

Abstract
The 5-year survival rate is very low when breast cancer becomes metastatic. The metastatic process is governed by a network of molecules of which SLUG is known to play a major role as a regulator of epithelial-to-mesenchymal transition (EMT). Prostate-derived ETS factor (PDEF) has been proposed as a tumor suppressor, possibly through inhibition of invasion and metastasis; therefore, understanding the mechanism of PDEF regulation may help to better understand its role in breast cancer progression. This study shows for the first time that the transcription factor SLUG is a direct target of PDEF in breast cancer. We show that the expression of PDEF is able to suppress/dampen EMT through the negative regulation of SLUG. In addition, we show that PDEF is also able to regulate downstream targets of SLUG, namely E-cadherin, in both SLUG-dependent and -independent manners, suggesting a critical role for PDEF in regulating EMT.
Keywords: ETS transcription factors, SLUG, breast cancer, epithelial-to-mesenchymal transition, PDEF
Introduction
Breast cancer is the leading cause of death among women in the Western world. At the time of diagnosis, 10% of breast cancers have progressed to the most lethal stage (metastatic), and after initial treatment of primary tumors, metastatic disease often occurs, indicating rapid disease progression to a dedifferentiated state (http://seer.cancer.gov/statfacts/html/breast.html). An increased understanding of the molecular etiology of breast cancer and the metastatic stage is a priority if mortality rates are to be reduced.
The E twenty-six (ETS) family of transcription factors functions in most biological pathways involved in normal cell as well as tumor cell growth and development (see reviews1,2). Prostate-derived ETS factor (PDEF) is an epithelial-specific member of the ETS family. Several studies have identified PDEF mRNA overexpression in breast and prostate tumor samples, which initially led to reports suggesting an oncogenic role for PDEF in cancer progression.3,4 However, molecular examination at the level of protein demonstrates a correlation between PDEF protein loss and increased aggressiveness in breast, prostate, ovarian, and colon cancer, suggesting tumor-suppressive functions.5-11
Although one study identified PDEF as a stimulator of cell migration in combination with colony-stimulating factor receptor 1,12 the vast majority of papers demonstrate tumor-suppressive effects for PDEF. Examination in multiple immortalized cancer cell lines correlates PDEF protein loss with increased cell growth, migration, and invasion. Re-expression of PDEF in invasive cells reciprocally reduces these same processes.5,6,8-10 PDEF’s role as a potential tumor suppressor was further substantiated by studies demonstrating that PDEF-mediated inhibition of invasive potential is correlated with the reduced expression of cancer-promoting genes such as uPA, survivin, MMPs, and vimentin as well as the increased expression of cancer-suppressive genes such as maspin, E-cadherin, and p21.6,7,13,14 In vivo, orthotopic implantation of PDEF-expressing breast cancer cells into mouse mammary glands inhibits tumor formation and growth and partially blocks G1/S cell cycle progression.14 Reciprocally, RNAi-mediated loss of PDEF expression in breast cancer cells promotes tumor formation in xenograft mouse models.6 A possible mechanism that contributes to the disparity between PDEF RNA and protein levels was recently made clear by the description of microRNA (miRNA)–mediated inhibition of PDEF translation. Both miRNA-204 (miR-204) and miRNA-510 (miR-510) bind to the 3′ untranslated region (3′UTR) of the PDEF mRNA to inhibit its translation, resulting in the functional inhibition of cell growth, migration, and invasion.15 This indicates a possible mechanism to rapidly inhibit PDEF function, leading to tumor progression.
The epithelial-to-mesenchymal transition (EMT) was originally characterized in embryonic development studies in which epithelial cells acquire the mesenchymal characteristics of decreased cell-to-cell contacts, increased cell motility, and increased invasiveness.16 It is now apparent that tumor cells activate specific pathways associated with EMT in order to promote these same processes and increase their invasive potential. Studies demonstrate the ability of several transcription factors (e.g., SLUG [SNAI-2], SNAIL [SNAI-1], ZEB-1, ZEB-2, and TWIST) to induce a partial or sometimes full EMT during cancer progression (see review17). Altered expression of these factors often results in the loss of the cell adhesion molecule E-cadherin, leading to the expression of mesenchymal proteins such as vimentin and N-cadherin. These are identified as critical features of EMT in tumor progression.
SLUG is a member of the SNAIL superfamily and was first identified as a developmental protein critical for neural crest formation in chick embryos.18 SLUG expression is associated with biological aggressiveness in several tumor types, and high expression in breast tumors associates with the aggressive basal phenotype.19 SLUG expression is correlated with reduced cell adhesion and increased cell migration and invasion and is associated with lymph node invasion and metastatic progression in breast tumors. SLUG is inversely correlated with E-cadherin expression and is a critical event–promoting EMT in many tumor types.20 Studies from this laboratory and others associate PDEF transcriptional regulation with altered levels of the EMT-associated SLUG transcription factor. Microarray gene expression analysis in multiple invasive breast cancer cells identified SLUG as a potential PDEF transcriptional target in breast cancer cells.7,13 RNAi-mediated loss of PDEF in prostate cancer cells increases the expression of mesenchymal-associated genes including SLUG, promotes a mesenchymal morphology, and increases invasive potential reminiscent of an EMT.7
This study identifies PDEF as a direct transcriptional regulator of SLUG during breast cancer progression and associates PDEF with a SLUG-dependent and -independent effect on the EMT phenotype. Phenotypic rescue experiments identify SLUG repression by PDEF as a critical interaction in inhibiting the migratory phenotype.
Results
Modulated PDEF levels alter SLUG expression in invasive breast cancer cells
Invasive breast cancer cells MDA MB 231 were infected at low multiplicity of infection (MOI) with adenovirus expressing the full-length human PDEF protein, and the effect on SLUG mRNA levels was examined by Northern blot. Expression of PDEF in this context significantly reduced SLUG mRNA (Fig. 1A) and validated our microarray data, which originally identified the PDEF-mediated downregulation of SLUG in 3 invasive breast cancer cell lines.13 Adenoviral-mediated PDEF overexpression also reduced SLUG protein levels in the breast cancer cell line MDA MB 231 compared to controls infected with GFP-expressing virus (Fig. 1B). Immunofluorescence studies using a SLUG-specific antibody also demonstrate the reduced expression of SLUG protein upon doxycycline-induced PDEF re-expression in MDA MB 157 cells (Fig. 1C). In agreement with earlier published data, re-expression of PDEF in MDA MB 157 cells produced a more epithelial-like morphology and altered actin cytoskeletal organization reminiscent of a mesenchymal-to-epithelial transition (MET) (Fig. 1C).
