

Abstract
ESCRT-I is required for the sorting of integral membrane proteins to the lysosome, or vacuole in yeast, for cytokinesis in animal cells, and for the budding of HIV-1 from human macrophages and T lymphocytes. ESCRT-I is a heterotetramer of Vps23, Vps28, Vps37, and Mvb12. The crystal structures of the core complex and the ubiquitin E2 variant and Vps28 C-terminal domains have been determined, but internal flexibility has prevented crystallization of intact ESCRT-I. Here we have characterized the structure of ESCRT-I in solution by simultaneous structural refinement against small-angle X-ray scattering and double electron–electron resonance spectroscopy of spin-labeled complexes. An ensemble of at least six structures, comprising an equally populated mixture of closed and open conformations, was necessary to fit all of the data. This structural ensemble was cross-validated against single-molecule FRET spectroscopy, which suggested the presence of a continuum of open states. ESCRT-I in solution thus appears to consist of an approximately 50% population of one or a few related closed conformations, with the other 50% populating a continuum of open conformations. These conformations provide reference points for the structural pathway by which ESCRT-I induces membrane buds.
Keywords: hybrid methods, protein sorting, vesicle budding, multiprotein assemblies, ensemble refinement
The endosomal sorting complex required for transport (ESCRT) complexes are required for ubiquitin-dependent receptor down-regulation, multivesicular body (MVB) biogenesis, the budding of HIV-1 and most other enveloped viruses, and cytokinesis (1, 2). From yeast to humans, ESCRTs evaginate membrane buds away from the cytosol and cleave narrow necks from their interior face (3). ESCRT-I directly binds to ubiquitin through its ubiquitin E2 variant (UEV) domain (4) and functions with ESCRT-II to bud membranes away from cytosol (5) by an unknown mechanism. The major pathway for the egress of HIV-1 from infected cells is via ESCRT-I (6–9). Cytokinesis requires the recruitment of ESCRT-I to the midbody (10, 11), which is the final connection between daughter cells. These roles have made ESCRT-I a target for structural analysis and antiviral drug development (12, 13).
ESCRT-I consists of one copy each of Vps23, Vps28, Vps37, and Mvb12 in yeast (Fig. 1A). Its structure has been characterized on a piecemeal basis. Structures have been solved of the yeast (Fig. 1B) and human UEV domains bound to ubiquitin (14, 15), and the human UEV domain bound to peptides from HIV-1 and ESCRT-0 (13, 16). Yeast ESCRT-I binds to ESCRT-II via the C-terminal domain (CTD) of Vps28 (17), whose structure has been determined (18, 19) (Fig. 1C). Vps23, Vps28, and Vps37 assemble via a trimeric subcomplex known as the “headpiece” (17, 20) (Fig. 1D). Vps23, Vps37, and Mvb12 form an elongated stalk (21) (Fig. 1D). The stalk and headpiece collectively comprise the core. Four other regions are absent from the solved structures. A 60-residue Pro-rich linker connects the UEV and stalk portions of Vps23 and includes the midbody localization sequence (22). A 30-residue flexible linker connects the headpiece and CTD of Vps28. The N-terminal predicted helix (NTH; Fig. 1E) of Vps37 is basic, predicted to be mostly helical, and contributes to membrane binding (21). Flexible hydrophobic residues at the C terminus of Mvb12 may be a ubiquitin binding domain (23). Fundamental to understanding the mechanism of ESCRT-I-mediated membrane budding is a better understanding of the arrangement of the functional domains in three dimensions.
Fig. 1.

Structural domains and labeling sites. (A) Domain schematic of the four subunits of ESCRT-I, colored blue (Vps23), red (Vps28), gray (Vps37), and orange (Mvb12) showing a speculative model for their orientation relative to the neck of a membrane bud, adapted from ref. 3. (B) UEV domain of Vps23. (C) CTD of Vps28. (D) Core assembly. (E) NTH of Vps37. Engineered Cys residues are shown in a filled sphere model.
ESCRT-I is representative of a class of signaling, trafficking, and regulatory proteins in this size range that contain intrinsically disordered regions. This class presents a fundamental unsolved challenge to structural biologists. The presence of flexible regions has precluded crystallization of intact ESCRT-I, whereas the low molecular weight of the complex (108 kDa) has precluded single particle EM analysis.
