

Abstract
Background
Exposure to cadmium is associated with human pathologies and altered gene expression. The molecular mechanisms by which cadmium affects transcription remain unclear. It has been proposed that cadmium activates transcription by altering intracellular calcium concentration ([Ca2+]i) and disrupting calcium-mediated intracellular signaling processes. This hypothesis is based on several studies that may be technically problematic; including the use of BAPTA chelators, BAPTA-based fluorescent sensors, and cytotoxic concentrations of metal.
Methodology/Principal Finding
In the present report, the effects of cadmium on [Ca2+]i under non-cytotoxic and cytotoxic conditions was monitored using the protein-based calcium sensor yellow cameleon (YC3.60), which was stably expressed in HEK293 cells. In HEK293 constitutively expressing YC3.60, this calcium sensor was found to be insensitive to cadmium. Exposing HEK293::YC3.60 cells to non-cytotoxic cadmium concentrations was sufficient to induce transcription of cadmium-responsive genes but did not affect [Ca2+]i mobilization or increase steady-state mRNA levels of calcium-responsive genes. In contrast, exposure to cytotoxic concentrations of cadmium significantly reduced intracellular calcium stores and altered calcium-responsive gene expression.
Conclusions/Significance
These data indicate that at low levels, cadmium induces transcription independently of intracellular calcium mobilization. The results also support a model whereby cytotoxic levels of cadmium activate calcium-responsive transcription as a general response to metal-induced intracellular damage and not via a specific mechanism. Thus, the modulation of intracellular calcium may not be a primary mechanism by which cadmium regulates transcription.
Introduction
The transition metal cadmium is a persistent environmental toxicant. Diet, occupation, and smoking are the primary routes of cadmium exposure to the public. Exposure to this metal is associated with numerous human pathologies including kidney dysfunction, osteoporosis, respiratory ailments, and birth defects [1], [2], [3], [4]. In addition, cadmium is classified as a Type I human carcinogen, based on animal studies and data indicating that occupational exposure leads to an increased risk of lung cancer [5]. The prevalence of cadmium-associated diseases is increasing and cadmium-induced pathologies are appearing at levels below current OSHA standards [6], [7], [8].
In vivo and in vitro exposure to low concentrations of cadmium (1–5 µM) initiates an adaptive response that ameliorates the metal-induced toxicity. Toxic effects are reduced by increasing the levels of multiple stress-response proteins [9], [10], [11]. Analysis of transcriptome data from multiple species indicates that cadmium exposure alters the expression of hundreds of genes [9], [12], [13], [14]. Bioinformatic analyses of cadmium-transcriptomes identify the expected metal-responsive and stress-response processes/pathways including p38, extracellular signal-regulated kinase (ERK), and Jun N-terminal kinase (JNK)/mitogen-activated protein kinase (MAPK) pathways. Other pathways have been identified however, that cannot be directly associated with metal detoxification or the repair of metal-induced damage. In addition, the transcription of hundreds of additional genes is affected at higher, cytotoxic cadmium concentrations. An analogous process is observed in HepG2 cells treated with physiological and toxicological concentrations of copper [15].
The ability of cadmium to affect the expression of hundreds of functionally unrelated genes can be attributed to its capacity to modulate the activity of multiple signal transduction pathways. Cadmium activates p38, ERK, and JNK/MAPK pathways [16]. Activation of MAPK pathways affects the transcription of genes involved in the stress-response, as well as growth and development. In addition to the MAPK pathway, cadmium influences the activities of p53, NRF2, protein Kinase C, casein kinase 2, and calcium/calmodulin-dependent kinase II (CaMK II) [17], [18], [19], [20]. Cadmium may also influence gene expression by affecting the levels of second messengers, such as reactive oxygen species, cAMP and calcium.
It has been suggested that cadmium-activation of ERK, p38, and JNK results in part from an elevation of intracellular calcium concentration ([Ca2+]i) [21], [22]. While several studies indicate that exposure to cadmium causes increased [Ca2+]i, the mechanism by which cadmium affects [Ca2+]i remains poorly understood [18]. Several factors have made defining the precise effects of cadmium on [Ca2+]i problematic. A major issue has been the use of the calcium chelator 1, 2-bis(o-aminophenoxy)ethane-N,N,N',N'-tetraacetic acid (BAPTA) and BAPTA-based fluorescent calcium indicators. The BAPTA-based indicators and chelators are able to bind cadmium with high affinity. BAPTA binds calcium with a Kd ∼0.2 µM, however it also binds cadmium, but with Kd ∼1 pM. In addition, the fluorescent intensity of cadmium-bound Fura-2, a common BAPTA-based fluorescent dye used to monitor [Ca2+]i, is 70% greater for the cadmium-bound form compared to the calcium-bound form [23]. A second confounding factor is the use of cytotoxic concentrations of cadmium. LD50s for cadmium in mammalian cells are <10 µM, while studies examining the effects of cadmium on [Ca2+]i routinely expose cells to concentrations of metal in far excess of this level [18], [21], [24]. Thus, there is a need to better understand the relationship between cadmium exposure, calcium mobilization, and the subsequent effect on transcription.
In the current report, the effect of cadmium on [Ca2+]i is examined using the protein-based calcium sensor, yellow cameleon (YC)3.60, which is constitutively expressed in HEK293 cells (HEK293::YC3.60) [25]. The yellow cameleon does not respond to changes in intracellular cadmium concentration ([Cd2+]i). Exposing HEK293 cells to 1 µM cadmium for 4 h was sufficient to induce transcription of several cadmium-responsive genes, but did not affect cell viability, intracellular calcium levels, or the transcriptional activity of calcium-responsive genes. In contrast, exposure to cytotoxic levels of cadmium, 30 µM for 4 h, significantly decreased cell viability, reduced endoplasmic reticulum (ER) calcium stores, and significantly altered the transcriptional activity of calcium-responsive genes. These data indicate that non-cytotoxic concentrations of cadmium induce transcription independently of intracellular calcium mobilization. Furthermore, only when cells are exposed to cytotoxic cadmium concentrations is calcium mobilized from ER pools.
Results
Cadmium-inducible transcription in HEK293::YC3.60 cells
qRT-PCR of three well-characterized cadmium-inducible genes: mt-1 (metallothionein-1), c-fos, and grp-78 (78-kDa glucose-regulated protein/HSPA5) was used to quantify the transcriptional response of HEK293::YC3.60 cells exposed to cadmium or with altered [Ca2+]i. Exposure of HEK293::YC3.60 cells to 1 or 30 µM cadmium resulted in a rapid and significant increase in mt-1 mRNA levels (Fig. 1). This observation was consistent with previous studies where cadmium exposure produced an increase in the steady-state levels of mt-1 mRNA [11], [18]. Treatment with thapsigargin also caused an increase in mt-1 mRNA, but only following a 24 h exposure. Thapsigargin is a potent inhibitor of calcium ATPases of the endoplasmic reticulum (ER), leading to a depletion of ER calcium and a concurrent increase in [Ca2+]i [26], [27].
Figure 1. Effects of cadmium and thapsigargin on transcription.

Total RNA was isolated from HEK293::YC3.60 cells exposed to either 1 µM (square) or 30 µM (triangle) cadmium, or 2 µM thapsigargin (circle) for 1, 4, or 24 h. Steady-state mRNA levels of mt-1, c-fos, and grp-78 were measured using qRT-PCR. All measurements were normalized to mRNA levels of actin. Fold change was normalized to mRNA levels observed in control cells. Results were mean log2fold change ± SEM (n = 3) and were analyzed by one-way ANOVA followed by Dunnett's post-test; single (*) and double (**) asterisks indicate significant differences from controls at p<0.05 and p<0.001, respectively.
c-fos mRNA levels significantly increased following 1 h cadmium and thapsigargin exposures (Fig. 1). Longer exposures to 1 µM cadmium resulted in a gradual decrease in the steady-state level of c-fos mRNA, where at 24 h the mRNA level was not significantly different from control cells. The elevated levels of c-fos mRNA following 24 h exposures to 30 µM cadmium and thapsigargin may be the result of non-specific stress-responses (see below).