Figure 1.
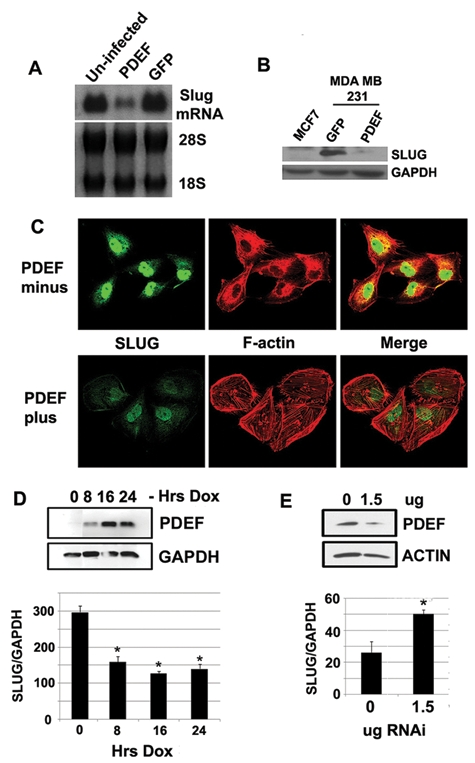
Modulated PDEF expression alters SLUG levels. (A) Northern blot analysis of SLUG expression levels in MDA MB 231 breast cancer cells with adenoviral-mediated PDEF and GFP expression. 28S and 18S were used as loading controls. (B) Western blot analysis of SLUG expression levels in MDA MB 231 breast cancer cells with adenoviral-mediated PDEF and GFP expression. GAPDH was used as a loading control. (C) Immunofluorescent staining of doxycycline-inducible MDA MB 157 cells probed for SLUG (left panel) and F-actin (middle panel) before and after induction of PDEF expression with 1 ug/mL doxycycline. The right panel shows a merged SLUG/F-actin image. (D) Western blot analysis of PDEF expression (upper panel) and quantitative real-time PCR of SLUG mRNA expression (lower panel) in MDA MB 157 cells treated with 1 ug/mL doxycycline for the times indicated. GAPDH was used as a loading control in the Western blot and for normalization in the PCR. *P < 0.005. (E) Western blot analysis of PDEF expression (upper panel) and quantitative real-time PCR of SLUG mRNA expression (lower panel) in MCF7 cells transfected with PDEF RNAi vector at the concentrations indicated. Actin was used as a loading control in the Western blot, and GAPDH was used for normalization in the PCR. *P < 0.05.
Using the same doxycycline-inducible expression system to express PDEF protein to examine the kinetics of response, we observed an optimal downregulation of SLUG expression levels after 8 hours in invasive MDA MB 157 breast cancer cells. This was not further decreased with increased PDEF expression (Fig. 1D). Using a previously described shRNA vector10 targeting endogenous PDEF expression in the noninvasive breast cancer cell line MCF7, we observed a concomitant increase in SLUG expression at the level of RNA as PDEF expression decreases (Fig. 1E).
PDEF directly alters SLUG expression through promoter occupancy
While the transcriptional function of SLUG in the context of an EMT is being intensively examined, relatively little is known about its own transcriptional regulation. To examine if the SLUG promoter is responsive to PDEF during cancer progression, we performed luciferase reporter assays in MCF7 cells. Cells were cotransfected with a reporter luciferase gene (pGL3) fused to 0.9 kb (−0.9) or 1.8 kb (−1.8) of the SLUG promoter and a vector containing control or PDEF shRNA vector. Compared to controls, PDEF expression was reduced over 75% by treatment with the shRNA vector as shown by Western blot (Fig. 2A). Basal luciferase activity was increased over 2-fold for the 0.9-kb region and a little under 2-fold for the 1.8-kb region upon the knockdown of PDEF expression (Fig. 2B). Sequence analysis of the first 2-kb region upstream of the SLUG translational start site (using the AliBaba 2.1. transcription factor prediction database: www.gene-regulation.com/pub/programs/alibaba2/index.html) identified 4 consensus ETS-binding sites (EBSs) situated 58, 179, 1,530, and 1,930 bases upstream of the SLUG translational start site (Fig. 2C). Chromatin immunoprecipitation (ChIP) was used to isolate endogenous PDEF-bound DNA fragments from total chromatin from MCF7 breast cancer cells. Using primer sets spanning the potential EBSs (Fig. 2C), conventional and real-time PCR amplification of the immunoprecipitated fragments demonstrated enrichment of the SLUG promoter, indicating that PDEF is bound to the promoter (Fig. 2D) at the EBS situated −179 bases upstream of the SLUG translational start site (Fig. 2C). A significant enrichment of PDEF-specific chromatin amplification was observed by conventional and real-time PCR analysis when compared to IgG and no DNA controls (Fig. 2D). No enrichment was observed for the remaining 3 EBSs or at a region lacking EBS within the SLUG coding region, demonstrating specificity for PDEF at the −179 EBS.
Figure 2.
