14%, while for low-dose cytarabine, it is 18%. But many hematologists view the hypomethylating agents as being more “au courant,” so more people are using them. In the community, more people are using Dacogen (SuperGen Inc., Dublin, CA) than Vidaza (Celgene) for AML because there is a perception that it is stronger than Vidaza (Celgene).
Histone Deacetylase (HDAC) Inhibitors
Vorinostat
Vorinostat is a new anticancer agent inhibiting histone deacetylase and approved for the treatment of cutaneous lymphoma. A phase II study of vorinostat in combination with idarubicin and cytarabine as front-line therapy for AML or MDS patients was reported. This combination was found to be safe, and overall response rates are very high with this combination, particularly in diploid and Flt-3 ITD patients. Longer follow-up is needed to assess the effect on survival. Studies specific for Flt-3–mutated patients and in combination with standard 7 + 3 therapy are ongoing.69 However, vorinostat as monotherapy demonstrated minimal activity in refractory and high-risk AML patients.70
Cytotoxics
Amonafide L-malate (AS1413)
Amonafide L-malate (amonafide, AS1413) is a unique DNA intercalator. In a phase II study, 88 patients with secondary AML were enrolled to receive amonafide and Ara-C. Overall CR + CRi rate was 42%. CR rates among age <60 years and ≥60 years were 39.4% and 43.6%, respectively; among tAML and prior MDS, the CR rates were 40% and 44.2%, respectively; for patients with intermediate and unfavorable cytogenetics, the CR rates were 61.1% and 23.8%, respectively. This study showed that amonafide in combination with cytarabine produced a high CR rate and durable responses in both older and younger patients with secondary AML.71
Gemtuzumab ozogamycin
Gemtuzumab ozogamycin (Mylotarg, Pfizer) is a monoclonal antibody GO against CD33 conjugated to calichemycin. Mylotarg (Pfizer) was granted accelerated approval in May 2000 as second-line therapy for patients 60 years or older with CD33+ve AML who were not candidates for chemotherapy. Pfizer recently withdrew the drug from the market because of a high death rate in postmarket studies. Besides, no benefit for progression-free survival or OS was observed with the addition of Mylotarg (Pfizer) to standard daunorubicin or Ara C induction.72
Cell Cycle Inhibitors
ON 01910
ON 01910.Na (Estybon, Onconova Therapeutics Inc.) is a small molecular weight compound that has a multitargeted mechanism of action, resulting in a selective mitotic block and cell death in cancer cells. In particular, the polo-like kinase (PLK) pathway is affected, causing polynumeric centrosomes and dysregulation of mitosis. At the molecular level, ON 01910.Na also inhibits PI-3 kinases (specifically the α and β isoforms). In ON 01910–treated cells, both the ERK (growth) and AKT (prosurvival) pathways are inhibited. Following G2/M arrest, cells undergo apoptosis via the caspase pathway. One of the remarkable activities noted for this compound is activity in drug-resistant cancer cells and in tumor cells with antiapoptotic barriers. PLKs now emerge as possible targets in future anticancer therapy. Interactions between PLK 2 and the AML/ETO hybrid molecule in t(8;21) AML seem to mediate antiapoptotic effects.73 A phase I/II study of ON 01910.Na is being conducted in patients with hematological malignancies. This study has shown that ON 01910.Na appears to be safe and well tolerated in patients with refractory or relapsed MDS and AML. ON 01910.Na has biological activity with reduction in bone marrow blasts, eradication of the MDS clone, and improvement in the peripheral blood counts in some patients in phase I and II trials. These effects are associated with increased survival, albeit in limited numbers of patients treated thus far.74 A pivotal phase III trial of ON 01910 in MDS patients is now underway (clintrial.org). A single-agent phase I study in refractory AML patients is evaluating single-agent activity as a prelude to combination therapy trials. Further study of ON 01910.Na is warranted to better define biological activity and appropriate target populations and to define mechanism of action.