The steady-state level of grp-78 mRNA increased in response to 1 and 30 µM cadmium and thapsigargin (Fig. 1). Thapsigargin had the fastest and largest effect on grp-78 mRNA levels, followed by 30 µM cadmium. Similar to c-fos, exposure to 1 µM cadmium caused a transient elevation in grp-78 which returned to baseline by 24 h.
The effect of cadmium and altered [Ca2+]i following thapsigargin on the steady-state levels of mt-1, c-fos, and grp-78 mRNAs in HEK293::YC3.60 cells were similar to those reported in previous studies [28]. These data indicated that this cell line would be an appropriate system to investigate mechanisms of cadmium-induced transcription and a potential contributory role of calcium.
Intracellular cadmium accumulation and cell viability
Cellular uptake of cadmium in HEK293::YC3.60 cells was monitored using fura-5F, which binds cadmium with high affinity [23]. Prior to cadmium exposure, intracellular calcium stores were depleted by incubating cells for 5 min with 10 µM ionomycin, in calcium-free HBSS. This would ensure that changes in fluorescence ratios represented cadmium influx rather than changes in [Ca2+]i. The addition of 2 mM cadmium resulted in a rapid and significant increase in the fluorescence ratio (Fig. 2A). This indicated that cadmium entered the cells and that fura-5F was capable of detecting changes in [Cd2+]i.
Figure 2. Cadmium uptake and cell viability in HEK 293::YC3.60 cells.

A, Fura-5F loaded cells were incubated with ionomycin for 10 min to deplete intracellular calcium stores and then 2 mM cadmium in calcium-free HBSS was added to the medium (solid line). In the experiment represented by the dashed line, similar conditions were used except cells were not exposed to ionomycin prior to cadmium addition. Traces are representative of typical responses observed in at least three independent experiments. B, HEK293::YC3.60 cells were exposed to 0, 1, 3, 10, and 30 µM cadmium for 4 (closed circle) and 24 (open circle) h. Following metal exposure, cells were incubated with fura-5F and then fluorescence ratios were determined. Asterisks indicate a significant (p<0.001) difference between control and cadmium exposed groups. Data were analyzed by two-way ANOVA followed by Tukey's post-test. Asterisks indicate a significant (p<0.001) difference between control and cadmium exposed groups. C, Cell viability of HEK293::YC3.60 cells exposed to 0, 1, 3, 10, and 30 µM cadmium for 4 (closed circle) and 24 (open circle) h. Data were expressed as the mean ± SEM and were analyzed by one-way ANOVA followed by Dunnett's post-test. Asterisks indicate significant (p<0.001) difference from control and cadmium exposed groups.
To assess intracellular cadmium concentration ([Cd2+]i) levels in chronically treated cells, cells were exposed to various concentrations of cadmium for either 4 or 24 h and then loaded with fura-5F (Fig. 2B). Cadmium accumulation was both time- and concentration-dependent. The fura-5F fluorescence ratio showed the largest increase in cells exposed to 30 µM cadmium for 24 h.
Alterations in [Ca2+]i occur during cadmium-induced cell death [21], [24], [29], [30], [31], [32], [33]. Therefore, the effect of cadmium on HEK293::YC3.60 cell viability was assessed (Fig. 2C). Exposure to 1 or 3 µM cadmium for 4 h or 24 h did not significantly affect the number of viable cells. These conditions were defined as non-cytotoxic cadmium concentrations. In contrast, exposure to higher cadmium concentrations for either 4 or 24 h significantly reduced cell viability compared to control cells and were therefore defined as cytotoxic cadmium concentrations. This suggests that changes in [Ca2+]i at 10 or 30 µM cadmium may be a consequence of on-going cell death and not a direct activity of cadmium to induce intracellular calcium release.
Effect of cadmium on YC3.60 expressed in HEK293 cells
The ability of YC3.60, a protein-based calcium ion sensor, to detect changes in [Cd2+]i was assessed in HEK293::YC3.60 cells. Figure 3 presents a time course of a typical biphasic calcium response in HEK293 cells expressing YC3.60 in calcium-free media. Similar to studies using fura-5F loaded HEK293::YC3.60 cells (Fig. 2A), the addition of ionomycin produced a transient increase in [Ca2+]i. In addition, supplementing the medium with external calcium produced an influx of calcium, as indicated by a rapid and sustained increase in the fluorescence ratio (Fig. 3). In contrast to studies using fura-5F-loaded cells, the addition of 2 mM cadmium following ionomycin treatment did not affect the fluorescence ratio. These results demonstrated that YC3.60 could distinguish between calcium and cadmium ions; thus it would be a useful tool to measure the effects of cadmium on [Ca2+]i.
Figure 3. Effect of cadmium on YC3.60 fluorescence in HEK293::YC3.60 cells.

Traces represent changes in YC3.60 fluorescence ratios following ionomycin exposure in calcium-free HBSS. At ∼15 min, the medium was supplemented with either 2 mM calcium (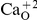 , black line) or 2 mM cadmium (
, black line) or 2 mM cadmium (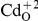 , red line). Traces are representative of typical responses observed in four independent experiments.
, red line). Traces are representative of typical responses observed in four independent experiments.
Effect of cadmium on ER calcium stores and calcium uptake
It has been proposed that cadmium activates transcription by depleting ER calcium stores to alter [Ca2+]i [34]. To determine if non-cytotoxic concentrations of cadmium had an effect on calcium homeostasis or ER calcium stores, HEK293::YC3.60 cells were treated with the SERCA pump inhibitor thapsigargin prior to cadmium addition. Treating HEK293::YC3.60 cells with thapsigargin resulted in a typical biphasic change in [Ca2+]i, which represented depletion of ER calcium stores and the re-entry of calcium across the plasma membrane by the activation of store-operated calcium entry (SOCE) (Fig. 4A) [35], [36]. The biphasic calcium response induced by thapsigargin allowed several aspects of calcium homeostasis to be investigated: (i) thapsigargin-mediated inhibition of SERCA pump activity; (ii) proper operation of plasma membrane calcium-ATPases; and (iii) the activation mechanism and permeation properties of SOCE [37]. To determine if cadmium exposure altered the activity of these homeostatic processes, the thapsigargin-induced biphasic calcium response in cells exposed to 1 µM cadmium for 4 h was compared to control cells. The biphasic calcium response in HEK293::YC3.60 cells exposed to 1 µM cadmium was not significantly different from control cells (Fig. 4B). This indicated that exposure to cadmium at a concentration sufficient to induce transcription did not disrupt calcium homeostasis.
Figure 4. panel A, Traces represent [Ca2+]i measured in control HEK293::YC3.60 cells (black line) or cells following exposure to 1 µM cadmium for 4 hr (red line).