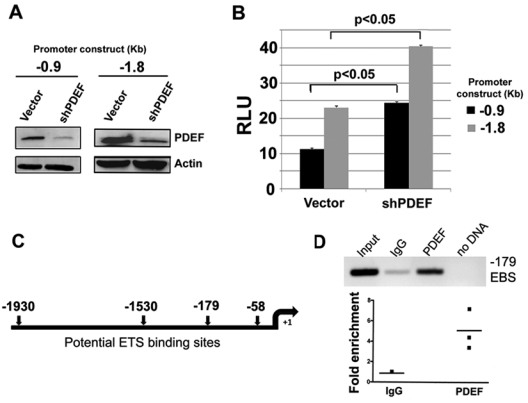
PDEF directly alters SLUG expression through promoter occupancy. (A) Western blot analysis of PDEF expression in MCF7 cells cotransfected with a reporter luciferase gene (pGL3) fused to 0.9 kb (−0.9) and 1.8 kb (−1.8) of the SLUG promoter and a vector containing control (Vector) or PDEF RNAi (shPDEF) vector. Actin was used as a loading control. (B) Luciferase activity in MCF7 cells cotransfected with a reporter luciferase gene (pGL3) fused to 0.9 kb (−0.9) and 1.8 kb (−1.8) of the SLUG promoter and a vector containing control (Vector) or PDEF RNAi (shPDEF) vector. Luciferase activity values are normalized to protein concentration. The luciferase data are expressed as the mean ± standard deviation for 3 experiments conducted in triplicate. (C) Schematic representation of the 4 EBSs found within 2 kb of the SLUG translational start site using the AliBaba database. (D) ChIP analysis of the EBS situated 179 bp upstream of the SLUG translational start site. Endogenous PDEF-bound DNA fragments were isolated from total chromatin from MCF7 breast cancer cells. Conventional (upper panel) and real-time PCR (lower panel) amplification using appropriate primer sets spanning the potential EBS was used to assess fragment enrichment over IgG control (P < 0.05).
Exogenous SLUG expression restores the migratory phenotype inhibited by PDEF occupancy of the SLUG promoter
The loss of PDEF expression increases cell migration in noninvasive breast cancer cells, while its re-expression inhibits the same process in invasive cells.8 A reciprocal pattern is observed for SLUG in which its elevated expression increases and its loss inhibits migratory ability. As PDEF is identified as a direct repressor of SLUG expression, phenotypic rescue experiments were used to examine the ability of exogenous SLUG to restore the migratory phenotype transcriptionally inhibited by PDEF expression (Fig. 3A). MDA MB 231 cells were transfected with pcDNA3 vector (control), pcDNA3-PDEF, or pCMV-3Tag-SLUG, respectively, and also dual transfected with pcDNA-PDEF and pCMV-3Tag-SLUG. Protein expression analysis by Western blot confirmed the PDEF and SLUG (M2-FLAG) expression levels in each transfection (Fig. 3B). Phenotypic analysis using transwell migration chambers confirmed the PDEF-mediated inhibition of migration and SLUG-mediated increase in migration compared to controls (Fig. 3C). Significantly, when exogenous PDEF and SLUG were expressed simultaneously, the exogenous SLUG expression restored the migratory phenotype back to that observed upon SLUG expression alone (Fig. 3C). SLUG is a well-characterized repressor of E-cadherin expression during EMT. Endogenous expression of E-cadherin in wild-type MDA MB 231 invasive breast cancer cells is not detectable by Western blot (Fig. 3B). However, re-expression of pcDNA-PDEF in this cell line promoted significant expression of the E-cadherin protein, and this expression was again lost upon exogenous expression of both PDEF and SLUG.
Figure 3.
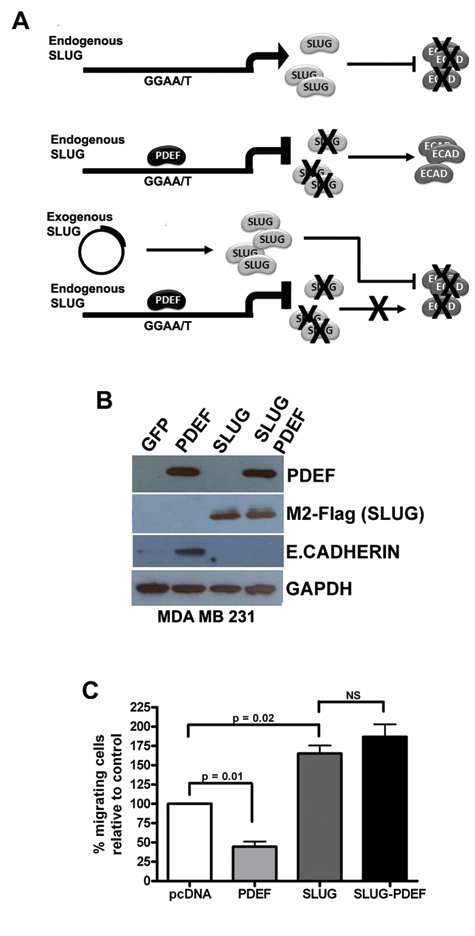
Exogenous SLUG expression restores the migratory phenotype inhibited by PDEF. (A) Schematic outline of the phenotypic rescue experiment. Endogenous SLUG expression inhibits the expression of E-cadherin. When PDEF is expressed, it binds to the SLUG promoter to inhibit its transcription and in turn to allow E-cadherin expression. The introduction of exogenous SLUG via transfection with an expression vector is expected to restore the inhibition of E-cadherin expression, as the vector promoter is not under PDEF regulation. (B) Western blot analysis of PDEF, M2 FLAG (SLUG), and E-cadherin expression in MDA MB 231 cells transfected with pcDNA vector (control), pcDNA-PDEF, pCMV-3Tag-SLUG, respectively, and also dual transfected with pcDNA-PDEF and pCMV-3Tag-SLUG. GAPDH was used as a loading control. (C) Transwell migration assays were used to assess the migratory ability of MDA MB 231 cells transfected with pcDNA vector (control), pcDNA-PDEF, pCMV-3Tag-SLUG, respectively, and also dual transfected with pcDNA-PDEF and pCMV-3Tag-SLUG.
PDEF expression alters the levels of EMT-associated proteins in MCF10A cells
MCF10A cells are nontransformed, spontaneously immortalized breast cells that, when plated at a low cell density (or sparse), undergo spontaneous morphological and phenotypic EMT-like changes.21 In contrast, MCF10A cells plated at a high cell density (or confluent) form a more epithelial compact shape. These changes are associated with a specific network of gene changes including cadherin switching (E- to N-cadherin expression) and the upregulation of SLUG and vimentin in sparse cultures. Because of the negative regulation of SLUG by PDEF observed in this study, we decided to explore the potential role of PDEF in density-dependent EMT. When cells were plated at low density (sparse), the mRNA levels of SLUG (Fig. 4B) and vimentin (Fig. 4C) were expressed at higher levels than those plated at high density (confluent). PDEF was able to inhibit both SLUG and vimentin expression when expressed under both sparse and confluent conditions (Fig. 4B and 4C). The inhibition of vimentin is also observed at the level of protein in sparse cultures overexpressing PDEF (Suppl. Fig. S1). In contrast, vimentin protein levels were undetectable in confluent cultures (Suppl. Fig. S1). Interestingly, PDEF was only able to increase the levels of E-cadherin when expressed under confluent conditions compared to control transfected cells (Fig. 4D). In contrast, a slight decrease in the expression levels of E-cadherin was observed when PDEF was expressed under sparse conditions (Fig. 4D). E-cadherin protein levels were undetectable in sparse cultures and remained relatively unchanged in confluent cultures upon PDEF overexpression (Suppl. Fig. S1).