Outlook and Summary
The major improvements in AML treatment during the last 2 decades have not been the introduction of new therapeutic agents but rather the more optimal use of well-known drugs (e.g., high-dose cytarabine therapy and the use of ATRA in maintenance therapy of acute promyelocytic leukemia). For younger patients with poor-risk cytogenetics and an available donor, HSCT offers the best opportunity for a cure. For older patients and for relapsed and refractory patients, there is an obvious need to develop better strategies with effective regimens. The limit of acceptable toxicity for standard chemotherapeutic drugs used in AML therapy has been reached. New therapeutic strategies are therefore needed. Although several deregulated proteins and genes have been identified, these are so diverse among AML cases that finding a substance with potential activity against all of them is challenging. Recently, several new agents have been explored and have shown promise in treating AML. However, it is unlikely that these agents will be curative when administered as monotherapy; it is more likely that they will be used in combination with other new agents or with conventional therapy.
It has long been appreciated that AML is a clinically heterogeneous disease with marked differences in survival following intensive chemotherapy based on age, blast cell morphology, cytogenetic abnormalities, and gene mutations. As described above, in many cases, one of the partners in a gene arrangement codes for a transcription factor. As a consequence, AML-associated fusion proteins often function as aberrant transcriptional regulators and ultimately interfere with the process of myeloid differentiation despite variations in gene expression changes induced by them.25 Similarly, class I mutations that activate signal transduction pathways and class II mutations that affect transcription factors or components of the cell cycle machinery also affect blast cell differentiation and elicit AML phenotype. These results suggest that mutation or upregulation in one pathway does not account for AML transformation. Blasts rely on multiple dysregulated pathways to emerge and survive and to ultimately develop resistance to therapy. Therefore, pursuing several molecular lesions in a concurrent or serial fashion may be a promising approach to targeted therapy.
Although many of the breakpoints involved in specific chromosomal translocations have been cloned and novel ones are still being discovered, in most cases, the molecular mechanisms and the central players leading to tumorigenesis are not elucidated. A number of genetically engineered mouse models have been employed to determine the molecular significance of the chromosomal abnormalities and to clarify the biological consequences upon disease states.75 The major contribution of these models has been the appreciation that AML is a multistep process requiring a number of synergistic mutations. However, the clinical relevance of these models has been limited. It is becoming exceedingly clear that a detailed knowledge of the molecular pathways influenced by the expression of these oncofusion proteins has an enormous potential and will lay the basis for diagnosis, prognosis, biomarker development, and new drug development. In this context, the use of genetically engineered mouse models that accurately mimic the genetic and biological progression of their equivalent AML subtype would not only facilitate understanding of the precise role of these molecular abnormalities but also serve in the development of novel therapeutics.
Acknowledgments
The author thanks Dr. Ramesh Kumar for his comments on the article.
Footnotes
The author(s) declared the following potential conflicts of interest with respect to the authorship and/or publication of this article: C. Chandra Kumar is an employee of Onconova Therapeutics Inc.
The author(s) received no financial support for the research and/or authorship of this article.
