

Abstract
The analysis of differentially expressed genes is a powerful approach to elucidate the genetic mechanisms underlying the morphological and evolutionary diversity among serially homologous structures, both within the same organism (e.g., hand vs. foot) and between different species (e.g., hand vs. wing). In the developing embryo, limb-specific expression of Pitx1, Tbx4, and Tbx5 regulates the determination of limb identity. However, numerous lines of evidence, including the fact that these three genes encode transcription factors, indicate that additional genes are involved in the Pitx1-Tbx hierarchy. To examine the molecular distinctions coded for by these factors, and to identify novel genes involved in the determination of limb identity, we have used Serial Analysis of Gene Expression (SAGE) to generate comprehensive gene expression profiles from intact, developing mouse forelimbs and hindlimbs. To minimize the extraction of erroneous SAGE tags from low-quality sequence data, we used a new algorithm to extract tags from phred-analyzed sequence data and obtained 68,406 and 68,450 SAGE tags from forelimb and hindlimb SAGE libraries, respectively. We also developed an improved method for determining the identity of SAGE tags that increases the specificity of and provides additional information about the confidence of the tag-UniGene cluster match. The most differentially expressed gene between our SAGE libraries was Pitx1. The differential expression of Tbx4, Tbx5, and several limb-specific Hox genes was also detected; however, their abundances in the SAGE libraries were low. Because numerous other tags were differentially expressed at this low level, we performed a ‘virtual‘ subtraction with 362,344 tags from six additional nonlimb SAGE libraries to further refine this set of candidate genes. This subtraction reduced the number of candidate genes by 74%, yet preserved the previously identified regulators of limb identity. This study presents the gene expression complexity of the developing limb and identifies candidate genes involved in the regulation of limb identity. We propose that our computational tools and the overall strategy used here are broadly applicable to other SAGE-based studies in a variety of organisms.
[SAGE data are all available at GEO (http://www.ncbi.nlm.nih.gov/geo/) under accession nos. GSM55 and GSM56, which correspond to the forelimb and hindlimb raw SAGE data.]
The developing vertebrate limb is a well-established model system for studying the genetic factors regulating growth, patterning, and cellular differentiation (Cohn and Tickle 1996; Johnson and Tabin 1997). Recent studies into the molecular determinants of forelimb/hindlimb identity have focused on several genes that show differential expression in the lateral plate mesoderm and the early developing limb bud, including Pitx1, Tbx4, and Tbx5 (Gibson-Brown et al. 1996; Szeto et al. 1996) as well as certain Hox genes (Peterson et al. 1994; Nelson et al. 1996; Cohn et al. 1997). The hypothesis that differential gene expression determines morphological fate was confirmed by experiments with Pitx1 and the Tbx genes.
Pitx1 and Tbx4 are expressed predominantly in the developing hindlimb, and Tbx5 is expressed predominantly in the forelimb. Studies in chicks have shown that Pitx1 and Tbx4 can exert a transformation of limb type when misexpressed in the developing wing (Gibson-Brown et al. 1998; Isaac et al. 1998; Logan and Tabin 1999). Similarly, Tbx5 expression in the developing leg results in the growth of a wing-like morphology (Rodriguez-Esteban et al. 1999; Takeuchi et al. 1999). Several Hox genes also appear to be regulated by the Pitx1-Tbx hierarchy (Logan and Tabin 1999); however, functional studies with these genes have not yet revealed any limb type transforming properties (Papenbrock et al. 2000).
Misexpression of Tbx5 in the chick hindlimb suppresses expression of Tbx4. Misexpression of Pitx1 in prospective forelimbs induces expression of Tbx4, Hoxc10, and Hoxc11 but has no effect on Tbx5 expression (Logan and Tabin 1999). Similarly, Tbx4 induces Hoxc9, Hoxc10, and Hoxc11 and suppresses Hoxd9 (Rodriguez-Esteban et al. 1999; Takeuchi et al. 1999). Therefore, these transcription factors mediate their limb-transforming properties, in part, by regulating each other, as well as specific downstream target genes.
In other work, engineered mice lacking Pitx1 develop hindlimbs with reduced Tbx4 gene expression and skeletal and muscle features more characteristic of forelimbs (Lanctôt et al. 1999; Szeto et al. 1999), showing the importance of Pitx1 in Tbx4 regulation and the necessity of Pitx1 for correct hindlimb morphogenesis. In addition to the mouse and chick, appendage-specific expression of these genes has also been observed in the developing embryos of Xenopus and Danio rerio (Tamura et al. 1999; Takabatake et al. 2000), indicating that the genetic determinants of tetrapod limb identity have ancient origins and that differences in final limb morphologies are likely to be related to target gene selection (Weatherbee and Carroll 1999).