Figure 4.
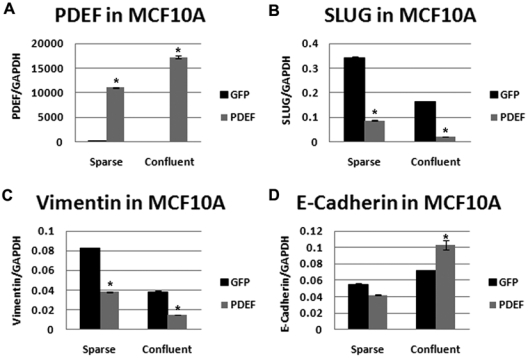
PDEF expression alters EMT markers in MCF10A cells. Quantitative real-time PCR of (A) PDEF, (B) SLUG, (C) vimentin, and (D) E-cadherin in MCF10A cells plated at low (sparse) and high (confluent) density and transfected with either GFP (black bars) or PDEF (gray bars) normalized to GAPDH. *P < 0.05.
PDEF alters the expression of EMT-associated proteins independent of SLUG expression
In order to examine whether PDEF is able to modulate EMT-related proteins in a SLUG-independent manner, we utilized the nontransformed, immortalized mouse cell line NMuMG. These cells undergo EMT-like changes when treated with TGFβ.22 Early TGFβ-induced EMT is reported to be SLUG independent.23 Indeed, E-cadherin loss is not an early EMT event, and in NMuMG cells, a dramatic decrease in E-cadherin expression is not observed until 6 to 9 days after TGFβ stimulation.24 Therefore, we expressed PDEF in these cells and evaluated TGFβ-induced EMT changes. As expected, upon induction of EMT in control cells, we observed an EMT-like morphological change (Fig. 5A) together with a slight reduction in E-cadherin and an increase in vimentin expression (Fig. 5B). However, PDEF-expressing cells retained their epithelial morphology (Fig. 5A), and E-cadherin levels were not reduced upon TGFβ stimulation, and the increase in vimentin levels was diminished (Fig. 5B). We examined SLUG mRNA levels, and although a modest increase (1.5-fold) was observed after 24 and 48 hours of treatment with TGFβ, this increase was not significantly changed (2.5-fold) upon PDEF overexpression (Fig. 5C). Similar observations were made for the transcription factor SNAIL. However, a significant increase in the mRNA levels of TWIST (6.9-fold) was observed after TGFβ stimulation, and these were significantly diminished (2.8-fold) in PDEF-expressing cells (Fig. 5C).
Figure 5.
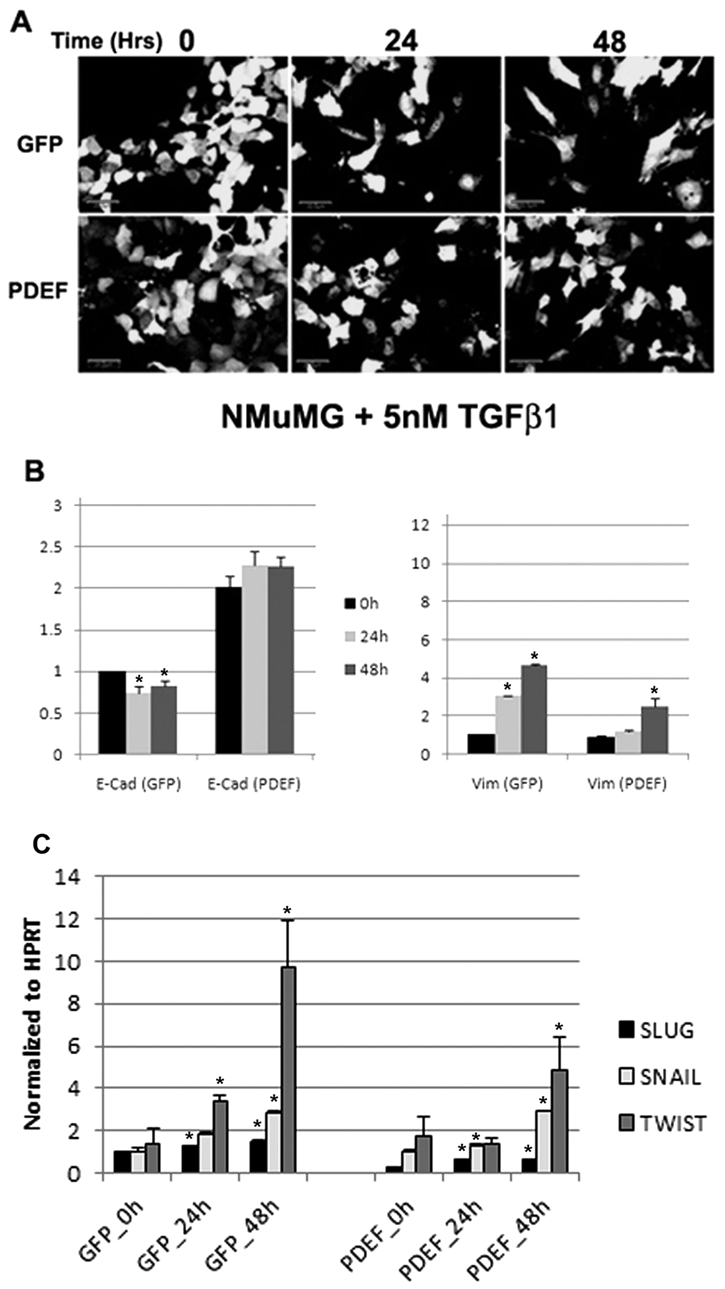
PDEF expression inhibits TGFβ-induced EMT in NMuMG cells. (A) Confocal images (scale bar represents 20 µM) of NMuMG cells expressing GFP alone or PDEF/GFP treated with 5 nM TGFβ for 0, 24, and 48 hours. Quantitative real-time PCR of (B) E-cadherin (E-Cad) and (C) vimentin (Vim) in NMuMG cells expressing GFP alone or PDEF/GFP treated with 5 nM TGFβ for 0 (black bars), 24 (light gray bars), or 48 (dark gray bars) hours normalized to GAPDH. (D) Quantitative real-time PCR of SLUG (black bars), SNAIL (light gray bars), and TWIST (dark gray bars) in NMuMG cells expressing GFP alone or PDEF/GFP treated with 5 nM TGFβ for 0, 24, or 48 hours normalized to HPRT. *P < 0.05.