References
- 1. Lowenberg B, Downing JR, Burnett A. Acute myeloid leukemia. N Engl J Med. 1999;341:1051-62 [DOI] [PubMed] [Google Scholar]
- 2. Estey E, Dohner H. Acute myeloid leukemia. Lancet. 2006;368:1894-907 [DOI] [PubMed] [Google Scholar]
- 3. Shipley JL, Butera JN. Acute myelogenous leukemia. Exp Hematol. 2009;37:649-58 [DOI] [PubMed] [Google Scholar]
- 4. Leone G, Voso MT, Sica S, Morosetti R, Pagano L. Therapy related leukemias: susceptibility, prevention and treatment. Leuk Lymphoma. 2001;41:255-76 [DOI] [PubMed] [Google Scholar]
- 5. Felix CA. Secondary leukemias induced by topoisomerase-targeted drugs. Biochim Biophys Acta. 1998;1400:233-55 [DOI] [PubMed] [Google Scholar]
- 6. Appelbaum FR. Acute myeloid leukemia in adults. In: Abeloff M, Armitage J, Kastan M, et al., editors. Clinical oncology, 3rd edition. Philadelphia: Elsevier Science; 2004. p. 2825-48 [Google Scholar]
- 7. Pedersen-Bjergaard J, Christiansen DH, Andersen MK, Skovby F. Causality of myelodysplasia and acute myeloid leukemia and their genetic abnormalities. Leukemia. 2002;16:2177-84 [DOI] [PubMed] [Google Scholar]
- 8. Licht JD. Reconstructing a disease: what essential features of the retinoic acid receptor fusion oncoproteins generate acute promyelocytic leukemia? Cancer Cell. 2006;9:73-4 [DOI] [PubMed] [Google Scholar]
- 9. Szotkowski T, Muzik J, Voglova J, et al. Prognostic factors and treatment outcome in 1,516 adult patients with de novo and secondary acute myeloid leukemia in 1999-2009 in 5 hematology intensive care centers in the Czech Republic. Neoplasma. 2010;57(6):578-89 [DOI] [PubMed] [Google Scholar]
- 10. Youn B-S, Mantel C, Broxmeyer HE. Chemokines, chemokine receptors and hematopoiesis. Immunol Rev. 2000;17:150-74 [DOI] [PubMed] [Google Scholar]
- 11. Farag SS, Ruppert AS, Mrozek K, et al. Outcome of induction and postremission therapy in younger adults with acute myeloid leukemia with normal karyotype: a Cancer and Leukemia Group B study. J Clin Oncol. 2005;23:482. [DOI] [PubMed] [Google Scholar]
- 12. Klepin HD, Balducci L. Acute myelogenous leukemia in older adults. Oncologist. 2009;14:222-32 [DOI] [PubMed] [Google Scholar]
- 13. Thomas X. Chemotherapy of acute leukemias in adults. Exp Opin Pharmacother. 2009;10:221-37 [DOI] [PubMed] [Google Scholar]
- 14. National Cancer Institute Acute myeloid leukemia. Available from: http://www.Seer.cancer.gov/statfacts/html/amyl.htm?statfacts_page=amyl.html&x=15&y=17
- 15. Jemal A, Siegel R, Xu J, Ward E. Cancer statistics 2010. CA Cancer J Clin. 2010;60;277-300 [DOI] [PubMed] [Google Scholar]
- 16. West RR, Stafford DA, White AD, Bowen DT, Padua RA. Cytogenetic abnormalities in the myelodysplastic syndromes and occupational or environmental exposure. Blood. 2000;95:2093-7 [PubMed] [Google Scholar]
- 17. Crane MM, Strom SS, Halabi S, et al. Correlation between selected environmental exposures and karyotype in acute myelocytic leukemia. Cancer Epidemiol Biomarkers Prev. 1996;5:639-44 [PubMed] [Google Scholar]
- 18. Preston DL, Kusumi S, Tomonaga M, et al. Cancer incidence in atomic bomb survivors: part III—leukemia, lymphoma and multiple myeloma, 1950-1987. Radiat Res. 1994;137:S68-97 [PubMed] [Google Scholar]
- 19. Frohling S, Scholl C, Gilliland DG, Levine RL. Genetics of myeloid malignancies: pathogenetic and clinical implications. J Clin Oncol. 2005;23:6285-95 [DOI] [PubMed] [Google Scholar]
- 20. Bennett JM, Catovsky D, Daniel MT, et al. Proposed revised criteria for the classification of acute myeloid leukemia: a report of the French-American-British Cooperative Group. Ann Intern Med. 1985;103:620-5 [DOI] [PubMed] [Google Scholar]