The traces are representative of typical responses observed in at least three independent experiments. panel B, Means of the peak values in thapsigargin response following exposure to 1 µM cadmium for 4 hr (gray bar) or non-cadmium treated (black bar). Data were expressed as the mean ± SEM and were analyzed by an unpaired t-test. There were no significant differences between control and the cadmium exposed groups. panel C, Traces represent [Ca2+]i measured in HEK 293::YC3.60 cells under control conditions (black line) or following a 4 h exposure to 1 µM cadmium (red line). Following incubation with cadmium, cells were treated with an ionomycin-BAPTA solution in calcium-free HBSS. The traces were representative of typical responses observed in at least three independent experiments. panel D, Mean peak values of ionomycin responses in HEK293::YC3.60 cells following exposure to 0, 1, 3, 10, and 30 µM cadmium for 4 h (black bar) or 24 h (gray bar). Data were expressed as the mean ± SEM and were analyzed by one-way ANOVA followed by Dunnett's post-test. Asterisks (*) indicate significant difference (p<0.001) between control and cadmium exposed groups.
Any decrease in ER calcium store content would likely be reflected in a diminished thapsigargin-induced [Ca2+]i peak. Exposure to cadmium however, did not significantly affect the thapsigargin-induced peak, which suggested that cadmium did not deplete ER calcium stores (Fig. 4B).
To further investigate the effect of cadmium on ER calcium stores, an ionomycin depletion protocol was utilized [38]. In the absence of extracellular calcium, ionomycin (10 µM) selectively triggers a rapid release of ER calcium stores [39]. The height of the ionomycin-induced [Ca2+]i peak is approximately proportional to the ER calcium content (Fig. 4C). By comparing peak heights, the effects of various concentrations and exposure times of cadmium on ER calcium stores were determined. Following 4 or 24 h exposures, concentrations below 30 µM cadmium did not significantly affect ER calcium stores. A decrease of ER calcium store content was observed, however, in cells treated with 30 µM cadmium (Fig. 4D). The levels of ER calcium stores were reduced by 36% and 57% following 4 and 24 h exposures, respectively.
These data suggest that low concentrations of cadmium (1–10 µM), which induced changes in gene transcription (Fig. 1), did not interfere with calcium signaling mechanisms nor deplete ER calcium stores.
Effects of cadmium and thapsigargin on cAMP/Ca2+ responsive gene expression
PCR arrays were used to assess the ability of cadmium to affect the steady-state levels of mRNAs encoded by 84 cAMP/calcium -responsive target genes. Consistent with the [Ca2+]i measurements (Fig. 4), 1 µM cadmium did not significantly affect the transcription of the majority of the cAMP/calcium-responsive genes (Tables 1 and 2). Exposure to 30 µM cadmium for 4 or 24 h, however, significantly affected the mRNA levels of many of the cAMP/calcium -responsive target genes. This response may be the result of 30 µM cadmium depleting ER calcium stores thus increasing [Ca2+]i (Fig. 4D), or a consequence of cadmium-mediated cell death (Fig. 2C), which also alters [Ca2+]i. To determine whether the ability of cadmium to induce transcription was due to changes in [Ca2+]i, the patterns of cadmium-responsive gene expression were compared to those produced following thapsigargin exposure (Table 3). Exposure to 1 µM cadmium resulted in the differential expression of four genes that were also affected by thapsigargin exposure (Fig. 5). For these genes (TNF, FOS, PTGS2, and ERG1) however, cadmium exposure caused a significant decrease in the steady-state level of mRNA, whereas thapsigargin exposure produced a significant increase in mRNA levels.
Table 1. SuperArray analysis of cAMP/calcium signaling in cells exposed to cadmium for 4 h.
| Gene Name | Fold Change | p value |
| 1 µM Cadmium (4 h) | ||
| PENK | 2.4512 | 0.023029 |
| TNF | -3.4233 | 0.012398 |
| 30 µM Cadmium (4 h) | ||
| FGF6 | 24.6539 | 0.005357 |
| DDIT3 | 9.0099 | 0.000048 |
| DUSP1 | 6.6408 | 0.000159 |
| PENK | 4.664 | 0.000512 |
| ATF3 | 4.6518 | 0.000101 |
| PPP1R15A | 4.6185 | 0.000054 |
| INHBA | 4.3349 | 0.000586 |
| CNN1 | 4.2822 | 0.002495 |
| EGR1 | 3.9973 | 0.016708 |
| AREG | 3.3761 | 0.002253 |
| HSPA5 | 3.0578 | 0.000963 |
| FOSB | 3.0299 | 0.001182 |
| TACR1 | 2.8573 | 0.005705 |
| GEM | 2.7639 | 0.000113 |
| SCG2 | 2.6789 | 0.029449 |
| EGR2 | 2.3043 | 0.018078 |
| FOS | 2.1596 | 0.033391 |
| PTGS2 | 2.1378 | 0.056764 |
| JUND | 2.1214 | 0.001347 |
| TNF | -19.1092 | 0.000215 |
| JUNB | -2.3095 | 0.000264 |
| ADRB1 | -2.1577 | 0.000071 |
| PER1 | -2.0993 | 0.00208 |
| PLN | -2.0285 | 0.09175 |
Table 2. SuperArray analysis of cAMP/calcium signaling in cells exposed to cadmium for 24 h.
| Gene Name | Fold Change | p value |
| 1 µM Cadmium (24 h) | ||
| TNF | -13.0436 | 0.000352 |
| FOS | -3.7234 | 0.003562 |
| PTGS2 | -3.3062 | 0.023548 |
| THBS1 | -2.3139 | 0.005863 |
| EGR1 | -2.0611 | 0.020815 |
| 30 µM Cadmium (24 h) | ||
| FOSB | 1994.706 | 0.000001 |
| CHGA | 1270.4422 | 0.000002 |
| EGR1 | 1247.6159 | 0.000005 |
| FGF6 | 740.5477 | 0.000597 |
| CGA | 226.4632 | 0.000004 |
| TH | 85.4584 | 0.00002 |
| EGR2 | 76.2662 | 0.000199 |
| PPP1R15A | 70.6985 | 0.000005 |
| DDIT3 | 61.3558 | 0.000042 |
| AREG | 57.1957 | 0.000005 |
| IL6 | 51.4587 | 0.000126 |
| FOS | 51.0661 | 0.000006 |
| S100A6 | 46.5164 | 0.000114 |
| TACR1 | 43.5103 | 0.000079 |
| GEM | 42.7737 | 0.000025 |
| JUND | 28.2554 | 0.006534 |
| PRL | 27.6541 | 0.000204 |
| CNN1 | 26.1911 | 0.000112 |
| INHBA | 25.5051 | 0.000085 |
| POU1F1 | 20.8117 | 0.000247 |
| CREM | 20.704 | 0.000779 |
| PTGS2 | 20.6576 | 0.000475 |
| HSPA5 | 20.0538 | 0.000024 |
| CALB1 | 19.4826 | 0.003432 |
| JUNB | 16.0708 | 0.000079 |
| SCG2 | 14.004 | 0.001741 |
| GIPR | 12.922 | 0.00001 |
| CALB2 | 12.3142 | 0.000762 |
| SGK1 | 10.5842 | 0.000023 |
| NR4A2 | 9.6651 | 0.00006 |
| THBS1 | 8.3133 | 0.000252 |
| SSTR2 | 7.538 | 0.001687 |
| MAF | 7.0063 | 0.001082 |
| NOS2A | 6.3728 | 0.002331 |
| HK2 | 5.6116 | 0.000267 |
| TNF | 5.5566 | 0.006301 |
| NPY | 4.7459 | 0.000043 |
| DUSP1 | 4.5155 | 0.000003 |
| PENK | 4.1311 | 0.005164 |
| CTF1 | 2.9341 | 0.021035 |
| BCL2 | 2.8517 | 0.000331 |
| CCNA1 | 2.7945 | 0.015029 |
| CALR | 2.5418 | 0.003024 |
| ATF3 | 2.5353 | 0.001326 |
| PLAT | 2.4699 | 0.046362 |
| PMAIP1 | 2.3666 | 0.002755 |
| CREB1 | -5.9272 | 0.052032 |
| NCAM1 | -2.4919 | 0.041313 |
| LDHA | -2.059 | 0.010214 |
| PRKAR1A | -2.0369 | 0.008681 |
Table 3. SuperArray analysis of cAMP/calcium signaling in cells exposed to thapsigargin.