Several observations support the hypothesis that additional regulators exist in the limb identity genetic pathway. First, Pitx1, Tbx4, and Tbx5 are transcription factors; however, the identity of their target genes as well as the upstream regulators that restrict expression to specific limbs are not known (Johnson and Tabin 1997; Niswander 1999). Second, limb-type transformations that occur in Pitx1, Tbx4, or Tbx5 misexpression experiments are incomplete. Incomplete transformations also occur in mice with loss of Pitx1 expression in the hindlimb. Although these incomplete transformations may simply reflect experimental limitations in the timing, domain, or level of expression, they highlight the need for further work. Third, even though Pitx1 is capable of inducing expression of Tbx4, Pitx1−/− mice express low levels of Tbx4, indicating the existence of alternative regulatory pathways. Finally, TBX5 mutations cause Holt-Oram syndrome in humans, resulting in upper limb and cardiac malformations, both regions of TBX5 expression in development (Basson et al. 1997; Li et al. 1997). The existence of numerous other inherited human malformation syndromes that involve predominantly either the upper or lower limbs (Margulies and Innis 2000a) indicates the potential for numerous unidentified genes with differential limb expression.
To identify new genes, integrate our findings with current knowledge, and foster new hypotheses about the relationships among limb-specific gene expression, morphology, and function, we used Serial Analysis of Gene Expression (SAGE) to generate comprehensive gene expression profiles from intact, developing mouse forelimbs and hindlimbs. We also developed several new computational tools for the analysis of SAGE data and used them to assemble a set of differentially expressed genes potentially involved in limb identity and development. Finally, to refine this set of genes, we compared our limb SAGE libraries with other nonlimb, mouse SAGE libraries. These studies not only document the complexity of gene expression in the developing limb, but also provide a molecular framework for exploring the evolutionary divergence of morphology and function in serially homologous structures.
RESULTS
Summary of Sequenced SAGE Libraries
SAGE was performed to obtain quantitative gene expression profiles from developing mouse forelimbs and hindlimbs. Table 1 summarizes the sequenced SAGE libraries, which have been submitted to the Gene Expression Omnibus (GEO) database at NCBI as accession numbers GSM55 and GSM56. The forelimb and hindlimb SAGE data have also been provided in the web supplement to this article (see the “Compare” table in the Access 97 database). This Compare table also includes the fold differences in gene expression for each tag and the calculated P values (Audic and Claverie 1997) for differential tag frequencies. Several measurements support the fact that these developing limb SAGE libraries are of high quality. First, the average GC content of the SAGE tags indicated there was no GC content bias (Margulies et al. 2001). Second, the frequency of duplicate DiTags and percent linker contamination was equivalent to or below that of other reported SAGE libraries (Velculescu et al. 1997). Third, Monte Carlo analysis showed that subpopulations of data from the same SAGE library were similar (Margulies et al. 2001; additional data not shown). Finally, 72% of the sequence data used to generate SAGE tags were analyzed with the phred base-calling algorithm, allowing us to determine a quantitative tag sequencing error rate.
Table 1.
Summary of SAGE Libraries
| SAGE library | ||
|---|---|---|
| Forelimb | Hindlimb | |
| Total SAGE tags excluding linkers1 | 68,406 | 68,450 |
| Unique transcripts | 23,140 | 22,329 |
| Transcripts observed in both libraries | 9,169 | |
| Linker contamination | 450 (0.65%) | 405 (0.64%) |
| Frequency of duplicate DiTags2 | 1.05% | 0.84% |
| Average GC content3 | 48.3 | 47.5 |
| Number of clones sequenced | 3,639 | |
| Averaged phred score per base | 38 | 38 |
Linker sequences removed from our SAGE libraries were TCCCTATTAA and TCCCCGTACA.
Upon examination of several duplicate DiTags in the limb SAGE libraries, all were found to be composed of high abundance SAGE Tags (data not shown), indicating that the duplicate DiTags in these SAGE libraries were not due to DiTag amplification bias.
For an explanation of this value, in the context of the quality of a SAGE library, see Margulies et al. (2001).