We report here an effort to construct a definitive solution structure of intact ESCRT-I and to develop an approach to the structural analysis of midsize protein complexes with regions of intrinsic disorder. Small-angle X-ray scattering (SAXS) and solution NMR are a powerful combination (24) because the global shape information from SAXS complements short-range NMR constraints. A natural counterpart of this approach for larger complexes is to combine SAXS with spectroscopic techniques that yield longer-range constraints by utilizing site-specific or selective labeling. These techniques include double electron–electron resonance (DEER) (25) and FRET spectroscopy (26). SAXS, DEER, and FRET data were collected for ESCRT-I in solution. A molecular simulation framework was devised for the joint refinement of protein complex structures against SAXS and spectroscopic data, leading to an improved model of the complete ESCRT-I structure. We show that ESCRT-I in solution is in an equilibrium between roughly equal populations of open and closed conformations. We describe these populations in terms of a set of six structures that serve as snapshots of a larger continuum of states.
Results
SAXS of ESCRT-I.
The solution structure of full-length Cys-free yeast ESCRT-I was analyzed by SAXS. The I(q) vs. q curve showed features to q = 0.5 Å-1 (Fig. 2A). The maximal protein dimension Dmax was determined to be 225 Å (Fig. 2C). SAXS data were initially analyzed by ab initio envelope generation (27). The envelope thus obtained (Fig. 2D) was elongated and had a large lobe at one end and a smaller lobe at the other. The envelope allowed us to make a visually satisfactory fit of the ESCRT-I core by placing the stalk into the elongated center of the envelope and the headpiece into one lobe (Fig. 2E). However, the envelope lacked sufficient detail to position the UEV, CTD, and NTH regions. Indeed, the Kratky plot (Fig. 2B) is characteristic of a partially unfolded or otherwise noncompact protein (28). Given the evidence for intrinsic flexibility in the connecting regions of ESCRT-I, the appropriateness of modeling these regions into the ab initio envelope in a unique conformation is not clear a priori.
Fig. 2.

SAXS of ESCRT-I. (A) Fit of the simulated scattering curves [I(q)] to the observed scattering of ESCRT-I. (B) Kratky plot of the same experimental data and simulated scattering shown in A. Experimental I(q) data points in A and B represent the mean of 10 consecutive measurements of the same sample, and the error bars represent the standard error of the mean. (C) Pair distribution function [P(r)] for ESCRT-I. (D) An ab initio molecular envelope for ESCRT-I. (E) Superposition of the two EROS-refined structures of ESCRT-I. (F) Fit of experimental SAXS data of A to values computed from the structures fit to both SAXS and DEER data.
Ensemble Refinement of ESCRT-I SAXS.
To sample physical configurations of ESCRT-I, we used a coarse-grained model for protein binding (29). The core, Vps23-UEV, Vps28-CTD, and Vps37-NTH were treated as rigid domains. The interactions between the domains were treated at the residue level with amino acid dependent pair potentials and Debye–Hückel-type electrostatic interactions. Flexible linker peptides connecting the four rigid domains are represented as amino acid beads on a polymer with appropriate stretching, bending, and torsion-angle potentials. Monte Carlo simulations of ESCRT-I were carried out using a starting model based on the crystal structures of yeast Vps23 UEV [Protein Data Bank (PDB) ID code 1UZX], yeast Vps28 CTD (PDB ID code 2J9U), the yeast ESCRT-I core (PDB ID code 2P22), a model of the NTH in a helical conformation, and linkers generated in sterically allowed but otherwise arbitrary conformations. The resulting 10,000 conformations were clustered and fit to the experimental I(q) curve by using the ensemble refinement of SAXS (EROS) program (30). An acceptable fit could be obtained with a minimum of two conformations, weighted equally (Fig. 2A). However, no single conformation was adequate to fit the I(q) data. One of the two conformations is more closed (black, Fig. 2E) and the other more open (yellow, Fig. 2E). In the more closed structure, both the UEV and CTD domains approach close to the stalk in “cis,” such that they are both on the same side of the stalk. The UEV and CTD do not touch one another even in the more closed conformation.
In the more open conformation, the CTD is farther from the stalk, but still on the cis face, whereas the UEV has swung away from the stalk to project past the tip of the stalk. In the more open conformation, there are no direct contacts between either the UEV or CTD and the core. Without interactions to hold these domains firmly in position, the open conformation is likely representative of an ensemble of structures that fill a similar scattering volume in solution. This analysis led us to conclude that at least half the population of ESCRT-I complexes in solution adopts a dynamic structure in which the UEV and CTD are mobile with respect to the core.
DEER EPR Analysis of ESCRT-I.