| Gene Name | Fold Change | p value |
| 2 µM Thapsigargin (4 h) | ||
| DDIT3 | 21.2583 | 0.00017 |
| PTGS2 | 15.4315 | 0.011724 |
| HSPA5 | 7.9023 | 0.00148 |
| INHBA | 7.8966 | 0.004143 |
| NR4A2 | 5.6134 | 0.004956 |
| IL6 | 5.0238 | 0.014326 |
| FOSB | 4.9708 | 0.079038 |
| PPP1R15A | 4.3266 | 0.001643 |
| GEM | 4.2286 | 0.001231 |
| EGR1 | 4.1209 | 0.048005 |
| PCK2 | 3.8763 | 0.001097 |
| ATF3 | 3.5312 | 0.000724 |
| CGA | 3.242 | 0.020191 |
| HK2 | 2.5842 | 0.133179 |
| PER1 | 2.541 | 0.018747 |
| FOS | 2.4214 | 0.003781 |
| TNF | 2.2131 | 0.048366 |
Figure 5. Venn diagram illustrating genes whose steady-state levels of expression change in HEK293::YC3.60 cells following exposure to 1 or 30 µM cadmium for 4 or 24 h, or 2 µM thapsigargin for 4 h.

The identity and description of the eleven common genes are presented in Table 4.
For 30 µM cadmium, the expression of eleven genes was significantly affected among the three exposure conditions: 4 and 24 h 30 µM cadmium and 4 h 2 µM thapsigargin (Fig. 5, Table 4). Gene Ontology analysis showed that these genes were involved in apoptosis, differentiation, and mitogenesis. Fifteen of the seventeen genes induced by thapsigargin were up-regulated in response to a 24 h exposure to 30 µM cadmium. The majority of the genes affected by cadmium, however, were not differentially expressed following thapsigargin exposure. In addition, the expression of TNF and PER1 was suppressed by cadmium exposure, but increased following thapsigargin exposure. Together, these results suggested that exposure to cytotoxic concentrations of cadmium affected transcription via a mechanism that may be independent of changes in [Ca2+]i.
Table 4. Genes affected by both cadmium and thapsigargin exposures.
| Gene | Name | Descriptiona |
| DDIT3 | DNA damage-inducible transcript 3 | Transcription factor that promotes cell death during ER stress |
| PTGS2 | Prostaglandin-endoperoxide synthase 2 | Responsible for the prostanoid biosynthesis involved in inflammation and mitogenesis |
| HSPA5 | Heat shock 70kDa protein 5 | Involved in the folding and assembly of proteins in the ER during stress |
| TNF | Tumor necrosis factor | Multifunctional proinflammatory cytokine that is mainly secreted by macrophages |
| INHBA | Inhibin beta A subunit | Member of the transforming growth factor-beta superfamily that may acts as both a growth/differentiation factor and a hormone |
| FOSB | FBJ murine osteosarcoma viral oncogene homolog B | Part of the transcription factor complex AP-1 and regulates cell proliferation, differentiation, and transformation |
| PPP1R15A | Protein phosphatase 1, regulatory (inhibitor) subunit 15A | Helps mediate apoptosis following stressful conditions |
| GEM | GTP binding protein over-expressed in skeletal musclehttp://www.genenames.org/data/hgnc_data.php?hgnc_id=4234 | Belongs to the RAD/GEM family of GTP-binding proteins, could play a role in receptor-mediated signal transduction |
| EGR1 | Early growth response 1http://www.genenames.org/data/hgnc_data.php?hgnc_id=3238 | Transcriptional regulator of genes required for differentiation and mitogenesis |
| ATF3 | Activating transcription factor 3 | Member of the CREB protein family and mediates pro-apoptotic effects of p38 |
| FOS | FBJ murine osteosarcoma viral oncogene homolog | Part of the transcription factor complex AP-1 and regulates cell proliferation, differentiation, and transformation |
Name and description were determined using GeneCards [59].
Discussion
Cadmium is a well-known toxicant that is continuously introduced into the environment. Environmental exposure to cadmium is a substantial public health concern. Recent studies suggest that incidences of cadmium-associated disease are escalating in populations exposed to low levels of cadmium [6], [8], [40], [41]. To more fully understand the relationship between cadmium and disease, it is imperative to understand the mechanism of cadmium-responsive transcription, under both adaptive and toxicological conditions. Toxicological effects at low-levels of exposure are prevented or repaired by altered expression of multiple stress-response proteins and their cognate genes. Alterations in gene expression have been observed in multiple systems at cadmium concentrations below those leading to measurable toxicological responses. In vitro exposure of HeLa or CCRF-CEM cells to non-toxic concentrations of cadmium affects the expression of ∼60 and 20 genes, respectively [42], [43]. Treatment of mice with non-toxic doses of cadmium causes the differential expression of 22 genes [44]. Likewise in C. elegans, ∼100 genes are differentially expressed following cadmium exposure under conditions that do not induce a general stress response [13].
At toxic concentrations of cadmium, the transcription of hundreds of genes is affected. Among the hundreds of cadmium-responsive genes, many of the cognate regulatory pathways have been identified. These pathways include MAPK, p53, NRF2, Protein Kinase C, casein kinase 2, and CaMK II [17], [18], [19], [20]. Although regulatory pathways have been identified, the molecular mechanisms by which cadmium initially activates these pathways to elicit specific transcriptional changes have not been defined.
One hypothesis that addresses how cadmium activates intracellular signaling pathways proposes that the metal modulates the level of [Ca2+]i [34]. Thus, calcium could be viewed as a second messenger that mediates cadmium-responsive transcription. While several mechanisms have been proposed by which cadmium may alter [Ca2+]i, the effects of cadmium on calcium signaling remain ambiguous. This ambiguity may be a consequence of technical approaches traditionally used to investigate calcium-mediated signaling processes. Specifically, there are potential problems in the interpretation of data from studies in which BAPTA or BAPTA-based fluorescent calcium indicators are used when examining the consequences of cadmium exposure on [Ca2+]i. Cadmium binds to these compounds with a >1000-fold higher affinity and can produce higher fluorescence than calcium making the interpretation of this data problematic [23]. A loss of a response during co-exposures of BAPTA with cadmium could be due to decreases in the effective cadmium concentrations, rather than effects on [Ca2+]i. Likewise, an increase in fluorescence in fura-loaded cells following exposure to cadmium could be due to the binding of cadmium to the dye, rather than a release of intracellular calcium from storage. To circumvent this problem, a protein-based calcium indicator, yellow cameleon 3.60 was used in the current studies. In HEK293::YC3.60 cells, cadmium exposure did not elicit a change in YC3.60 fluorescence (Fig. 3). Under similar experimental conditions however, cadmium produced significant increases in fura-5F fluorescence ratios (Fig. 2). This indicates that fura-5F and potentially other BAPTA-based fluorescent dyes can be used to measure [Cd2+]i, but are not appropriate when measuring the effects of cadmium on [Ca2+]i.