Estimation of Tag Sequencing Error Rate
To minimize the inclusion of SAGE tags arising from sequencing errors, we used a minimum acceptable phred quality value of 20, corresponding to an error rate of 1 in 100 (Ewing and Green 1998). SAGE tag sequences containing base calls with phred quality values <20 were not added to the database (Margulies and Innis 2000b). Much of our sequence data had phred scores >40 (error rate of 1 in 10,000). In fact, the average phred score for all phred-analyzed sequence data from which SAGE tags were extracted was 38. This phred score corresponds to an average sequencing error rate of 1 in 6300. Because there are 10 bp to a SAGE tag, it can be estimated that 1 in every 630 SAGE tags may have a sequencing error. Therefore, we estimated that only 220 out of the 136,856 total tags sequenced in both SAGE libraries arose from sequencing error artifacts. For these calculations, a similar error rate was assumed for sequences edited manually from the ALFexpress (comprising 28% of the total sequenced SAGE tags).
In contrast, extracting SAGE tags from the same trace data (2908 total sequence files), but instead using the default ABI-generated base calls, which have no associated quality information, increased the number of SAGE tags obtained by 23.1% (120,544 vs. 97,948), representing an increase in the number of unique transcripts by 20.1% (34,525 vs. 28,738). Ninety-five percent of the additional unique transcripts were represented by SAGE tags observed once. Therefore, using phred in combination with eSAGE allowed us to define the minimum acceptable sequence quality used in the SAGE tag extraction process, likely reducing the addition of erroneous SAGE tags and increasing the accuracy of the resulting SAGE library.
Distribution of SAGE Tags
Table 2 shows the proportion of tags matching genes or ESTs separated by abundance class. The proportion of SAGE tags matching genes was highly skewed toward the abundant tags. Eighty-six percent of the unique tags in the highest abundance class matched a gene whereas only 3.2% of all tags in the lowest abundance class matched a gene. Overall, 33.1% of the unique SAGE tags matched genes or ESTs. Excluding the lowest abundance class of tags observed once, this proportion increased to 58%. A comprehensive table containing our limb SAGE data linked to the ehm-tag-Mapping data is presented in the web supplement to this article (see the “ehmTagID” table in the Access 97 database).
Table 2.
Distribution of SAGE Tags Matching Genes or ESTs, Sorted by Abundance Class
| Copies per cell12 | Percent abundance1 | Range of tag frequency | Matching gene3 | EST | No match | Total |
|---|---|---|---|---|---|---|
| >100 | >0.27 | >382 | 18 | 1 | 2 | 21 |
| 50–100 | 0.132–0.27 | 183–363 | 51 | 13 | 7 | 71 |
| 5–50 | 0.0135–0.132 | 19–180 | 440 | 199 | 162 | 801 |
| 1–5 | 0.0025–0.0135 | 4–18 | 1,280 | 2,077 | 1,570 | 4,927 |
| 0.3–1 | 0.0015–0.0022 | 2–3 | 776 | 2,195 | 3,463 | 6,434 |
| <0.3 | <0.00075 | 1 | 775 | 3,563 | 19,708 | 24,046 |
| All transcripts | 3,711 | 8,296 | 24,293 | 36,300 | ||
| (10.2%) | (22.9%) | (66.9%) |
This table represents information calculated from the sum of both SAGE libraries (136,856 total tags representing 36,300 unique transcripts.)
Unless otherwise noted, ranges are given as > the first value and ≤ the second value.
Copies per cell was calculated assuming, or average, 500,000 transcripts in a cell (Hastie and Bishop 1976).
If a tag matches a gene and an EST, it was counted only as a gene. See Methods for tag-to-UniGene cluster matching algorithm.
Figure 1 depicts the overall distribution of gene expression between the forelimb and hindlimb SAGE libraries, relative to the 1% and 5% confidence intervals from the test statistic developed by Audic and Claverie (1997). Most genes fell within the “funnel,” indicating that the gene expression profiles between these two SAGE libraries are very similar. Also depicted in Figure 1 is the wide range in the levels of gene expression that span more than three orders of magnitude, corresponding to a minimum and maximum abundance of 0.27 and 342 copies/cell, respectively. Finally, Figure 1 shows that most of the gene expression complexity (number of unique transcripts) was observed in the lowest abundance class, in agreement with other SAGE experiments (e.g., see Zhang et al. 1997), as well as earlier COt/ROt analyses (Hastie and Bishop 1976).
Figure 1.