Four sets of unique Cys pairs were engineered into the Cys-free yeast ESCRT-I construct and labeled with the spin probe (1-oxyl-2,2,5,5-tetramethyl-∆3-pyrroline-3-methyl) methanethiosulfonate (MTSL). Cys pairs were selected using the EROS-refined structures to constrain the most ambiguous interdomain distances. Solvent-exposed residues in crystallized regions were selected to avoid interference with complex assembly. The pair Vps28 Cys27—Vps37 Cys173 was chosen within the crystallized headpiece region to serve as a control. The remaining three pairs were designed to measure the position of one of the major functional domains relative to the core. Vps23 Cys108—Vps23 Cys256 allowed measurement of the UEV-core separation. Vps28 Cys65—Vps28 Cys 151 and Vps23 Cys223–Vps37 Cys12 were used to measure the CTD-core and NTH-core separations, respectively.
Each labeled ESCRT-I sample yielded interpretable DEER spectra (Fig. 3). In a Gaussian fit, the control pair Vps28 Cys27—Vps37 Cys173 produced a sharply peaked P(r) distribution centered at 52 Å (Table S1). The positions of the MTSL labels were modeled onto the structures using the appropriate distribution of MTSL conformers (31). For the control pair, the P(r) distribution calculated from the modeled MTSL coordinates is in excellent agreement with experiment (Fig. 3, Top). Thus this MTSL label faithfully reported a known intracomplex distance in ESCRT-I. The Vps23 Cys108—Vps23 Cys256 and Vps23 Cys223– Vps37 Cys12 samples yielded P(r) distributions in Gaussian fits that were about twice as broad as the intracore P(r) described above (Table S1). From the broad P(r) distributions for the UEV-, CTD-, and NTH-core pairs, we conclude that all of these domains are mobile with respect to the core.
Fig. 3.

DEER of ESCRT-I. (Left) Experimentally observed (solid) modulation of V(t) together with points calculated from six structural clusters. (Right) Histograms of MTSL distances computed from the models. The green curves in the top panels show the results from Gaussian regularization.
Combined SAXS and DEER Refinement.
In order to avoid introducing regularization-dependent artifacts into the structural refinement, the structures were refined directly against the measured V(t) DEER data. The EROS procedure (30) was modified to allow simultaneous fitting of SAXS and DEER data with a minimal ensemble of structures. In the ensemble refinement, the structure weights wk were varied to improve the agreement between the computed, ensemble-averaged quantities I(q), V(i,j)(t) and the measured Iobs(q),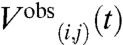 signals. To find a minimum set of structures that jointly account for both the SAXS and DEER data, the function
signals. To find a minimum set of structures that jointly account for both the SAXS and DEER data, the function 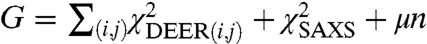 was minimized numerically using a Monte Carlo search algorithm in which the structure weights were varied between wk = 0 and wk = 1 under the constraint
was minimized numerically using a Monte Carlo search algorithm in which the structure weights were varied between wk = 0 and wk = 1 under the constraint 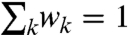 . In the formula above, the parameter μ controls the number n of structures with nonzero weights wk > 0, allowing us to identify a minimal set of representative structures. For small parameters μ ≪ 1, we obtained very good fits to the experimental data (χ2 < 1) with many structures in the refined ensemble (n≫1). For large parameters μ > 1, the ensemble contained very few structures with nonzero weights, but the agreement with experimental data was not satisfactory (χ2 was significantly larger than 1). The balance was struck at μ = 0.2, when we obtained good fits to all experimental datasets (χ2 ≈ 1) with only n = 6 structures (Figs. 2F and 3).
. In the formula above, the parameter μ controls the number n of structures with nonzero weights wk > 0, allowing us to identify a minimal set of representative structures. For small parameters μ ≪ 1, we obtained very good fits to the experimental data (χ2 < 1) with many structures in the refined ensemble (n≫1). For large parameters μ > 1, the ensemble contained very few structures with nonzero weights, but the agreement with experimental data was not satisfactory (χ2 was significantly larger than 1). The balance was struck at μ = 0.2, when we obtained good fits to all experimental datasets (χ2 ≈ 1) with only n = 6 structures (Figs. 2F and 3).