Discussion
Cancer death is due in large part to metastases.25,26 One of the more interesting challenges is to understand the cellular changes that occur during progression towards a more invasive cancer phenotype. ETS proteins represent one of the largest families of transcription factors with diverse functions and activities that activate or repress the expression of genes that are involved in various biological processes, including cellular proliferation, differentiation, development, transformation, and apoptosis.1,2 The ETS family gene, PDEF, is expressed in normal epithelial tissues including prostate, breast, and colon.27 In normal and noninvasive cancers, PDEF protein and mRNA levels are easily detectable by Northern and Western blot. However, during progression to a more invasive phenotype, PDEF protein is reduced or lost. Understanding the consequences of PDEF loss during breast cancer progression will lead to a better insight into the transcriptional networks in cells and may provide novel therapeutic targets.
We show here that PDEF is a negative regulator of SLUG expression at the level of RNA and protein. Furthermore, we show that PDEF is a direct regulator of the SLUG promoter by luciferase and ChIP assays. Previous studies in prostate have also shown that PDEF is a direct negative regulator of the SLUG promoter using luciferase assay.7 However, this is the first study to show a direct interaction in vivo using ChIP analysis. Furthermore, we show that the negative regulation of SLUG by PDEF is localized to the −200-bp region of the promoter.
E-cadherin is a calcium-dependent, cell-cell adhesion glycoprotein, comprised of 5 extracellular cadherin repeats, a transmembrane region, and a highly conserved cytoplasmic tail. Loss of function is thought to contribute to progression in cancer by increasing proliferation, invasion, and/or metastasis.28,29 E-cadherin loss in tumors contributes to metastatic dissemination by inducing wide-ranging transcriptional and functional changes.30 E-cadherin is also known to be a transcriptional target of SLUG during cancer progression. Importantly, cadherin switching (E-cadherin levels decrease, and N-cadherin levels increase) has been associated with the progression of cancer and to be necessary for EMT. MCF10A cells are often used in breast cancer studies, as they are a nontransformed, immortalized human cell line. Interestingly, MCF10A cell morphology is dependent on cell density in 2-dimensional culture. That is, in low density (sparse) cultures, the cells appear more mesenchymal, but in high density (confluent) cultures, they appear more epithelial.24 The study by Maeda et al.24 showed that the mesenchymal appearance in sparse cultures was reminiscent of that observed in TGFβ-mediated EMT. Indeed, we observed that vimentin and SLUG levels were higher in sparse cultures and, more importantly, that these levels were reduced by the re-expression of PDEF. Interestingly, although it was shown that SLUG regulates E-cadherin in SW 480 colon cancer cells in a cell density–dependent manner,31 we did not observe an increase in E-cadherin levels upon PDEF re-expression in sparse MCF10A cultures, even though a significant decrease in SLUG expression was observed. These data suggest that PDEF is able to relieve the repression of E-cadherin through direct inhibition of SLUG when SLUG is expressed at low levels and perhaps suggest an alternate or additional mechanism for E-cadherin regulation in cells expressing high levels of SLUG.
NMuMG cells are used as a model system to study the role of TGFβ-induced EMT in breast cancer. Upon stimulation with TGFβ, these cells undergo a rapid morphological change and a genetic EMT reprogramming (e.g., increase in mesenchymal markers vimentin, ZO-1, and N-cadherin and decrease in the epithelial marker E-cadherin). Interestingly, the expression of PDEF in these cells was able to block the morphological change in these cells as well as dampen the genetic reprogramming, as evidenced by the expression levels of vimentin and E-cadherin. In this model, the increase in vimentin is an early event in EMT, whereas cadherin switching comes later; therefore, the reduction in E-cadherin levels, although significant, is only slight.23
Furthermore, it has been demonstrated that the early TGFβ1-induced EMT in NMuMG cells is a SLUG-independent process, suggesting that PDEF is able to inhibit breast cancer progression through the elevation of E-cadherin in both SLUG-dependent and -independent manners. In fact, the data in this study suggest PDEF may inhibit EMT through a TWIST-dependent manner, although further examination of this connection is required. Taken together, these data suggest that PDEF may be a more appealing therapeutic target in breast cancers, as a recent study shows that SLUG expression is not always associated with downregulation of E-cadherin.32 More recently, studies have begun to report on the relationship between EMT and stem cells.33,34 Specifically, studies have shown that the induction of EMT in breast cells results in the expression of stem cell markers.35 In addition, they showed that stem-like cells isolated from mammary carcinomas expressed EMT markers. Another study has shown that EMT traits in human breast cancer cell lines parallel the CD44high CD24low stem cell phenotype in breast cancer.36 These data have important implications to the work described here in that inhibition of the EMT phenotype via direct regulation of important drivers of this process, namely SLUG, may represent a novel mechanism of breast cancer stem cell inhibition by targeting the important tumor-initiating cells in breast cancer. Future studies directed towards understanding the exact role of PDEF in these processes are currently underway.
Materials and Methods
Cell culture
Human breast epithelial cell lines were obtained from American Type Culture Collection (Manassas, VA) and maintained at 37°C with 5% CO2 in medium supplemented with 10% fetal bovine serum and 100 U of penicillin/streptomycin. MDA MB 231, MDA MB 157, and NMuMG were grown in DMEM. MCF7 was also grown in DMEM supplemented with 1 mM sodium pyruvate, 1 mM sodium bicarbonate, 2 mM L-glutamine, 0.1 mM nonessential amino acids, and 0.01 mg/mL insulin. MCF10A cells were grown in DMEM:F12 (50:50) supplemented with 2 mM L-glutamine, 5% horse serum, 10 µg/mL insulin, 20 ng/mL epidermal growth factor (EGF), 500 ng/mL hydrocortisone, and 10 µg/mL cholera toxin. MDA MB 157 stable clones expressing doxycycline-inducible human PDEF were grown in DMEM containing 150 ug/mL hygromycin and 200 ug/mL G418. Stable cell lines were grown in growth media supplemented with 200 ug/mL G418. All tissue culture reagents were purchased from Invitrogen (Carlsbad, CA).