Scatter plot of tag abundance distributions in the forelimb and hindlimb. Note the log scale. Each point represents a tag with a given frequency in the forelimb (X-axis) and hindlimb (Y-axis). The number at each point indicates the number of unique tags with a given frequency. (Dashed and solid lines) 0.05 and 0.01 P values for probability of differential expression, respectively. Tags falling outside these lines are differentially expressed according to the test statistic developed by Audic and Claverie (1997). Seventy SAGE tags fall outside the 1% confidence interval and 317 SAGE tags fall outside the 5% confidence interval. (Dotted line) Equal expression in the forelimb and hindlimb. Zero is plotted as 0.5 to be observed on a log scale.
Among the most abundant transcripts were several expected housekeeping genes (e.g., translation elongation factor 1α and glyceraldehyde-3-phosphate dehydrogenase). The most abundant SAGE tag represented a B2 repeat element. This element is known to be expressed widely in developing and adult mouse tissues (Taylor and Piko 1987) and has also been observed at high abundance in other mouse SAGE libraries (e.g., see Chrast et al. 2000). Interestingly, ancient B2 fragments are found in a number of 3′ UTRs (Maichele et al. 1993), making it likely that, in our SAGE libraries, the B2 repeat SAGE tag was present in multiple transcripts.
Sixty-six percent of the unique tags were observed only once and correspond to genes expressed, on average, less than 0.3 copies per cell. This highly complex abundance class comprised only 17.6% of all sequenced SAGE tags (24,046 out of 136,856 tags). Unique SAGE tags continued to accumulate at a rate of 16% toward the end of our sequencing effort (Fig. 2), indicating that we had likely sampled >85% of the unique transcripts present in the developing limb. This observation was consistent with an analysis of 3.5 million SAGE tags from 19 different human tissues (Velculescu et al. 1999). Interestingly, 45% of all identified transcription factors (113 out of 251) were expressed in the lowest abundance classes, on average one copy per cell or less (data not shown).
Figure 2.

Fraction of unique SAGE tags added to the database. (X-axis) Cumulative total number of SAGE tags in our combined forelimb/hindlimb database; (Y-axis) average fraction of unique SAGE tags added to the database. eSAGE calculates the fraction of unique SAGE tags extracted from each sequence file. The data points were calculated by averaging sequence file information over the cumulative SAGE tag interval (just after the previous data point and including the current data point). For example, of the last 17,000 tags added to the database (from ∼120,000 to ∼137,000 tags), an average of 16% were unique (or novel entries).
Quality of the ehm-Tag-Mapping Method
From the mouse UniGene build # 85 on December 18, 2000 (containing 83,862 different UniGene clusters), the ehm-tag-Mapping method identified 65,245 tag-UniGene cluster matches, representing 55,113 UniGene clusters and 52,355 unique tags. We found that 86.4% of the tags matched only one UniGene cluster, and 85% of the UniGene clusters had only one matching SAGE tag. These percentages increased to 96.3% and 97.1% by including tags matching two UniGene clusters and UniGene clusters with two matching SAGE tags, respectively. There were several SAGE tags with either low sequence complexity or matching a B2 repetitive element (data not shown) that made up the bulk of the remaining tag-to-UniGene cluster matches.
Manual analysis on a limited number of tag-UniGene cluster matches indicates that the ehm-tag-Mapping method produces a correct tag-to-UniGene cluster match ∼90% of the time (32 out of 35 were correct). Ambiguous tag matches were the result of minimal and/or low quality sequence data in the UniGene cluster and could all be identified by a lower Fit value (see next paragraph). This indicates that the limiting factor for this tag matching method, as well as others relying on EST data, is the quality of sequences submitted to public databases and the ability to group them correctly into different gene clusters.
Because the scope of manual tag confirmations was performed on a relatively small subset of tag-UniGene cluster matches, we developed a quantitative measure (denoted as a Fit value) that correlates with the accuracy of a given tag-UniGene cluster match (see Methods), and analyzed the distribution of Fit values for all tag-UniGene cluster matches produced by the ehm-tag-Mapping method. Fit values of 1 (the highest possible value) indicate that all other SAGE tags matching the UniGene cluster were each observed once. More than 80% of the Fit values for all tag-UniGene cluster matches were between 0.9 and 1 (Fig. 3), indicating that other tag matches present in the UniGene clusters, and subsequently removed from the final ehm-tag-Mapping file, were usually observed only once. Because this is the first tag-Mapping method to supply these quantitative measurements, it is not possible to compare these values across the different methods.