Qualitatively, the ensemble of six structures (Fig. 4) resembles the open and closed conformations used to fit the SAXS data and fills the same overall region of space. The most closed of the six SAXS-EPR conformations resembles the closed SAXS conformation (Fig. 2E). Another structure has the UEV, CTD, and NTH displaced from the core in a highly extended state. The remaining four have one or two of the domains in more closed states. The UEV folds back against the cis side of the stalk. In each of the three structures with a closed UEV conformation, the UEV fills a similar region of space, but different surfaces of the UEV interact with slightly different portions of the core. In one of the structures, the ubiquitin binding site (14) is occluded. The NTH folds back along the stalk such that similar regions of space are filled. Again, in each case the precise angle between the NTH and the stalk varies, as do the details of molecular contacts. Two of the three closed states of the CTD involve packing against the distal tip of the headpiece, which is formed by the N-terminal half of the Vps28 subunit. These closed conformers fill the same region of space and contact the same part of the headpiece, but the CTD four helix bundle is rotated 120° apart in the two states. In the third state, the CTD also contacts the N-terminal half of Vps28, but in this case a slightly different region of space is filled, and the CTD points back toward the cis side of the core. The precise closed state conformations of each of the three domains vary enough that reliable conclusions about the detailed nature of the closed state, or even whether there is a single closed conformation, cannot be drawn. Notably, even in the most open conformations, the space on the “trans” side of the stalk is avoided. Thus even in the context of a highly flexible and dynamic structure, there are distinct preferences for domains to occupy some regions of space over others.
Fig. 4.

Solution structure of ESCRT-I. Structures are shown with their relative weights obtained from fitting, indicated as percentages. Subunits are colored as in Fig. 1.
Bulk FRET Measurements.
We sought to cross-validate the refined ESCRT-I structure with independent data excluded from the refinement. Bulk FRET lifetime data were obtained on a sample labeled with Atto488 and Atto594 on Cys137 and Cys223 of Vps23, which are on the UEV domain and stalk, respectively. This pair provides an independent check on the UEV-core separation, distinct from the MTSL-labeled Cys108-Cys256 pair used in refinement. The donor lifetime was found to be τD = 3.7 ns, which was reduced in the presence of the acceptor to τDA = 3.4 ns (Fig. S1). The FRET efficiency calculated from the structures (see SI Methods) was E = 0.11, which is in good agreement with the mean efficiency E = 0.09 measured in the bulk FRET experiment.
Single-Molecule FRET.
To provide an independent check on the Vps28 Cys65–Cys151 pair, the pair was labeled with Alexa488 and Alexa594. Fig. 5 A and B compares the measured FRET efficiency histograms (light brown wide bars) with the histogram predicted from the model structures (black narrow bars). To verify the assembly of ESCRT-I at the 40-pM concentration used for these experiments, FRET was also measured between the intersubunit pair Vps28 Cys27—Vps37 Cys173 (Fig. S2). The experimental FRET efficiencies, 〈Eapp〉 = nA/(nA + nD), calculated from 2-ms bins containing more than 50 photons (Fig. 5A) or more than 110 photons (Fig. 5B), were converted to the true FRET efficiencies, 〈E〉, after correcting for background and donor leakage into the acceptor channel, by using a γ-factor to correct for differences in the detection efficiencies of the donor and acceptor channels and differences in the quantum yields of the donor and acceptor dyes as 〈E〉 = nA/(nA + γnD), with γ = 2 determined from donor lifetime measurements (32).
Fig. 5.

Single-molecule FRET. Comparison of experimental (light brown wide bars) and calculated (black narrow bars) FRET efficiency histograms, using the interdye distances for the model structures, for a threshold number of photons (nT) of (A) 50 and (B) 110 with a bin time (Tbin) of 2 ms. In the experimental histogram, the FRET efficiency of each bin was γ-corrected after subtraction of the average background photons (donor: 0.6 ms-1; acceptor: 1.1 ms-1) and the correction for leak of donor fluorescence into the acceptor channel (6%). The fluorescence bursts from the donor-only-labeled molecules (E < 0.18) were excluded and the data were prefiltered to remove bursts of photons in which either blinking or bleaching occurred. (C) Inferred model for an open state of ESCRT-I (Top) consisting of a continuum of conformations, in equilibrium with a closed state (Bottom) consisting of one or a few related conformations.
Model Validation Against FRET Measurements.
FRET efficiencies were calculated for the Vps28 Cys65/Cys151 double mutant and are in good overall agreement with experiment (Fig. 5 A and B). The low efficiency tail of the measured distribution is not observed in the predicted histogram. More importantly, the predicted histogram contains two distinct peaks at E ∼ 0.60 and E ∼ 0.85 that are enveloped by the single broad peak of the experimental histogram. This difference is more pronounced in the distribution obtained from the bins with a higher photon threshold (nT) of 110 photons where the shot noise width is narrower (Fig. 5B). Possible origins of this difference in the measured and predicted histograms are (i) interconversion among the clusters of conformations during the 2-ms bin time that result in bins with FRET efficiencies intermediate between the closed and open conformations, (ii) orientational motion of the dyes is comparable to or slower than the bin time due to dyes sticking to the protein to produce a range of values for the orientation factor κ2, and (iii) the predicted structures do not include conformations with intermediate FRET efficiencies.