Modulation of PDEF expression
The construction and experimental conditions for exogenous adenoviral and inducible PDEF expression have been described previously.5,8 For PDEF gene knockdown, MCF7 cells at 70% confluence were transfected with a short hairpin RNA (shRNA) vector construct in pSuppressor (Imgenex, San Diego, CA) by using Lipofectamine 2000 (Invitrogen), as per the manufacturer’s instructions. A 19-nucleotide loop (GCTATGGCCGCTTCATTAG) located at position 1,206 to 1,224 of the open reading frame (ORF) region of the PDEF gene (Gen-Bank accession no. NP 036523) was used to target PDEF mRNA.10 Cells were cultured for 48 hours before RNA and protein collection. Controls consisted of cells transfected with 1.5 ug of vector alone. Each transfection contained 1.5 ug of total DNA, consisting of the indicated amounts of PDEF shRNA vector adjusted to total amount with the vector DNA. GFP expression from a bicistronic promoter was used to assess transfection efficiency.
Expression constructs and transfection protocol
SLUG cDNA coding regions were amplified by PCR using the primers detailed in Supplementary Table S1 and ligated into pCMV-3Tag (Stratagene, Santa Clara, CA) by using appropriate restriction sites. Plasmid DNA from sequence-verified positive clones was isolated using the ENDO-free plasmid purification kit (Qiagen, Valencia, CA). Plasmid DNA (1 ug) was transfected into cells using Lipofectamine 2000 (Invitrogen), as per the manufacturer’s instructions, and then incubated for 48 hours before RNA and protein collection and/or assessment of migration and localization.
Northern blot analysis
Total RNA (20 µg) was fractionated on 1% agarose gels containing 0.66 M formaldehyde. RNA quality was assessed by ethidium bromide visualization of 28S and 18S rRNA. RNA was transferred to nylon membranes (Duralon, Stratagene) in 0.1 M sodium phosphate buffer (pH 6.8), UV crosslinked, and hybridized for 2 hours at 65°C in QuikHyb (Stratagene). SLUG α-32P-dCTP–labeled probes were prepared by random-primed synthesis using PrimeIt (Stratagene). Washed membranes were exposed to X-ray film for autoradiography.
Western blot
Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer, containing protease inhibitors (complete protease inhibitors; Roche, Nutley, NJ) and phosphatase inhibitors (Sigma-Aldrich, St. Louis, MO). Equal amounts of total protein (as determined using bicinchoninic acid protein assay kit; Pierce Chemical, Rockford, IL) were resolved by SDS–polyacrylamide gel electrophoresis and subjected to Western blot analysis by using enhanced chemiluminescence (Pierce Chemical). Blots were probed using commercially available antibodies as detailed. Human PDEF antibody was generated as described previously5 and was purified against truncated N-terminal PDEF protein (amino acids 1-141) lacking its pointed domain and ETS domain. Commercial antibodies used were SLUG rabbit polyclonal (Calbiochem, San Diego, CA), E-cadherin mouse monoclonal (BD Transduction Laboratories, San Diego, CA), vimentin mouse monoclonal (MAB 3400, Millipore, Billerica, MA), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) rabbit polyclonal (Abcam, Cambridge, MA). Anti-rabbit, anti-mouse, and anti-goat horseradish peroxidase-conjugated secondary antibodies were purchased from GE Healthcare (Piscataway, NJ).
Transwell migration assay
Treated or untreated control cells were seeded in triplicate into the upper chamber of a transwell insert (BD Biosciences, San Diego, CA) precoated with 5 ug/mL fibronectin (Thermo Fisher Scientific, Hudson, NH) in serum-free media at a density of 50,000 cells per well. Media containing 10% serum were placed in the lower chamber to act as a chemoattractant, and cells were further incubated for 6 hours. Nonmigratory cells were removed from the upper chamber by scraping, and the cells remaining on the lower surface of the insert were stained using Diff-quick (Dade Behring, Newark, DE). Cells were quantified as the number of cells found in 10 random microscope fields. Error bars represent the standard deviation from 3 separate experiments.
Immunofluorescence
Cells were seeded onto sterile coverslips (18 mm in diameter) coated with 5 ug/mL fibronectin and allowed to attach overnight. Cells were fixed with 2% formaldehyde, permeabilized with 0.1% Triton X-100, and blocked in 2% bovine serum albumin (BSA) for 1 hour at room temperature. SLUG localization was examined using the antibodies detailed above and visualized using appropriate (480 and 540 nm) Alexa-Fluor secondary antibodies (Invitrogen). Immunofluorescence was examined using an Olympus IX70 confocal microscope (Center Valley, PA).
Reverse transcription and real-time PCR
Total RNA from cancer cell lines was extracted using the RNeasy Plus Mini Kit (Qiagen). Also, 1 µg total RNA was reverse transcribed in a 20-µL reaction using iScript (Bio-Rad, Hercules, CA). Real-time PCR for gene expression was performed with 5 µL of a 1:20 dilution of reverse-transcribed cDNA using the Universal Probe Library (UPL) system (Roche) in a LightCycler 480 (Roche). The cycling conditions were performed as per the manufacturer’s instructions. Primer sequences and probe numbers are described (Suppl. Table S1). Triplicate reactions were run for each cDNA sample. The relative expression of each gene was quantified on the basis of Ct value measured against an internal standard curve for each specific set of primers (Suppl. Table S1) using the software provided by the instrument manufacturer (Roche). These data were normalized to GAPDH or HPRT.