Figure 3.

Distribution of Fit values for all tag-UniGene cluster matches. Fit values were calculated as described in Equation 1 and represent the proportion of tag-UniGene cluster matches that come from the given tag sequence plus the count of all other tag matches in the given UniGene cluster. In our experience, Fit values >0.9 generally result from UniGene clusters with numerous EST sequencing errors and usually are indicative of only one SAGE tag matching a UniGene cluster.
Shared Gene Expression between Forelimbs and Hindlimbs
Most genes were similarly expressed between forelimbs and hindlimbs. In fact, 93.4% of the unique transcripts were twofold different or less (data not shown). We used a Venn analysis on the limb SAGE libraries to identify sets of genes expressed only in forelimbs, only in hindlimbs, and in both limbs (Fig. 4A). Of the tags observed in only one limb library, >99% had a frequency of three or less (Fig. 4B). Because it is difficult to assess gene expression accurately from this low-abundance class at this depth of sequencing, it is likely that many of these transcripts are present in both libraries but were not observed as such because of the random sampling properties of the SAGE method.
Figure 4.


Venn diagram representation of the forelimb and hindlimb SAGE libraries. This figure is not drawn to scale. (A) (Light gray circle) Number of unique tags observed in the forelimb; (black circle) number in the hindlimb; (overlapping fraction) number of unique tags observed in both SAGE libraries; (bold numbers) tags observed more than once; (numbers in parentheses) unique tags, including those observed once. (B) Fraction of unique tags (Y-axis) from the subset of 27,131 unique tags observed in only one SAGE library (light gray and black, not including the overlapping fraction), with a cumulative frequency indicated (X-axis). For example, 97% of the unique tags in only one library were observed two times or less.
We identified the corresponding SAGE tags, in both limb SAGE libraries, for a number of genes that regulate normal limb development (Table 3). Of the 21 Hox genes that had Tag-to-UniGene cluster matches, 10 were identified in the forelimb and/or hindlimb SAGE libraries. SAGE tags for several additional Hox genes were determined by sequencing the 3′ ends of the genes, or by database mining (see legend of Table 3). We also detected 12 fibroblast growth factors and related genes as well as 251 transcription factors that included 138 zinc finger, 23 homeodomain, 15 LIM, 4 POU, and 4 forkhead domain-containing proteins. Included in these numbers were UniGene clusters that also represent ESTs with homology with those classes of genes.
Table 3.
Hox Genes Identified in the Forelimb and Hindlimb SAGE Libraries
| Gene | SAGE tag | Tag count | |
|---|---|---|---|
| forelimb | hindlimb | ||
| Hoxa1 | TCTGTAATAA | 0 | 1 |
| Hoxa7 | AAGTGGAAGA | 1 | 1 |
| Hoxa9 | AAACTGCTCT | 5 | 2 |
| Hoxa10 | CATAAAAGGG | 13 | 10 |
| Hoxa11 | TGAAATAATA | 0 | 1 |
| Hoxa11, antisense1 | CAATTGAGGC | 9 | 7 |
| Hoxa11, antisense1 | CATCAGGGTA | 0 | 4 |
| Hoxa132 | GTGGATTAAC | 2 | 4 |
| Hoxb8 | CGCGCTGTGA | 0 | 1 |
| Hoxc9 | TACGGCTCGC | 0 | 2 |
| Hoxc103 | TAGCTTCCTT | 0 | 4 |
| CAAAGTTGAG | 0 | 5 | |
| Hoxc114 | TGCGTGAGTG | 0 | 1 |
| Hoxd105 | TTTCTGAAAA | 1 | 0 |
| Hoxd11 | AGTCACTGTC | 17 | 15 |
| Hoxd132 | GGCCTCTCAG | 6 | 3 |
Unless otherwise noted, all SAGE tags in this table were initially identified by the ehmtag-mapping method and subsequently verified manually.1There are numerous polyadenylated antisense transcripts produced from this locus (Hsieh-Li et al. 1995). Both of these SAGE tags are valid matches to different antisense transcripts.
The SAGE tags for these genes were identified by cloning and sequencing the 3′ ends of the genes.
Obtained from an analysis of GenBank sequence data (GI:51413). There are two different poly(A) signals that would generate the two different SAGE tags listed here, the second of which was generated from an ATTAA poly(A) signal and poly(A) rich region 1 kb upstream (at 5752 bp) of the reported putative poly(A) signal (Peterson et al. 1992).