The first possibility was evaluated using a simple kinetic model of reversible conformational changes between two states, with each of the three clusters within either the open or closed set of conformations considered as one state. This model was compared to FRET efficiency histograms constructed with varying bin times as described in SI Analysis, from which we conclude that dynamics are most probably not responsible for broadening the experimental histogram. The second possibility was addressed by measuring the polarization anisotropy values for the donor and acceptor dyes. The anisotropy value of the donor (ra) is higher than expected from the donor lifetime (τD) and the reorientational correlation time (τc) of approximately 0.74 ns for freely reorienting dyes determined from time-resolved anisotropy measurements (33) (see Table S2). The reorientational correlation time of the dyes (τc) calculated from 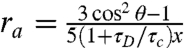 and θ = 0 (the angle between the absorption and emission dipoles) is 1–2 ns, indicating that the dye dynamics do not add width to the FRET efficiency histogram in excess of the shot noise contribution (SI Analysis), but could, because κ2 ≠ 2/3, produce a small shift in the FRET efficiency peaks compared to those corresponding to freely reorienting dyes. The conclusion from the above analysis is that the single broad peak of the measured histogram probably reflects the presence of additional conformations not yet accounted for in the modeling.
and θ = 0 (the angle between the absorption and emission dipoles) is 1–2 ns, indicating that the dye dynamics do not add width to the FRET efficiency histogram in excess of the shot noise contribution (SI Analysis), but could, because κ2 ≠ 2/3, produce a small shift in the FRET efficiency peaks compared to those corresponding to freely reorienting dyes. The conclusion from the above analysis is that the single broad peak of the measured histogram probably reflects the presence of additional conformations not yet accounted for in the modeling.
Discussion
Here we have applied a combination of techniques—SAXS, DEER, and FRET—that probe overall shape along with distances between specific residue pairs within ESCRT-I.
The observations here support the expectation that each complementary technique adds to the overall information content and defines the solution structure more sharply. A set of three RH values for ESCRT-I constructs could previously be interpreted in terms of a single open structure (21). The SAXS data could be fit by a minimum of two structures, one open and one closed. The SAXS and DEER data together could be fit by a minimum of six, spanning a spectrum of more open and closed conformations of the UEV, CTD, and NTH relative to the core. In the earlier hydrodynamic model, ESCRT-I was entirely in an open state. Indeed, the information content of the previous hydrodynamic study, with only three data points, was insufficient to define more than a single state for the three domains. The major insight to emerge from the present analysis is the unanticipated existence of a roughly 50% population of closed conformations.
One of the most unexpected aspects of the analysis was the constraint imposed by the distribution information in the DEER spectra. For the three domain-core pairs, these spectra each led to a broad P(r) distribution in Gaussian fits. Although these distributions were not used in the refinement, the broad collection of six structures emerging from the refinement reflects the same underlying conformational heterogeneity. Single-molecule FRET histograms provide a direct measurement of the conformational space sampled. The core-CTD label pair showed a single very broad peak in the histogram that had excellent overlap with the calculated histogram, giving us confidence in the model derived from SAXS and DEER. The broad and relatively featureless character of the FRET efficiencies highlights the likely presence of intermediate conformations in between the six structures used to fit the SAXS and DEER data. It is important to note that there is no evidence that there are precisely six (or some other particular number) discrete conformations. We view these six structures as snapshots that span and represent the larger conformational space sampled by ESCRT-I (Fig. 5C), a concept supported by the FRET histograms and the broad P(r) distributions from the DEER.