Chromatin immunoprecipitation
Chromatin was prepared, and immunoprecipitation was performed using MCF7 cells (express endogenous PDEF) in a 2-step crosslinking protocol as described previously.37 (Nowak et al. [2005]). Chromatin was fragmented into 500- to 1,000-bp fragments by sonicating the cells 8 times for 10 seconds at level 3 in an ethanol ice bath by using a Virsonic 475 sonicator (Virtis, Gardiner, NY). Soluble chromatin was quantified (absorbance at 260 nM), and 10 absorbance units were incubated with 2 ug of PDEF rabbit polyclonal antibody or immunoglobulin G (IgG) alone for 4 hours. Collection, washing, and reverse crosslinking of immune complexes were as described previously.37 (Nowak et al. [2005]). Primers (Suppl. Table S1) spanning EBSs (Fig. 2C) were used to examine PDEF occupancy by conventional and real-time PCR. For real-time PCR, 3 uL of purified chromatin was used in each real-time reaction, which was conducted using a LightCycler (Roche Diagnostics, Basel, Switzerland) with the Platinum SYBR Green qPCR SuperMix (Invitrogen), as per the manufacturers’ instructions. Primers were used at a concentration of 250 nM, and the cycling conditions were as follows: preincubation, 50°C for 10 minutes, 95°C for 2 minutes, followed by 40 to 50 cycles of denaturation at 95°C; annealing at 58°C and extension at 72°C, all for 20 seconds, with a single data acquisition at the end of each extension. Melting curve analysis was carried out as per the manufacturer’s recommendations. All primer sequences are listed in Supplementary Table S1. Relative enrichment was expressed as the percentage of total input.
Phenotypic rescue
MDA MB 231 invasive breast cancer cells were transfected with 1) pcDNA and pCMV-3Tag vector (control), 2) pcDNA expressing PDEF, 3) pCMV-3Tag expressing SLUG, and 4) a combination of pcDNA expressing PDEF and pCMV-3Tag expressing SLUG, respectively. After transfection, cells were incubated for 48 hours in normal growth media before harvesting by trypsinization. Rescue of the migratory phenotype was examined using transwell migration assays as described.
Luciferase constructs
A 2,259-bp fragment of the human SLUG (Snai2) promoter (−2,068/+191) was amplified from HUVEC DNA by PCR with primers listed in Supplementary Table S1.38,39 The resultant PCR product was isolated and cloned into the pCR2.1 TOPO vector (Invitrogen), and the sequence was validated by automated sequencing (MUSC Sequencing Facility). The human SLUG reporter plasmids were constructed using the pCR2.1 TOPO SNAI2 vector described above by digestion with AvaI (−1.8 kb) or SspI-AvaI (−0.9 kb). After filling in with Klenow polymerase, the resultant fragments were cloned at the SmaI site of the pGL3 basic vector (Promega, Madison, WI) to generate pGL3 SLUG 1.8 and pGL3 SLUG 0.9, respectively. The insert ends were sequenced to identify clones with proper orientation.
Luciferase assays
Cells were plated at 10,000 cells per well in a 24-well plate. The pGL3 reporter constructs (0.5 ug, firefly luciferase) were cotransfected with GFP (0.1 ug) using Lipofectamine 2000 (Invitrogen), as per the manufacturer’s instructions. The media were changed the next day, and luciferase activity was measured after 48 hours using the luciferase reporter assay system (Promega). Firefly luciferase activity was normalized to protein concentration for each transfected well.
Statistical analysis
For statistical testing, 2-sided paired Student t tests were done using an Excel spreadsheet (Microsoft, Redmond, WA). P values are given for each individual experiment, but in general, P < 0.05 was considered statistically significant. Error bars represent standard deviations of 3 independent experiments unless indicated otherwise.
Supplementary Material
Footnotes
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
This work was supported in part by the National Institutes of Health [grant number PO1-CA78582 (D.K.W.)].
Supplementary material for this article is available on the Genes & Cancer website at http://ganc.sagepub.com/supplemental.
References
- 1. Seth A, Watson DK. ETS transcription factors and their emerging roles in human cancer. Eur J Cancer. 2005;41(16):2462-78 [DOI] [PubMed] [Google Scholar]
- 2. Watson DK, Turner DP, Scheiber MN, Findlay VJ, Watson PM. ETS transcription factor expression and conversion during prostate and breast cancer progression. Open Cancer J. 2010;3:24-39 [Google Scholar]
- 3. Ghadersohi A, Sood AK. Prostate epithelium-derived Ets transcription factor mRNA is overexpressed in human breast tumors and is a candidate breast tumor marker and a breast tumor antigen. Clin Cancer Res. 2001;7(9):2731-8 [PubMed] [Google Scholar]
- 4. Thompson HG, Harris JW, Wold BJ, Lin F, Brody JP. p62 overexpression in breast tumors and regulation by prostate-derived Ets factor in breast cancer cells. Oncogene. 2003;22(15):2322-33 [DOI] [PubMed] [Google Scholar]
- 5. Feldman RJ, Sementchenko VI, Gayed M, Fraig MM, Watson DK. Pdef expression in human breast cancer is correlated with invasive potential and altered gene expression. Cancer Res. 2003;63(15):4626-31 [PubMed] [Google Scholar]
- 6. Ghadersohi A, Pan D, Fayazi Z, Hicks DG, Winston JS, Li F. Prostate-derived Ets transcription factor (PDEF) downregulates survivin expression and inhibits breast cancer cell growth in vitro and xenograft tumor formation in vivo. Breast Cancer Res Treat. 2007;102(1):19-30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gu X, Zerbini LF, Otu HH, et al. Reduced PDEF expression increases invasion and expression of mesenchymal genes in prostate cancer cells. Cancer Res. 2007;67(9):4219-26 [DOI] [PubMed] [Google Scholar]
- 8. Turner DP, Moussa O, Sauane M, Fisher PB, Watson DK. Prostate-derived ETS factor is a mediator of metastatic potential through the inhibition of migration and invasion in breast cancer. Cancer Res. 2007;67(4):1618-25 [DOI] [PubMed] [Google Scholar]