Obtained from the sequence presented in Figure 1 of Hostikka and Capecchi (1998).
This SAGE tag was identified from a genomic clone (see Methods for details).
Identification of Differentially Expressed SAGE Tags
Detection of an Overall Difference
The extent of similarity between comprehensive gene expression profiles of serially homologous structures has not been reported previously. Monte Carlo simulations were used in a formal statistical analysis to determine if an overall difference existed between the forelimb and hindlimb SAGE libraries. These simulations showed that there was an overall statistically significant difference (data not shown) and, in this context, validated our efforts to search for specific pair-wise differences.
Detection of Known Differentially Expressed Genes
Several genes shown previously to be differentially expressed between forelimbs and hindlimbs, including Pitx1, Tbx4, Tbx5, and several Hox genes, were also differentially expressed between the limb SAGE libraries (Table 4). The identification of these genes provided a good positive control for the ability of this experimental system to detect differentially expressed genes. We have confirmed Pitx1, Tbx4, and Tbx5 differences independently by semi-quantitative RT-PCR and whole-mount in situ hybridizations (data not shown).
Table 4.
A List of Genes Known to be Differentially Expressed between Forelimbs and Hindlimbs
| Gene | SAGE tag | Tag count | |
|---|---|---|---|
| forelimb | hindlimb | ||
| Pitx1 | TACGTCTATT | 0 | 26 |
| Pitx1 (IP)1 | TCGCCGGGCG | 0 | 14 |
| Tbx42 | GACATTTTGT | 0 | 9 |
| Tbx52 | TTCCCCGATT | 3 | 0 |
| Hoxc9 | TACGGCTCGC | 0 | 2 |
| Hoxc103 | TAGCTTCCTT | 0 | 4 |
| CAAAGTTGAG | 0 | 5 | |
| Hoxc114 | TGCGTGAGTG | 0 | 1 |
| Hoxd95 | GGTTGGAAAA | 0 | 0 |
This SAGE tag appears to represent a cDNA generated from an internally primed poly(A) rich region of Pitx1. Further analysis showed that this SAGE tag also matched at the 11th base-pair relative to the Pitx1 sequence data (GI:1616804, CATG at 1256 bp).
This SAGE tags were identified by cloning and sequencing the 3′ ends of these genes (see Methods for details).
Obtained from an analysis of GenBank sequence data (GI:51413). There are two different poly(A) signals that would generate the two different SAGE Tags listed here, the second of which was generated from an ATTAAA poly(A) signal (Peterson et al. 1992).
Obtained from the sequence presented in Figure 1 of Hostikka and Capecchi (1998).
A tag with a 1 bp mismatch was observed 1 and 0 (GGTTGGAAGA) and may represent Hoxd9.
Statistical Analysis
Using the statistical test developed by Audic and Claverie (1997), Pitx1 and Tbx4 had statistically significant differentially expressed SAGE tags (P value = 1.5 × 10−8 and 0.002, respectively) and were the two most differentially expressed genes in the hindlimb. However, at the current depth of sequencing, Tbx5 and most of the Hox genes were not differentially expressed with statistical significance (P value > 0.05). Nevertheless, their limb-specific tag frequencies support what is known about the limb-specific expression of these genes. This statistical test was most useful for detecting gene expression differences in the higher-abundance classes, but it lacked the ability to accurately assess low-abundance SAGE tags (Man et al. 2000).
Only 70 SAGE tags were differentially expressed at a P value ≤ 0.01, and only 19 SAGE tags were differentially expressed at a P value ≤ 0.001 (see Compare table in the web supplement). These numbers are above what would be expected by chance alone, calculated as the P value × 3075 (the number of tests performed). Because the statistical test by Audic and Claverie (1997) was shown to have low power for low tag frequencies (Man et al. 2000), only SAGE tags in which the total tag count from both libraries was seven or higher were tested (3075).
Fold Differences Analysis
The three tags with the greatest fold difference in the hindlimb represent Pitx1 and Tbx4, genes known previously to have hindlimb-specific expression patterns. A total of 96 SAGE tags were at least fivefold different and represented only in the forelimb or hindlimb SAGE library (tag frequencies that were five or higher in one limb library and zero in the other limb library). Because the tag frequencies of other genes identified previously as differentially expressed (Tbx5 and several Hox genes) were low, and in this fold difference analysis tag counts of zero were treated as one, they appeared to be only twofold different. One of the limitations of using fold difference as a measure of differential expression is that at low abundance levels, it is impossible to discern between genes that are not expressed (“true” zero) and those that are at such low levels they simply have not been observed in the SAGE sample (a “nonzero” element). Therefore, the genes in this low range are all potential candidates for novel regulators of limb identity.