To assess the effects of cadmium on intracellular calcium homeostasis, HEK293 cells that stably expressed YC3.60 were used. YC3.60 provides a direct measure of [Ca2+]i without interference from cadmium (Fig. 3). A second consideration in the current experimental design is the use of non-cytotoxic concentrations of metal. Exposing HEK293::YC3.60 cells to 1 µM cadmium for 4 h was sufficient to increase steady-state mRNA levels of three well-characterized cadmium-responsive genes (Fig. 1). In addition, exposure to 1 µM cadmium for 4 or 24 h did not produce any significant toxicological responses (Fig. 2C). Based on these results, subsequent calcium homeostasis and signaling experiments were performed using HEK293::YC3.60 cells exposed to 1 µM cadmium. To replicate previous studies and gain an understanding of how cytotoxic levels of cadmium affect [Ca2+]i, cells were also exposed for 4 and 24 h to 30 µM cadmium, which is approximately three-times the 24 h LC50 for this cell line.
Using this experimental design, low-dose cadmium exposures did not interfere with calcium homeostasis nor deplete ER calcium store content (Fig. 4). Only high concentrations of cadmium (30 µM) depleted ER calcium stores. This is similar to that reported by Biagioli et al., who observed a significant depletion of ER calcium stores in NIH 3T3 cells treated with 15 µM cadmium for 12 h [34]. The reported LC50s for cadmium in 3T3 cells range from 1–5 µM [45]. These results suggest that as cells succumb to metal toxicity, calcium is released from intracellular stores.
Cadmium exposure increases the activity of MAPK and CaMK II regulated pathways [16], [17], [20], [21], [46]. Since MAPKs and CaMK II are considered integrators of calcium signaling, the effect of cadmium on the expression of calcium responsive genes was investigated using cAMP/calcium signaling focused arrays. Exposure of HEK293::YC3.60 cells to non-cytotoxic levels of cadmium, 1 µM for 4 or 24 h, did not affect the expression of a significant number of genes. One gene was commonly affected by both non-cytotoxic cadmium conditions and thapsigargin-induced intracellular calcium release. Following 24 h exposure to1 µM cadmium, an additional three genes were affected by both cadmium and thapsigargin. However, among the commonly affected genes; TNF, FOS and EGR1; cadmium caused a significant decrease in their steady-state mRNA levels while an increase in [Ca2+]i had the opposite effect (Table 2). These results are consistent with the lack of a significant effect on [Ca2+]i in cells exposed to low concentrations of cadmium (Fig. 4). These metal concentrations are associated with adaptive responses and do not deplete ER calcium stores nor interfere with intracellular calcium signaling. Thus under these conditions calcium does not function as a second messenger mediating cadmium-responsive transcription.
Exposure to cytotoxic levels of cadmium affected the steady-state mRNA levels of ∼60% of the genes, in contrast to non-cytotoxic conditions that affected ∼2% (Fig. 5, Table 1 and 2). These results are similar to previous studies demonstrating concentration-dependent increases in the number of affected genes when cells are exposed to environmental toxicants; i.e., as the concentration of toxicant increases from adaptive to cytotoxic, there is a concomitant increase in the number of genes whose steady-state level of expression change. This was observed in HepG2 cells exposed to copper; where at physiological copper concentrations (200 µM) the expression of 30 genes was affected, but at toxicological concentrations (600 µM) the number of affected genes increased to 790 [15].
Exposure to 30 µM cadmium for 24 h affected the expression of 50 genes. This result was consistent with the [Ca2+]i measurements in which 30 µM cadmium affected ER calcium stores (Fig. 4). The majority of the thapsigargin-inducible genes were also affected by 30 µM cadmium. However, three-times as many genes were affected by cadmium as thapsigargin, 17 vs. 53 (Fig. 5). In addition, the steady-state mRNA levels of TNF and PER1 increased in response to intracellular calcium release, but decreased following cadmium exposure. This suggests that the overlap among affected genes may be the result of a general activation of transcription by cadmium rather than a specific calcium-mediated effect.
Exposure to 30 µM cadmium caused a significant decrease in cell viability and depletion of ER calcium stores. At cytotoxic concentrations, calcium release may not be a specific cadmium-induced response; rather it could be a secondary or tertiary response, or non-specific affect. For example, cadmium exposure in rodents causes an increase in cAMP levels by increasing adenylate cyclase and decreasing cAMP phosphodiesterase activities, which ultimately leads to the activation of cAMP-dependent protein kinase regulated genes [47]. Similarly, the activation of DNA damage response is due to cadmium-induced DNA damage via oxidative stress and inhibition of DNA repair, and not a direct interaction between cadmium and p53 [48]. In addition, the activation/suppression of transcription could be a consequence of metal-induced membrane damage and cell death. As a consequence of the breakdown of intracellular structures, calcium would be released from membrane-bound intracellular stores. Cadmium-induced oxidative stress and lipid peroxidation occur within minutes of exposure to toxic concentrations of metal and prior to any measurable cytotoxicity [49], [50], [51]. Metal-induced damage could activate multiple processes. The activation of signaling proteins and cognate regulatory pathways would affect the expression of dozens of genes including the calcium/cAMP responsive genes on the array.
Low-level exposure to cadmium is relevant to human health as the general population is constantly exposed to low levels of this metal. Exposure to non-cytotoxic levels of cadmium is sufficient to affect gene expression, but does not alter calcium homeostasis. In addition, the transcription of calcium/cAMP responsive genes is unaffected by non-cytotoxic levels of cadmium. These data strongly suggest that cadmium-activated transcription is independent of intracellular calcium signaling. The results also support the hypothesis that at cytotoxic concentrations of cadmium, calcium-regulated signaling is affected as part of a general downstream response to cadmium-induced intracellular damage, and not a specific effect of cadmium on calcium homeostasis. They also suggest that further examination of the molecular mechanisms regulating cadmium-responsive transcription should be conducted at non-cytotoxic metal concentrations, which are environmentally relevant, and confirm that experimental reagents do not interact with cadmium.
Materials and Methods
Cell culture
HEK293 cells stably expressing the calcium sensitive protein yellow cameleon 3.60 (HEK293::YC3.60) were generated by transfecting HEK293 cells with YC3.60 cDNA using lipofectamine 2000 according to manufacturer's instructions (Invitrogen/Life Technologies, Carlsbad, CA). Details about the YC3.60 plasmid and its construction can be found in Nagai, et al. [25]. Following a 48 h recovery period, transfected cells were sorted on a FACSVantage SE Flow Cytometer (BD Bioscience, San Jose, CA) to select and enrich the cell population for cells expressing YC3.60. The sorting and enriching procedure was repeated three times to establish a population of cells homogeneously expressing YC3.60. Subsequently, HEK293::YC3.60 cells were maintained in culture at 37°C in a 5% CO2 atmosphere in Dulbecco's modified Eagle's medium supplemented with 2 mM glutamine, 10% fetal bovine serum, and 75 µg/ml G-418 (Invitrogen/Life Technologies). Cells stably expressing YC3.60 were used for all cell culture experiments.
Cell viability assays
Sensitivity of HEK293::YC3.60 cells to increasing concentrations and exposure times to cadmium was determined by using the neutral red assay as previously described [52]. HEK293::YC3.60 cells were exposed to 0, 1, 3, 10, or 30 µM cadmium for 4 or 24 h. Quadruplicate experiments were performed at each concentration. Results are presented as a percentage of control values, mean ± standard mean error (SEM) (n = 4). Data were analyzed by one-way ANOVA followed by Dunnett's Multiple Comparison post-test.
Isolation of total RNA and qRT-PCR
To assess the effects of cadmium and thapsigargin on transcription in HEK293::YC3.60 cells, steady-state levels of mt-1, c-fos, and grp-78 mRNA were determined using quantitative real time PCR (qRT-PCR). Cells were treated with cadmium (1 or 30 µM) or thapsigargin (2 µM) for the last 1, 4, or 24 h of 48 h incubations. Total RNA was then isolated from both treated and control cells using RNeasy Mini Kits following manufacturer's instructions (Qiagen, Inc., Valencia, CA).