- 21. Harris N, Jaffe E, Diebold J, et al. WHO classification of neoplastic disease of the hematopoietic and lymphoid tissues: report of the clinical advisory committee meeting-Airlie House, Virginia. J Clin Oncol. 1999;17:3835-49 [DOI] [PubMed] [Google Scholar]
- 22. Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114:937-51 [DOI] [PubMed] [Google Scholar]
- 23. Martens JHA, Stunnenberg HG. The molecular signature of oncofusion proteins in acute myeloid leukemia. FEBS Lett. 2010;584:2662-9 [DOI] [PubMed] [Google Scholar]
- 24. Byrd JC, Mrózek K, Dodge RK, et al. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: results from Cancer and Leukemia Group B (CALGB 8461). Blood. 2002;100:4325-36 [DOI] [PubMed] [Google Scholar]
- 25. Mrózek K, Radmacher MD, Bloomfield CD, Marcucci G. Molecular signatures in acute myeloid leukemia. Curr Opin Hematol. 2009;16:64-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Melnick A, Licht JD. Deconstructing a disease: RARalpha, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood. 1999;93:3167-215 [PubMed] [Google Scholar]
- 27. Look AT. Oncogenic transcription factors in the human acute leukemias. Science. 1997;278:1059-64 [DOI] [PubMed] [Google Scholar]
- 28. Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer. 2007;7:233-45 [DOI] [PubMed] [Google Scholar]
- 29. Mitelman F, Johansson B, Mertens F, editors. Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer. 2010. Available from: http://cgap.nci.nih.gov/Chromosomes/Mitelman
- 30. Cameron ER, Neil JC. The Runx genes: lineage-specific oncogenes and tumor suppressors. Oncogene. 2004;23:4308-14 [DOI] [PubMed] [Google Scholar]
- 31. de Bruijn MF, Speck NA. Core-binding factors in hematopoiesis and immune function. Oncogene. 2004;23:4238-48 [DOI] [PubMed] [Google Scholar]
- 32. Davis JN, McGhee L, Meyers S. The ETO (MTG8) gene family. Gene. 2003;303:1-10 [DOI] [PubMed] [Google Scholar]
- 33. Meyers S, Lenny N, Hiebert SW. The t (8;21) fusion protein interferes with AML-1B-dependent transcriptional activation. Mol Cell Biol. 1995;15:1974-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Frank R, Zhang J, Uchida H, Meyers S, Hiebert SW, Nimer SD. The AML1/ETO fusion protein blocks transactivation of the GM-CSF promoter by AML1B. Oncogene. 1995;11:2667-74 [PubMed] [Google Scholar]
- 35. Lutterbach B, Hiebert SW. Role of the transcription factor AML-1 in acute leukemia and hematopoietic differentiation. Gene. 2000;245:223-35 [DOI] [PubMed] [Google Scholar]
- 36. Lutterbach B, Hou Y, Durst KL, Hiebert SW. The inv(16) encodes an acute myeloid leukemia 1 transcriptional corepressor. Proc Natl Acad Sci U S A. 1999;96:12822-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Eguchi M, Eguchi-Ishimae M, Greaves M. Molecular pathogenesis of MLL-associated leukemias. Int J Hematol. 2005;82:9-20 [DOI] [PubMed] [Google Scholar]
- 38. Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer. 2007;7:823-33 [DOI] [PubMed] [Google Scholar]
- 39. Mrózek K, Dohner H, Bloomfield CD. Influence of new molecular prognostic markers in patients with karyotypically normal acute myeloid leukemia: recent advances. Curr Opin Hematol. 2007;14:106-14 [DOI] [PubMed] [Google Scholar]
- 40. Patscka P, Marucci G, Rupport AS, et al. Adverse prognostic significance of KIT mutations in adult acute myeloid leukemia with inv(16) and t(8:21): a Cancer and Leukemia Group B study. J Clin Oncol. 2006;24:3904-11 [DOI] [PubMed] [Google Scholar]