Comparison to Other Mouse SAGE Libraries
The majority of differentially expressed SAGE tags between forelimbs and hindlimbs represented transcripts expressed, on average, less than one copy per cell and could not be analyzed accurately by statistical measures or a fold difference analysis. Therefore, a “virtual” subtraction approach was used to define a subset of genes expressed exclusively in the limb. SAGE tags from six additional mouse SAGE libraries (Table 5) were pooled together for a total of 362,344 SAGE tags representing 81,424 unique transcripts from nonlimb SAGE libraries.
Table 5.
SAGE Libraries Represented in the Pooled, Non-Limb Library
| SAGE library | Total tags | Unique transcripts |
|---|---|---|
| Whole brains1 | 152,791 | 45,855 |
| T-cells2 | 183,599 | 44,331 |
| 3T3 cells3 | 25,954 | 10,217 |
| Total | 362,344 | 81,4244 |
The sum of three different SAGE libraries from whole brains of adult male, female, and Ts65Dn mice (Chrast et al. 2000).
The sum of two separate T-cell libraries (Shires et al., in prep).
Prepared by V.E. Velculescu and available at http://www.sagenet.org.
The total number of unique transcripts represents the union of the six pooled libraries.
The pooled, nonlimb SAGE library was compared with our forelimb and hindlimb SAGE libraries (Fig. 5). Of the unique transcripts in the limb SAGE libraries, 50.3% were not present in the pooled, nonlimb library. Furthermore, only 26% (810) of the SAGE tags with frequencies of two or higher in one limb library and zero in the other limb library were not present in the pooled, nonlimb library (cf. bold numbers in light gray and black, Fig. 5, with Fig. 4). A data table of these limb-specific, differentially expressed SAGE tags that matched a UniGene cluster with the ehm-tag-Mapping method is provided as a web supplement to this article (see the “LimbSpecificKnownDiffExp” table in the Access 97 database).
Figure 5.

Venn diagram comparing the forelimb and hindlimb SAGE libraries with other libraries listed in Table 5. This figure is not drawn to scale. (Light gray circles) Forelimb SAGE library; (black circle) hindlimb SAGE library (as in Fig. 4); (white circle) pooled, nonlimb SAGE tags from six additional SAGE libraries; (bold numbers) tags observed more than once (tags represented in more than one library are inherently observed more than once); (numbers in parentheses) unique tags, including those observed once. For example, 407 tags were observed two times or more in the forelimb and not observed in either the hindlimb or pooled, nonlimb SAGE libraries.
Finally, of the 251 identified transcription factors and homologous ESTs, only 43 representative tags were specific to the limb libraries (Table 6). This latter candidate subset of genes may be valuable for identifying the genetic basis of inherited limb malformation syndromes as well as novel regulators of limb identity.
Table 6.
Limb-Specific Transcription Factors and Homologous ESTs
| SAGE tag | Tag count | Copies per cell1 | Percent abund. | UniGene ID | Description | Tag matching values2 | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| fore | hind | sum | Num | Sum | Count | Fit | |||||
| General transcription factors | |||||||||||