For qRT-PCR, total RNA from three independent experiments was isolated from treated and control cells. Each biological replicate was measured in triplicate by two-step qRT-PCR using SuperScript First-Strand Synthesis System for RT-PCR (Invitrogen) and SYBR Green (Qiagen) as previously described [53]. Fold changes in mRNA levels were calculated using the ΔΔCt method with β-actin as reference mRNA [54], [55]. Changes in gene expression for cadmium-treated cells were compared to the level of expression in non-treated cells. Changes in gene expression for thapsigargin-treated cells were compared to DMSO vehicle-treated cells. Results are presented as mean log2 (fold change) ± SEM. Sequences for primers used in the qRT-PCR are presented in Table S1.
Measurements of intracellular calcium concentration
For [Ca2+]i measurements, log-phase cells were transferred onto 30 mm round glass coverslips and allowed to attach in a small volume of medium for 4–6 h. Additional DMEM was then added and the cells incubated for 24–36 h before [Ca2+]i measurements.
Fura-5F-based calcium measurements
Cells were loaded in the dark with fura-5F by incubating coverslips, mounted in a Teflon incubation chamber, with 1 µM fura-5F/AM in DMEM at 37°C for 25 min. Immediately before [Ca2+]i measurements, cells were washed three times and incubated for 15–30 min at room temperature in HEPES-buffered salt solution (HBSS; 120 mM NaCl, 5.4 mM KCl, 0.8 mM MgCl2, 1.8 mM CaCl2, 10 mM glucose in 20 mM HEPES, pH 7.4). Teflon chambers were then mounted onto a Nikon TS-100 inverted microscope equipped with a 20X objective (0.75 NA). In experiments where cells were incubated in nominally calcium-free buffer, HBSS with no added CaCl2 was used. Fluorescence images were recorded and analyzed, as previously described [56]. Changes in intracellular calcium are represented by changes in the ratio of the fura-5F fluorescence at 340 nm to that at 380 nm (F340/F380). Ratio changes were obtained from multiple regions of interest (ROI), where each ROI represented an individual cell. Typically, 25 to 35 ROIs were monitored per experiment. Ratio values were corrected for autofluorescence, which was determined after treating cells with 10 µM ionomycin and 20 mM MnCl2 [37]. Because BAPTA-based calcium indicators bind cadmium with high affinity, fura-5F-loaded HEK293::YC3.60 cells were also used to monitor the accumulation of [Cd2+]i.
YC3.60-based calcium measurements
HEK293::YC3.60 cells were mounted in Teflon incubation chambers and maintained in HBSS. Chambers were mounted on a Zeiss LSM 510 confocal microscope equipped with a 20X objective (NA 0.8). The YC3.60 FRET signals were monitored by exciting at 458 nm and collecting emission images at 490 nm for CFP and 530 nm for YFP [57]. After correcting for background fluorescence, [Ca2+]i levels were monitored by calculating the fluorescence ratio of YFP to CFP emissions (F530/F490). An increase in [Ca2+]i was observed as an increase in the F530/F490 ratio. Typically, fluorescence signals from 25–30 ROIs were monitored for a single experiment.
Effects of cadmium on intracellular calcium pools and store-operated calcium entry
The status of intracellular calcium pools following exposure to 0, 1, 3, 10 or 30 µM cadmium for 4 or 24 h was assessed by exposing HEK293 cells to 10 µM ionomycin in the presence of 3 mM BAPTA [37]. Following incubation in cadmium containing buffer, cells were mounted in a Teflon incubation chamber and maintained in HBSS for ∼10 min before being treated with 10 µM ionomycin in the presence of 3 mM BAPTA [38]. The transient calcium response was indicative of the size of intracellular calcium pool and could be quantified by calculating the peak YC3.60 response. The peak YC3.60 response was defined by the maximal response minus the resting fluorescence ratio.
To investigate the effects of cadmium on SOCE, a “calcium re-addition” protocol was used in HEK293::YC3.60 cells treated with 2 µM thapsigargin [37]. In cases where the cadmium concentration was cytotoxic, YC3.60 fluorescence was monitored only in recognizably viable cells. All data were analyzed by one-way ANOVA followed by Dunnett's Multiple Comparison post-test.
Human cAMP/calcium Signaling RT2 ProfilerTM PCR Array
Human cAMP/calcium Signaling RT2 ProfilerTM PCR Arrays (SABiosciences, Frederick, MD) were used to examine the effect of cadmium on calcium-responsive transcription in HEK293::YC3.60 cells. These arrays contain 84 genes that are reported to be responsive to changes in cAMP and calcium levels. The layout and description of the genes on the array are presented in Table S2.
Cells were grown and RNA was isolated as described above. The purity and quality of RNA was assessed using the RNA 6000 LabChip and Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clare, CA, USA). Procedures for qRT-PCR and array analysis were performed according to manufacturer's instructions. Array data was normalized to the average threshold cycle (Ct) value of three housekeeping genes: β-2-microglobulin (B2M), hypoxanthine phosphoribosyltransferase (HPRT1), and ribosomal protein L13a (RPL13A). These genes were chosen for normalization because their Ct values did not differ by more than one cycle among all of the samples and treatments. The average Ct value for the three housekeeping genes did differ by more than one cycle in cells treated with 30 µM cadmium for 24 h.
To make a meaningful biological analysis of differentially expressed genes in cells treated with 30 µM cadmium for 24 h, a normalization factor was calculated for each of the three independent experiments (0.91, 0.88, and 0.89) so that the normalized expression ratio of the average Ct value for each housekeeping gene was equal across compared samples (cadmium treated vs. control). This normalization factor was then used to appropriately scale the expression value for each of the 84 calcium specific genes within each array for cells treated with 30 µM cadmium for 24 h. These normalized expression values were then used to determine fold change. Data was then processed with SABiosciences web-based RT2 ProfilerTMPCR Array Data Analysis to calculate Ct and relative gene expression values according to the ΔΔ Ct method [58]. A list of differentially expressed genes was identified using a Student's t-test. Only two-fold or greater changes in gene expression with a p<0.05 were considered significant.
Supporting Information
Sequences of primers used for qRT-PCR.
(DOCX)
Functional gene grouping of human cAMP/calcium PCR Array.
(DOCX)
Acknowledgments
The authors would like to thank Dr. Atsushi Miyawaki, RIKEN Brain Institute, Japan, for the generous gift of yellow cameleon 3.60.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This work was supported in part by the Intramural Research Program of the NIH, and NIEHS (Z01ES102045). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. No additional external funding received for this study.