In the current model of ESCRT-driven membrane budding, multiple copies of ESCRT-I and -II assemble to form a ring at the bud neck (3). ESCRT-I localization to bud necks in vitro has been directly observed (5), but the nature and structure of the putative membrane-bound ESCRT-I-II assembly is unknown. In yeast, the intralumenal vesicles (ILVs) in MVBs are 22–26 nm in diameter (34). Assuming the bud neck is of similar dimensions to the bud itself implies a bud neck circumference of approximately 75 nm. With a maximum dimension of 22 nm seen here, at least four ESCRT-I complexes aligned end to end would be needed to span the bud neck. The internal flexibility observed for approximately 50% of ESCRT-I complexes in solution (Fig. 5C, Top) would allow the complex to adjust its structure along the pathway of bud biogenesis. Once incorporated with ESCRT-II into a membrane-bound assembly, a single conformation is predicted, possibly one corresponding to the closed state seen in the other approximately 50% of the solution population (Fig. 5C, Bottom). The solution structure will be useful as a reference point for the structural pathway of ESCRT-I-II assembly responsible for bud neck induction. Although SAXS is not feasible in the membrane setting due to scattering from liposomes, the DEER and FRET techniques can be carried over. Thus a comparison of the spectra in solution and in membrane-bound settings could provide a first step to understanding conformational changes involved in forming the bud neck assembly. The role of conformational changes in ESCRT-I-mediated HIV-1 budding is an even more urgent question. These spectroscopic techniques should also be portable to the HIV-1 setting, once the HIV budding reaction can be reconstituted in vitro.
The successes and limitations of this analysis suggest lessons for the integration of global and site-specific structural data on partially flexible complexes. EROS analysis of the SAXS data revealed an ensemble of structures capable of fitting the data, but lacked information about the distinct behavior of individual domains. Site-specific labeling provided powerful complementary information. Site-specific spin labeling and DEER spectroscopy provided direct evidence that each individual domain was conformationally heterogeneous relative to the core, a conclusion that we could not have drawn from the SAXS analysis alone. We used another site-specific structural probe, FRET spectroscopy, to validate conclusions drawn from the two other techniques and to reveal the presence of conformational intermediates that were not detected by the other techniques. Multisubunit complexes represent a major challenge in structural biology, and the integration of restraints from multiple methods is likely to be critical in developing a molecular understanding of function.
Materials and Methods
In the ensemble refinement, the number N and weight wk of structures k in the ensemble were obtained by simultaneous fits to SAXS and DEER data. The scattering intensity Ik(q) of structure k was calculated as in ref. 30 and averaged over the ensemble,
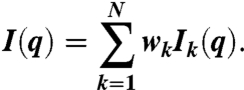 |
Deviations from the measured SAXS intensity Iobs(q) were quantified by
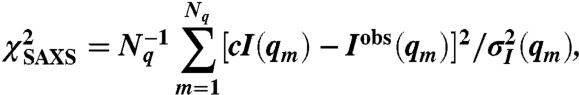 |
summed over the Nq q points in the intensity curve, with c a constant determined by the condition 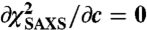 . To refine the DEER data, possible conformations of the six MTSL labels on ESCRT-I domains were generated by the multiscale modeling of macromolecular system Matlab module (31). By appropriate translation and rotation of the MTSL coordinates, the rotamers were positioned on the rigid domains of all the ESCRT-I structures in the ensemble. For each MTSL label pair (i,j) and configuration k of the ESCRT-I complex in the ensemble, the dipolar evolution function
. To refine the DEER data, possible conformations of the six MTSL labels on ESCRT-I domains were generated by the multiscale modeling of macromolecular system Matlab module (31). By appropriate translation and rotation of the MTSL coordinates, the rotamers were positioned on the rigid domains of all the ESCRT-I structures in the ensemble. For each MTSL label pair (i,j) and configuration k of the ESCRT-I complex in the ensemble, the dipolar evolution function
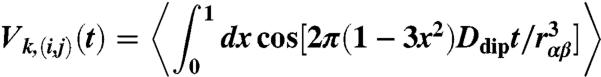 |
was calculated with Ddip = 52.04 MHz nm3. The average is over all MTSL label conformations, with rαβ the distance between the spin labels α and β attached to residues i and j, respectively. To validate the DEER model, MTSL labels were attached to residues 27 and 173 on the same rigid domain. The computed dipolar evolution function V(27,173)(t) is thus the same for each structure k and is found to be in good agreement with the measured DEER signal (see Fig. 3, Top). For the other three label pairs, (i,j) = (108,256), (12, 223), and (65, 151), the dipolar evolution function was averaged over the ensemble,
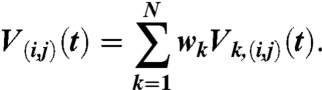 |
Deviations from the measured DEER signals 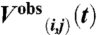 were quantified by
were quantified by
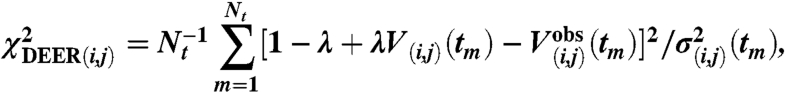 |
which is summed over the Nt points in the 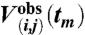 curve, with the modulation depth λ obtained from
curve, with the modulation depth λ obtained from 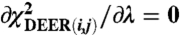 . The statistical error
. The statistical error 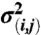 was estimated from the noise level in the experimental data.
was estimated from the noise level in the experimental data.