- 41. Small D. Targeting FLT3 for the treatment of leukemia. Semin Hematol. 2008;45:S17-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stirewalt DL, Radich JP. The role of FLT3 in haematopoietic malignancies. Nat Rev Cancer. 2003;3:650-65 [DOI] [PubMed] [Google Scholar]
- 43. Tyner JW, Erikson H, Deininger MW, et al. High-throughput sequencing screen reveals novel, transforming RAS mutations in myeloid leukemia patients. Blood. 2009;113:1749-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nerlov C. C/EBPalpha mutations in acute myeloid leukaemias. Nat Rev Cancer. 2004;4:394-400 [DOI] [PubMed] [Google Scholar]
- 45. Ernst P, Wang J, Korsmeyer SJ. The role of MLL in hematopoiesis and leukemia. Curr Opin Hematol. 2002;9:282-7 [DOI] [PubMed] [Google Scholar]
- 46. Grisendi S, Mecucci C, Falini B, Pandolfi PP. Nucleophosmin and cancer. Nat Rev Cancer. 2006;6:493-505 [DOI] [PubMed] [Google Scholar]
- 47. Ley TJ, Ding L, Walter MJ, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363:2424-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Goldstone AH, Burnett AK, Avivi I, et al. Secondary AML has a worse outcome than de novo AML even taking into account cytogenetics and age. Blood. 2002;100:88 [Google Scholar]
- 49. Farag SS, Archer KJ, Mrozek K, et al. Pretreatment cytogenetics add to other prognostic factors predicting complete remission and long-term outcome in patients 60 years of age or older with acute myeloid leukemia: results from Cancer and Leukemia Group B 8461. Blood. 2006;108:63-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. de Lima M, Strom S, Keating M, et al. Implications of potential cure in acute myelogenous leukemia: development of subsequent cancer and return to work. Blood. 1997;90:4719-24 [PubMed] [Google Scholar]
- 51. Tallman MS, Gilliland DG, Rowe JM. Drug therapy of acute myeloid leukemia. Blood. 2005;106:1154-63 [DOI] [PubMed] [Google Scholar]
- 52. National Comprehensive Cancer Network Home page. Available from: www.nccn.org
- 53. Erba HP. Has there been progress in the treatment of older patients with acute myeloid leukemia? Best Pract Res Clin Haematol. 2010;23:495-501 [DOI] [PubMed] [Google Scholar]
- 54. Schlenk RF, Frohling S, Hartmann F, et al. Intensive consolidation versus oral maintenance therapy in patients 61 years or older with acute myeloid leukemia in first remission: results of second randomization of AML HD98-B treatment trial. Leukemia. 2006;20:748-50 [DOI] [PubMed] [Google Scholar]
- 55. Kohrt HE, Coutre SE. Optimizing therapy for acute myeloid leukemia. J Natl Compr Canc Netw. 2008;6:1003-16 [DOI] [PubMed] [Google Scholar]
- 56. Jones CV, Copelan EA. Treatment of acute myeloid leukemia with hematopoietic stem cell transplantation. Future Oncol. 2009;5:559-68 [DOI] [PubMed] [Google Scholar]
- 57. Deschler B, de Witte T, Mertelsmann R, Lübbert M. Treatment decision-making for older patients with high-risk myelodysplastic syndrome or acute myeloid leukemia: problems and approaches. Haematologica. 2006;91:1513-22 [PubMed] [Google Scholar]
- 58. Kell J. Treatment of relapsed acute myeloid leukemia. Rev Recent Clin Trials. 2006;1:103-11 [DOI] [PubMed] [Google Scholar]
- 59. Stapnes C, Gjertsen BG, Reikvam H, Bruserud O. Targeted therapy in acute myeloid leukaemia: current status and future directions. Expert Opin Investig Drugs. 2009;18:433-55 [DOI] [PubMed] [Google Scholar]
- 60. Stone RM, DeAngelo DJ, Klimek V, et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood. 2005;105:54-60 [DOI] [PubMed] [Google Scholar]
- 61. Fathi AT, Levis M. Lestaurtinib: a multi-targeted FLT3 inhibitor. Expert Rev Hematol. 2008;2:17-26 [DOI] [PubMed] [Google Scholar]