References
- 1.Paniagua-Castro N, Escalona-Cardoso G, Madrigal-Bujaidar E, Martinez-Galero E, Chamorro-Cevallos G. Protection against cadmium-induced teratogenicity in vitro by glycine. Toxicol In Vitro. 2008;22:75–79. doi: 10.1016/j.tiv.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 2.Waalkes MP, Coogan TP, Barter RA. Toxicological principles of metal carcinogenesis with special emphasis on cadmium. Crit Rev Toxicol. 1992;22:175–201. doi: 10.3109/10408449209145323. [DOI] [PubMed] [Google Scholar]
- 3.Hogervorst J, Plusquin M, Vangronsveld J, Nawrot T, Cuypers A, et al. House dust as possible route of environmental exposure to cadmium and lead in the adult general population. Environ Res. 2007;103:30–37. doi: 10.1016/j.envres.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 4.Friberg L, Elinder CG. Geneva: World Health Organization; 1992. Environmental Health Criteria 134: Cadmium. [Google Scholar]
- 5.IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Lyon, France Geneva: IARC; Distributed for the International Agency for Research on Cancer by the Secretariat of the World Health Organization; 1993. Beryllium, cadmium, mercury, and exposures in the glass manufacturing industry. p. 444 p. [Google Scholar]
- 6.Akesson A, Lundh T, Vahter M, Bjellerup P, Lidfeldt J, et al. Tubular and glomerular kidney effects in Swedish women with low environmental cadmium exposure. Environ Health Perspect. 2005;113:1627–1631. doi: 10.1289/ehp.8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engstrom A, Skerving S, Lidfeldt J, Burgaz A, Lundh T, et al. Cadmium-induced bone effect is not mediated via low serum 1,25-dihydroxy vitamin D. Environ Res. 2009;109:188–192. doi: 10.1016/j.envres.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 8.Schutte R, Nawrot TS, Richart T, Thijs L, Vanderschueren D, et al. Bone resorption and environmental exposure to cadmium in women: a population study. Environ Health Perspect. 2008;116:777–783. doi: 10.1289/ehp.11167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hsiao CJ, Stapleton SR. Early sensing and gene expression profiling under a low dose of cadmium exposure. Biochimie. 2009;91:329–343. doi: 10.1016/j.biochi.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 10.Klaassen CD, Liu J, Choudhuri S. Metallothionein: an intracellular protein to protect against cadmium toxicity. Annu Rev Pharmacol Toxicol. 1999;39:267–294. doi: 10.1146/annurev.pharmtox.39.1.267. [DOI] [PubMed] [Google Scholar]
- 11.Waisberg M, Joseph P, Hale B, Beyersmann D. Molecular and cellular mechanisms of cadmium carcinogenesis. Toxicology. 2003;192:95–117. doi: 10.1016/s0300-483x(03)00305-6. [DOI] [PubMed] [Google Scholar]
- 12.Andrew AS, Warren AJ, Barchowsky A, Temple KA, Klei L, et al. Genomic and proteomic profiling of responses to toxic metals in human lung cells. Environ Health Perspect. 2003;111:825–835. doi: 10.1289/ehp.111-1241504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cui Y, McBride SJ, Boyd WA, Alper S, Freedman JH. Toxicogenomic analysis of Caenorhabditis elegans reveals novel genes and pathways involved in the resistance to cadmium toxicity. Genome Biol. 2007;8:R122. doi: 10.1186/gb-2007-8-6-r122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kawata K, Shimazaki R, Okabe S. Comparison of gene expression profiles in HepG2 cells exposed to arsenic, cadmium, nickel, and three model carcinogens for investigating the mechanisms of metal carcinogenesis. Environ Mol Mutagen. 2009;50:46–59. doi: 10.1002/em.20438. [DOI] [PubMed] [Google Scholar]
- 15.Song MO, Li J, Freedman JH. Physiological and toxicological transcriptome changes in HepG2 cells exposed to copper. Physiol Genomics. 2009;38:386–401. doi: 10.1152/physiolgenomics.00083.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chuang SM, Wang IC, Yang JL. Roles of JNK, p38 and ERK mitogen-activated protein kinases in the growth inhibition and apoptosis induced by cadmium. Carcinogenesis. 2000;21:1423–1432. [PubMed] [Google Scholar]
- 17.Adams TK, Saydam N, Steiner F, Schaffner W, Freedman JH. Activation of gene expression by metal-responsive signal transduction pathways. Environ Health Perspect. 2002;110(Suppl 5):813–817. doi: 10.1289/ehp.02110s5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beyersmann D, Hechtenberg S. Cadmium, gene regulation, and cellular signaling in mammalian cells. Toxicol Appl Pharmacol. 1997;144:247–261. doi: 10.1006/taap.1997.8125. [DOI] [PubMed] [Google Scholar]
- 19.Liu Y, Templeton DM. Cadmium activates CaMK-II and initiates CaMK-II-dependent apoptosis in mesangial cells. FEBS Lett. 2007;581:1481–1486. doi: 10.1016/j.febslet.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 20.Watkin RD, Nawrot T, Potts RJ, Hart BA. Mechanisms regulating the cadmium-mediated suppression of Sp1 transcription factor activity in alveolar epithelial cells. Toxicology. 2003;184:157–178. doi: 10.1016/s0300-483x(02)00577-2. [DOI] [PubMed] [Google Scholar]
- 21.Kim J, Sharma RP. Calcium-mediated activation of c-Jun NH2-terminal kinase (JNK) and apoptosis in response to cadmium in murine macrophages. Toxicol Sci. 2004;81:518–527. doi: 10.1093/toxsci/kfh221. [DOI] [PubMed] [Google Scholar]
- 22.Misra UK, Gawdi G, Akabani G, Pizzo SV. Cadmium-induced DNA synthesis and cell proliferation in macrophages: the role of intracellular calcium and signal transduction mechanisms. Cell Signal. 2002;14:327–340. doi: 10.1016/s0898-6568(01)00268-6. [DOI] [PubMed] [Google Scholar]
- 23.Hinkle PM, Shanshala ED, 2nd, Nelson EJ. Measurement of intracellular cadmium with fluorescent dyes. Further evidence for the role of calcium channels in cadmium uptake. J Biol Chem. 1992;267:25553–25559. [PubMed] [Google Scholar]
- 24.Lemarie A, Lagadic-Gossmann D, Morzadec C, Allain N, Fardel O, et al. Cadmium induces caspase-independent apoptosis in liver Hep3B cells: role for calcium in signaling oxidative stress-related impairment of mitochondria and relocation of endonuclease G and apoptosis-inducing factor. Free Radic Biol Med. 2004;36:1517–1531. doi: 10.1016/j.freeradbiomed.2004.03.020. [DOI] [PubMed] [Google Scholar]
- 25.Nagai T, Yamada S, Tominaga T, Ichikawa M, Miyawaki A. Expanded dynamic range of fluorescent indicators for Ca2+ by circularly permuted yellow fluorescent proteins. Proc Natl Acad Sci U S A. 2004;101:10554–10559. doi: 10.1073/pnas.0400417101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thastrup O, Dawson AP, Scharff O, Foder B, Cullen PJ, et al. Thapsigargin, a novel molecular probe for studying intracellular calcium release and storage. 1989. Agents Actions. 1994;43:187–193. doi: 10.1007/BF01986687. [DOI] [PubMed] [Google Scholar]
- 27.Treiman M, Caspersen C, Christensen SB. A tool coming of age: thapsigargin as an inhibitor of sarco-endoplasmic reticulum Ca2+-ATPases. Trends Pharmacol Sci. 1998;19:131–135. doi: 10.1016/s0165-6147(98)01184-5. [DOI] [PubMed] [Google Scholar]
- 28.Templeton DM, Wang Z, Miralem T. Cadmium and calcium-dependent c-fos expression in mesangial cells. Toxicol Lett. 1998;95:1–8. doi: 10.1016/s0378-4274(98)00015-0. [DOI] [PubMed] [Google Scholar]