Supplementary Material
Acknowledgments.
We thank B. Beach and M. Kostelansky for preparing the Cys-free ESCRT-I construct and A. Bax for discussions. SAXS data were collected at the Stanford Synchrotron Radiation Laboratory (SSRL), a national user facility operated by Stanford University on behalf of the US Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program. B.R. was supported by a Marie Curie International Outgoing Fellowship within the 7th European Community Framework Programme. This work was supported by the Intramural Program of the National Institutes of Health (NIH), National Institute of Diabetes and Digestive and Kidney Diseases (to W.A.E., G.H., and J.H.H.), the Intramural AIDS Targeted Anti-viral Program of the Office of the Director, NIH (to J.H.H.), NIH Grant GM072694 (to D.S.C.), and the Ministry of Education, Youth and Sports of the Czech Republic Grant MSM0021620835 (to J.V.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1101763108/-/DCSupplemental.
References
- 1.McDonald B, Martin-Serrano J. No strings attached: The ESCRT machinery in viral budding and cytokinesis. J Cell Sci. 2009;122:2167–2177. doi: 10.1242/jcs.028308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raiborg C, Stenmark H. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature. 2009;458:445–452. doi: 10.1038/nature07961. [DOI] [PubMed] [Google Scholar]
- 3.Hurley JH, Hanson PI. Membrane budding and scission by the ESCRT complexes: It’s all in the neck. Nat Rev Mol Cell Biol. 2010;11:556–566. doi: 10.1038/nrm2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Katzmann DJ, Babst M, Emr SD. Ubiquitin-dependent sorting into the multivesicular body pathway requires the function of a conserved endosomal protein sorting complex, ESCRT-I. Cell. 2001;106:145–155. doi: 10.1016/s0092-8674(01)00434-2. [DOI] [PubMed] [Google Scholar]
- 5.Wollert T, Hurley JH. Molecular mechanism of multivesicular body biogenesis by ESCRT complexes. Nature. 2010;464:864–873. doi: 10.1038/nature08849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garrus JE, et al. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell. 2001;107:55–65. doi: 10.1016/s0092-8674(01)00506-2. [DOI] [PubMed] [Google Scholar]
- 7.VerPlank L, et al. Tsg101, a homologue of ubiquitin-conjugating (E2) enzymes, binds the L domain in HIV type 1 Pr55(Gag) Proc Natl Acad Sci USA. 2001;98:7724–7729. doi: 10.1073/pnas.131059198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martin-Serrano J, Zang T, Bieniasz PD. HIV-I and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat Med. 2001;7:1313–1319. doi: 10.1038/nm1201-1313. [DOI] [PubMed] [Google Scholar]
- 9.Demirov DG, Ono A, Orenstein JM, Freed EO. Overexpression of the N-terminal domain of TSG101 inhibits HIV-1 budding by blocking late domain function. Proc Natl Acad Sci USA. 2002;99:955–960. doi: 10.1073/pnas.032511899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carlton JG, Martin-Serrano J. Parallels between cytokinesis and retroviral budding: A role for the ESCRT machinery. Science. 2007;316:1908–1912. doi: 10.1126/science.1143422. [DOI] [PubMed] [Google Scholar]
- 11.Morita E, et al. Human ESCRT and ALIX proteins interact with proteins of the midbody and function in cytokinesis. EMBO J. 2007;26:4215–4227. doi: 10.1038/sj.emboj.7601850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tavassoli A, et al. Inhibition of HIV budding by a genetically selected cyclic peptide targeting the Gag-TSG101 interaction. ACS Chem Biol. 2008;3:757–764. doi: 10.1021/cb800193n. [DOI] [PubMed] [Google Scholar]
- 13.Im YJ, et al. Crystallographic and functional analysis of the ESCRT-1/HIV-1 Gag PTAP interaction. Structure. 2010;18:1536–1547. doi: 10.1016/j.str.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Teo H, Veprintsev DB, Williams RL. Structural insights into endosomal sorting complex required for transport (ESCRT-I) recognition of ubiquitinated proteins. J Biol Chem. 2004;279:28689–28696. doi: 10.1074/jbc.M400023200. [DOI] [PubMed] [Google Scholar]
- 15.Sundquist WI, et al. Ubiquitin recognition by the human TSG101 protein. Mol Cell. 2004;13:783–789. doi: 10.1016/s1097-2765(04)00129-7. [DOI] [PubMed] [Google Scholar]