- 62. Knapper S, Burnett AK, Littlewood T, et al. A phase 2 trial of the FLT3 inhibitor lestaurtinib (CEP701) as first-line treatment for older patients with acute myeloid leukemia not considered fit for intensive chemotherapy. Blood. 2006;108:3262-70 [DOI] [PubMed] [Google Scholar]
- 63. Zarrinkar PP, Gunawardane RN, Cramer MD, et al. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood. 2009;114:2984-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Al-Kali A, Jones D, Cortes J, et al. Patterns of molecular response to and relapse after combination of sorafenib, idarubicin, and cytarabine in patients with newly diagnosed FLT3-mutant acute myeloid leukemia (AML). Blood. 2009;114 Abstract 2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Serve H, Wagner R, Sauerland C, et al. Sorafenib in combination with standard induction and consolidation therapy in elderly AML patients: results from a randomized, placebo-controlled phase II trial. Blood. 2010;116:333 [Google Scholar]
- 66. Lancet JE, Cortes JE, Hogge DE, et al. Phase 2B randomized study of CPX-351 vs. cytarabine (CYT) + daunorubicin (DNR) (7 + 3 regimen) in newly diagnosed AML patients aged 60-75. Program and abstracts from the 52nd American Society of Hematology Annual Meeting and Exposition. Blood. 2010;116:655 [Google Scholar]
- 67. Faderl S, Ravandi F, Garcia-Manero G, et al. Frontline therapy for older patients (pts) with acute myeloid leukemia (AML): clofarabine plus low-dose cytarabine induction followed by prolonged consolidation with clofarabine plus low-dose cytarabine alternating with decitabine. Blood. 2010;116:336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sekeres MA, Gundacker H, Lancet J, et al. A phase II study of lenalidomide for previously untreated deletion (del) 5q acute myeloid leukemia (AML) patients age 60 or older who are not candidates for remission induction chemotherapy (Southwest Oncology Group Study S0605). Blood. 2010;116 Abstract 332 [Google Scholar]
- 69. Garcia-Manero G, Tambaro FP, Bekele NB, et al. Final report of a phase II trial of vorinostat, idarubicin and cytarabine in previously untreated acute myelogenous leukemia (AML) or high risk myelodysplastic syndrome (MDS). Blood. 2010;116:2189 [Google Scholar]
- 70. Schaefer EW, Loaiza-Bonilla A, Juckett M, et al. A phase 2 study of vorinostat in acute myeloid leukemia. Haematologica. 2009;94:1375-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Erba HP, O’Donnell M, Allen SL, et al. Amonafide L-malate (AS1413) in combination with cytarabine is equally effective in older and younger patients with secondary acute myeloid leukemia (AML): final data from a phase II study. Blood. 2009;114 Abstract 1047 [Google Scholar]
- 72. Löwenberg B, Beck J, Graux C, et al. Gemtuzumab ozogamicin as postremission treatment in AML at 60 years of age or more: results of a multicenter phase 3 study. Blood. 2010;115:2586-91 [DOI] [PubMed] [Google Scholar]
- 73. Didier C, Cavelier C, Quaranta M, et al. Evaluation of Polo-like kinase 1 inhibition on the G2/M checkpoint in acute myelocytic leukaemia. Eur J Pharmacol. 2008;591:102-5 [DOI] [PubMed] [Google Scholar]
- 74. Silverman LR, Navada SC, Odchimara-Reissig R, et al. Evaluation of ON01910.Na in patients with a myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML) relapsed or refractory to hypomethylating agents: a phase I study. Blood. 2010;116:2944 [Google Scholar]
- 75. McCormack E, Bruserud O, Gjertsen BT. Review: genetic models of acute myeloid leukemia. Oncogene. 2008;27:3765-79 [DOI] [PubMed] [Google Scholar]