- 29.Joseph P, Muchnok TK, Klishis ML, Roberts JR, Antonini JM, et al. Cadmium-induced cell transformation and tumorigenesis are associated with transcriptional activation of c-fos, c-jun, and c-myc proto-oncogenes: role of cellular calcium and reactive oxygen species. Toxicol Sci. 2001;61:295–303. doi: 10.1093/toxsci/61.2.295. [DOI] [PubMed] [Google Scholar]
- 30.Lee MJ, Nishio H, Ayaki H, Yamamoto M, Sumino K. Upregulation of stress response mRNAs in COS-7 cells exposed to cadmium. Toxicology. 2002;174:109–117. doi: 10.1016/s0300-483x(02)00045-8. [DOI] [PubMed] [Google Scholar]
- 31.Li M, Kondo T, Zhao QL, Li FJ, Tanabe K, et al. Apoptosis induced by cadmium in human lymphoma U937 cells through Ca2+-calpain and caspase-mitochondria- dependent pathways. J Biol Chem. 2000;275:39702–39709. doi: 10.1074/jbc.M007369200. [DOI] [PubMed] [Google Scholar]
- 32.Shih YL, Lin CJ, Hsu SW, Wang SH, Chen WL, et al. Cadmium toxicity toward caspase-independent apoptosis through the mitochondria-calcium pathway in mtDNA-depleted cells. Ann N Y Acad Sci. 2005;1042:497–505. doi: 10.1196/annals.1338.043. [DOI] [PubMed] [Google Scholar]
- 33.Wang SH, Shih YL, Ko WC, Wei YH, Shih CM. Cadmium-induced autophagy and apoptosis are mediated by a calcium signaling pathway. Cell Mol Life Sci. 2008;65:3640–3652. doi: 10.1007/s00018-008-8383-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Biagioli M, Pifferi S, Ragghianti M, Bucci S, Rizzuto R, et al. Endoplasmic reticulum stress and alteration in calcium homeostasis are involved in cadmium-induced apoptosis. Cell Calcium. 2008;43:184–195. doi: 10.1016/j.ceca.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 35.Inesi G, Sagara Y. Thapsigargin, a high affinity and global inhibitor of intracellular Ca2+ transport ATPases. Arch Biochem Biophys. 1992;298:313–317. doi: 10.1016/0003-9861(92)90416-t. [DOI] [PubMed] [Google Scholar]
- 36.Putney JW., Jr Pharmacology of capacitative calcium entry. Mol Interv. 2001;1:84–94. [PubMed] [Google Scholar]
- 37.Bird GS, DeHaven WI, Smyth JT, Putney JW., Jr Methods for studying store-operated calcium entry. Methods. 2008;46:204–212. doi: 10.1016/j.ymeth.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bird GS, Putney JW., Jr Capacitative calcium entry supports calcium oscillations in human embryonic kidney cells. J Physiol. 2005;562:697–706. doi: 10.1113/jphysiol.2004.077289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morgan AJ, Jacob R. Ionomycin enhances Ca2+ influx by stimulating store-regulated cation entry and not by a direct action at the plasma membrane. Biochem J. 1994;300(Pt 3):665–672. doi: 10.1042/bj3000665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Menke A, Muntner P, Silbergeld EK, Platz EA, Guallar E. Cadmium levels in urine and mortality among U.S. adults. Environ Health Perspect. 2009;117:190–196. doi: 10.1289/ehp.11236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Satarug S, Moore MR. Adverse health effects of chronic exposure to low-level cadmium in foodstuffs and cigarette smoke. Environ Health Perspect. 2004;112:1099–1103. doi: 10.1289/ehp.6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Th Tsangaris G, Botsonis A, Politis I, Tzortzatou-Stathopoulou F. Evaluation of cadmium-induced transcriptome alterations by three color cDNA labeling microarray analysis on a T-cell line. Toxicology. 2002;178:135–160. doi: 10.1016/s0300-483x(02)00236-6. [DOI] [PubMed] [Google Scholar]
- 43.Yamada H, Koizumi S. DNA microarray analysis of human gene expression induced by a non-lethal dose of cadmium. Ind Health. 2002;40:159–166. doi: 10.2486/indhealth.40.159. [DOI] [PubMed] [Google Scholar]
- 44.Zhou T, Jia X, Chapin RE, Maronpot RR, Harris MW, et al. Cadmium at a non-toxic dose alters gene expression in mouse testes. Toxicol Lett. 2004;154:191–200. doi: 10.1016/j.toxlet.2004.07.015. [DOI] [PubMed] [Google Scholar]
- 45.Morton KA, Jones BJ, Sohn MH, Schaefer AE, Phelps RC, et al. Uptake of cadmium is diminished in transfected mouse NIH/3T3 cells enriched for metallothionein. J Biol Chem. 1992;267:2880–2883. [PubMed] [Google Scholar]
- 46.Liu F, Inageda K, Nishitai G, Matsuoka M. Cadmium induces the expression of Grp78, an endoplasmic reticulum molecular chaperone, in LLC-PK1 renal epithelial cells. Environ Health Perspect. 2006;114:859–864. doi: 10.1289/ehp.8920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Merali Z, Kacew S, Singhal RL. Response of hepatic carbohydrate and cyclic AMP metabolism to cadmium treatment in rats. Can J Physiol Pharmacol. 1975;53:174–184. doi: 10.1139/y75-024. [DOI] [PubMed] [Google Scholar]
- 48.Jin YH, Clark AB, Slebos RJ, Al-Refai H, Taylor JA, et al. Cadmium is a mutagen that acts by inhibiting mismatch repair. Nat Genet. 2003;34:326–329. doi: 10.1038/ng1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pourahmad J, O'Brien PJ. A comparison of hepatocyte cytotoxic mechanisms for Cu2+ and Cd2+. Toxicology. 2000;143:263–273. doi: 10.1016/s0300-483x(99)00178-x. [DOI] [PubMed] [Google Scholar]
- 50.Hsiao CJ, Stapleton SR. Characterization of Cd-induced molecular events prior to cellular damage in primary rat hepatocytes in culture: activation of the stress activated signal protein JNK and transcription factor AP-1. J Biochem Mol Toxicol. 2004;18:133–142. doi: 10.1002/jbt.20018. [DOI] [PubMed] [Google Scholar]
- 51.Xu J, Maki D, Stapleton SR. Mediation of cadmium-induced oxidative damage and glucose-6-phosphate dehydrogenase expression through glutathione depletion. J Biochem Mol Toxicol. 2003;17:67–75. doi: 10.1002/jbt.10062. [DOI] [PubMed] [Google Scholar]
- 52.Mattie MD, Freedman JH. Protective effects of aspirin and vitamin E (α-tocopherol) against copper- and cadmium-induced toxicity. Biochem Biophys Res Commun. 2001;285:921–925. doi: 10.1006/bbrc.2001.5259. [DOI] [PubMed] [Google Scholar]
- 53.McElwee MK, Song MO, Freedman JH. Copper activation of NF-κB signaling in HepG2 cells. J Mol Biol. 2009;393:1013–1021. doi: 10.1016/j.jmb.2009.08.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods (Duluth) 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 55.Song MO, Freedman JH. Expression of copper-responsive genes in HepG2 cells. Mol Cell Biochem. 2005;279:141–147. doi: 10.1007/s11010-005-8286-0. [DOI] [PubMed] [Google Scholar]
- 56.Jones BF, Boyles RR, Hwang SY, Bird GS, Putney JW. Calcium influx mechanisms underlying calcium oscillations in rat hepatocytes. Hepatology. 2008;48:1273–1281. doi: 10.1002/hep.22461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miyawaki A, Griesbeck O, Heim R, Tsien RY. Dynamic and quantitative Ca2+ measurements using improved cameleons. Proc Natl Acad Sci U S A. 1999;96:2135–2140. doi: 10.1073/pnas.96.5.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative CT method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 59.Safran M, Solomon I, Shmueli O, Lapidot M, Shen-Orr S, et al. GeneCards 2002: towards a complete, object-oriented, human gene compendium. Bioinformatics. 2002;18:1542–1543. doi: 10.1093/bioinformatics/18.11.1542. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sequences of primers used for qRT-PCR.
(DOCX)
Functional gene grouping of human cAMP/calcium PCR Array.
(DOCX)