- 16.Pornillos O, Alam SL, Davis DR, Sundquist WI. Structure of the Tsg101 UEV domain in complex with the PTAP motif of the HIV-1 p6 protein. Nat Struct Biol. 2002;9:812–817. doi: 10.1038/nsb856. [DOI] [PubMed] [Google Scholar]
- 17.Kostelansky MS, et al. Structural and functional organization of the ESCRT-I trafficking complex. Cell. 2006;125:113–126. doi: 10.1016/j.cell.2006.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pineda-Molina E, et al. The crystal structure of the C-terminal domain of Vps28 reveals a conserved surface required for Vps20 recruitment. Traffic. 2006;7:1007–1016. doi: 10.1111/j.1600-0854.2006.00440.x. [DOI] [PubMed] [Google Scholar]
- 19.Gill DJ, et al. Structural insight into the ESCRT-I/-II link and its role in MVB trafficking. EMBO J. 2007;26:600–612. doi: 10.1038/sj.emboj.7601501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Teo HL, et al. ESCRT-I core and ESCRT-II GLUE domain structures reveal role for GLUE in linking to ESCRT-I and membranes. Cell. 2006;125:99–111. doi: 10.1016/j.cell.2006.01.047. [DOI] [PubMed] [Google Scholar]
- 21.Kostelansky MS, et al. Molecular architecture and functional model of the complete yeast ESCRT-I heterotetramer. Cell. 2007;129:485–498. doi: 10.1016/j.cell.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee HH, et al. Midbody targeting of the ESCRT machinery by a noncanonical coiled coil in CEP55. Science. 2008;322:576–580. doi: 10.1126/science.1162042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shields SB, et al. ESCRT ubiquitin binding domains function cooperatively during MVB cargo sorting. J Cell Biol. 2009;185:213–224. doi: 10.1083/jcb.200811130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grishaev A, et al. Refined solution structure of the 82-kDa enzyme malate synthase G from joint NMR and synchrotron SAXS restraints. J Biomol NMR. 2008;40:95–106. doi: 10.1007/s10858-007-9211-5. [DOI] [PubMed] [Google Scholar]
- 25.Fanucci GE, Cafiso DS. Recent advances and applications of site-directed spin labeling. Curr Opin Struct Biol. 2006;16:644–653. doi: 10.1016/j.sbi.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 26.Brunger AT, et al. Three-dimensional molecular modeling with single molecule FRET. J Struct Biol. 2011;173:497–505. doi: 10.1016/j.jsb.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Volkov VV, Svergun DI. Uniqueness of ab initio shape determination in small-angle scattering. J Appl Crystallogr. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Putnam CD, Hammel M, Hura GL, Tainer JA. X-ray solution scattering (SAXS) combined with crystallography and computation: Defining accurate macromolecular structures, conformations and assemblies in solution. Q Rev Biophys. 2007;40:191–285. doi: 10.1017/S0033583507004635. [DOI] [PubMed] [Google Scholar]
- 29.Kim YC, Hummer G. Coarse-grained models for simulations of multiprotein complexes: Application to ubiquitin binding. J Mol Biol. 2008;375:1416–1433. doi: 10.1016/j.jmb.2007.11.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rozycki B, Kim YC, Hummer G. SAXS ensemble refinement of ESCRT-III CHMP3 conformational transitions. Structure. 2011;19:109–116. doi: 10.1016/j.str.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Polyhach Y, Bordignon E, Jeschke G. Rotamer libraries of spin labelled cysteines for protein studies. Phys Chem Chem Phys. 2011;13:2356–2366. doi: 10.1039/c0cp01865a. [DOI] [PubMed] [Google Scholar]
- 32.Merchant KA, et al. Characterizing the unfolded states of proteins using single-molecule FRET spectroscopy and molecular simulations. Proc Natl Acad Sci USA. 2007;104:1528–1533. doi: 10.1073/pnas.0607097104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nettels D, Hoffmann A, Schuler B. Unfolded protein and peptide dynamics investigated with single-molecule FRET and correlation spectroscopy from picoseconds to seconds. J Phys Chem B. 2008;112:6137–6146. doi: 10.1021/jp076971j. [DOI] [PubMed] [Google Scholar]
- 34.Nickerson DP, West M, Odorizzi G. Did2 coordinates Vps4-mediated dissociation of ESCRT-III from endosomes. J Cell Biol. 2006;175:715–720. doi: 10.1083/jcb.200606113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.