- 9. Ghadersohi A, Odunsi K, Zhang S, et al. Prostate-derived Ets transcription factor as a favorable prognostic marker in ovarian cancer patients. Int J Cancer. 2008;123(6):1376-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Moussa O, Turner DP, Feldman RJ, et al. PDEF is a negative regulator of colon cancer cell growth and migration. J Cell Biochem. 2009;108(6):1389-98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Johnson TR, Koul S, Kumar B, et al. Loss of PDEF, a prostate-derived Ets factor is associated with aggressive phenotype of prostate cancer: regulation of MMP 9 by PDEF. Mol Cancer. 2010;9:148. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12. Gunawardane RN, Sgroi DC, Wrobel CN, Koh E, Daley GQ, Brugge JS. Novel role for PDEF in epithelial cell migration and invasion. Cancer Res. 2005;65(24):11572-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Turner DP, Findlay VJ, Kirven AD, Moussa O, Watson DK. Global gene expression analysis identifies PDEF transcriptional networks regulating cell migration during cancer progression. Mol Biol Cell. 2008;19(9):3745-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schaefer JS, Sabherwal Y, Shi HY, et al. Transcriptional regulation of p21/CIP1 cell cycle inhibitor by PDEF controls cell proliferation and mammary tumor progression. J Biol Chem. 2010;285(15):11258-69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Findlay VJ, Turner DP, Moussa O, Watson DK. MicroRNA-mediated inhibition of prostate-derived Ets factor messenger RNA translation affects prostate-derived Ets factor regulatory networks in human breast cancer. Cancer Res. 2008;68(20):8499-506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Trelstad RL, Hay ED, Revel JD. Cell contact during early morphogenesis in the chick embryo. Dev Biol. 1967;16(1):78-106 [DOI] [PubMed] [Google Scholar]
- 17. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871-90 [DOI] [PubMed] [Google Scholar]
- 18. Nieto MA. The snail superfamily of zinc-finger transcription factors. Nat Rev Mol Cell Biol. 2002;3(3):155-66 [DOI] [PubMed] [Google Scholar]
- 19. Storci G, Sansone P, Trere D, et al. The basal-like breast carcinoma phenotype is regulated by SLUG gene expression. J Pathol. 2008;214(1):25-37 [DOI] [PubMed] [Google Scholar]
- 20. Jethwa P, Naqvi M, Hardy RG, et al. Overexpression of Slug is associated with malignant progression of esophageal adenocarcinoma. World J Gastroenterol. 2008;14(7):1044-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sarrio D, Rodriguez-Pinilla SM, Hardisson D, Cano A, Moreno-Bueno G, Palacios J. Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. 2008;68(4):989-97 [DOI] [PubMed] [Google Scholar]
- 22. Bhowmick NA, Ghiassi M, Bakin A, et al. Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol Biol Cell. 2001;12(1):27-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lindley LE, Briegel KJ. Molecular characterization of TGFbeta-induced epithelial-mesenchymal transition in normal finite lifespan human mammary epithelial cells. Biochem Biophys Res Commun. 2010;399(4):659-64 [DOI] [PubMed] [Google Scholar]
- 24. Maeda M, Johnson KR, Wheelock MJ. Cadherin switching: essential for behavioral but not morphological changes during an epithelium-to-mesenchyme transition. J Cell Sci. 2005;118(Pt 5):873-87 [DOI] [PubMed] [Google Scholar]
- 25. Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9(4):274-84 [DOI] [PubMed] [Google Scholar]
- 26. Talmadge JE, Fidler IJ. AACR Centennial Series. The biology of cancer metastasis: historical perspective. Cancer Res. 2010;70(14):5649-69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Feldman RJ, Sementchenko VI, Watson DK. The epithelial-specific Ets factors occupy a unique position in defining epithelial proliferation, differentiation and carcinogenesis. Anticancer Res. 2003;23(3A):2125-31 [PubMed] [Google Scholar]
- 28. Oka H, Shiozaki H, Kobayashi K, et al. Expression of E-cadherin cell adhesion molecules in human breast cancer tissues and its relationship to metastasis. Cancer Res. 1993;53(7):1696-701 [PubMed] [Google Scholar]
- 29. Umbas R, Isaacs WB, Bringuier PP, et al. Decreased E-cadherin expression is associated with poor prognosis in patients with prostate cancer. Cancer Res. 1994;54(14):3929-33 [PubMed] [Google Scholar]
- 30. Onder TT, Gupta PB, Mani SA, Yang J, Lander ES, Weinberg RA. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008;68(10):3645-54 [DOI] [PubMed] [Google Scholar]
- 31. Conacci-Sorrell M, Simcha I, Ben-Yedidia T, Blechman J, Savagner P, Ben-Ze’ev A. Autoregulation of E-cadherin expression by cadherin-cadherin interactions: the roles of beta-catenin signaling, Slug, and MAPK. J Cell Biol. 2003;163(4):847-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Alves CC, Carneiro F, Hoefler H, Becker KF. Role of the epithelial-mesenchymal transition regulator Slug in primary human cancers. Front Biosci. 2009;14:3035-50 [DOI] [PubMed] [Google Scholar]
- 33. Feng XH. The changing faces of cancer cells. Nat Rev Mol Cell Biol. 2010;11(7):466. [DOI] [PubMed] [Google Scholar]
- 34. Radisky DC, LaBarge MA. Epithelial-mesenchymal transition and the stem cell phenotype. Cell Stem Cell. 2008;2(6):511-2 [DOI] [PubMed] [Google Scholar]
- 35. Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Blick T, Hugo H, Widodo E, et al. Epithelial mesenchymal transition traits in human breast cancer cell lines parallel the CD44(hi/)CD24 (lo/-) stem cell phenotype in human breast cancer. J Mammary Gland Biol Neoplasia. 2010;15(2):235-52 [DOI] [PubMed] [Google Scholar]
- 37. Nowak DE, Tian B, Brasier AR. Two-step cross-linking method for identification of NF-kappaB gene network by chromatin immunoprecipitation. Biotechniques. 2005;39(5):715-25 [DOI] [PubMed] [Google Scholar]
- 38. Deveaux S, Filipe A, Lemarchandel V, Ghysdael J, Romeo PH, Mignotte V. Analysis of the thrombopoietin receptor (MPL) promoter implicates GATA and Ets proteins in the coregulation of megakaryocyte-specific genes. Blood. 1996;87(11):4678-85 [PubMed] [Google Scholar]
- 39. Mignotte V, Vigon I, Boucher de Crevecoeur E, Romeo PH, Lemarchandel V, Chretien S. Structure and transcription of the human c-mpl gene (MPL). Genomics. 1994;20(1):5-12 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.