| CTCTTATTTT | 2 | 0 | 2 | 0.55 | 0.0015 | 38850 | pre B-cell leukemia transcription factor 1 | 2 | 4 | 3 | 1.00 |
| CTGATTTATT | 0 | 2 | 2 | 0.55 | 0.0015 | 7103 | pre B-cell leukemia transcription factor 2 | 5 | 23 | 8 | 0.52 |
| CCAAGAAACA | 1 | 0 | 1 | 0.27 | 0.00073 | 4563 | metal response element binding transcription factor 1 | 2 | 5 | 3 | 0.80 |
| CATAATAAAT | 0 | 1 | 1 | 0.27 | 0.00073 | 8012 | Mus musculus Ets transcriptions factor Spi-B, partial cds | 4 | 4 | 1 | 1.00 |
| GACAAGATGT | 0 | 1 | 1 | 0.27 | 0.00073 | 4795 | transcription factor AP-2 beta | 3 | 9 | 7 | 1.00 |
| ACGCCCCGGCT | 1 | 0 | 1 | 0.27 | 0.00073 | 41124 | ESTs, highly similar to transcription factor TFE3 (Homo sapiens) | 1 | 1 | 1 | 1.00 |
| TCTCTCATTT | 1 | 0 | 1 | 0.27 | 0.00073 | 25856 | ESTs, moderately similar to AF133123_1 transcription factor IIIC102 (H. sapiens) | 3 | 11 | 9 | 1.00 |
| TTTCAGTGGG | 0 | 1 | 1 | 0.27 | 0.00073 | 46503 | M. musculus ets family transcription factor ELF2B1 (ELF2) mRNA, complete cds, alternatively spliced. | 7 | 20 | 8 | 0.70 |
| AACAAGAAAG | 1 | 0 | 1 | 0.27 | 0.00073 | 897 | POU domain, class 2, associating factor 1 | 10 | 18 | 7 | 0.89 |
| Homeodomain containing | |||||||||||
| TACGTCTATT | 0 | 26 | 26 | 7.1 | 0.019 | 4832 | paired-like homeodomain transcription factor 1 | 2 | 3 | 2 | 1.00 |
| CAGAACAAGT | 7 | 7 | 14 | 3.8 | 0.010 | 4346 | ESTs, highly similar to MOX2_MOUSE HOMEOBOX PROTEIN MOX-2 (M. musculus) | 16 | 23 | 7 | 0.96 |
| TCGCCGGGCG | 0 | 14 | 14 | 3.8 | 0.010 | 4832 | paired-like homeodomain transcription factor 1 | 1 | 3 | 2 | 0.67 |
| TGAATGTTCG | 2 | 0 | 2 | 0.55 | 0.0015 | 5039 | sine oculis-related homeobox 2 homolog (Drosophila) | 1 | 2 | 2 | 1.00 |
| TTATACAGAA | 2 | 0 | 2 | 0.55 | 0.0015 | 1385 | paired-like homeodomain transcription factor 2 | 4 | 4 | 1 | 1.00 |
| TCTGTGTATG | 1 | 0 | 1 | 0.27 | 0.00073 | 39039 | iroquois related homeobox 3 (Drosophila) | 19 | 19 | 1 | 1.00 |
| TACAGGACTT | 1 | 0 | 1 | 0.27 | 0.00073 | 5194 | distal-less homeobox 3 | 1 | 1 | 1 | 1.00 |
| TATCAGTTTT | 0 | 1 | 1 | 0.27 | 0.00073 | 3404 | mesenchyme homeobox 1 | 3 | 3 | 1 | 1.00 |
| TATCAGTTTT | 0 | 1 | 1 | 0.27 | 0.00073 | 141812 | ESTs, highly similar to MOX1_MOUSE HOMEOBOX PROTEIN MOX-1 (M. musculus) | 24 | 29 | 5 | 0.97 |
| TTGGCTTCTC | 0 | 1 | 1 | 0.27 | 0.00073 | 147618 | EST, moderately similar to HEX1_MOUSE ANTERIOR-RESTRICTED HOMEOBOX PROTEIN (M. musculus) | 1 | 1 | 1 | 1.00 |
| ATGGATAGAC | 1 | 0 | 1 | 0.27 | 0.00073 | 153716 | mesenchyme homeobox 2 | 1 | 1 | 1 | 1.00 |
| Zinc finger containing | |||||||||||
| CGATCAGCAA | 4 | 0 | 4 | 1.1 | 0.0029 | 23452 | M. musculus early B-cell factor associated zinc finger transcription factor (Ebfaz) mRNA, complete cds | 3 | 10 | 8 | 1.00 |
| CACTATCTCC | 0 | 2 | 2 | 0.55 | 0.0015 | 29525 | ESTs, weakly similar to T33754 O/E-1-associated zinc finger protein Roaz-rat (Rattus norvegicus) | 9 | 28 | 8 | 0.57 |
| GTGGGATGGA | 1 | 1 | 2 | 0.55 | 0.0015 | 129369 | ESTs, weakly cimilar to I48208 zinc finger protein 30-mouse (M. musculus) | 4 | 5 | 2 | 1.00 |
| AAATAAATCT | 0 | 2 | 2 | 0.55 | 0.0015 | 31016 | ESTs, weakly similar to Z33A_HUMAN ZINC FINGER PROTEIN 33A (H. sapiens) | 5 | 5 | 1 | 1.00 |
| CCCATAAAAG | 1 | 0 | 1 | 0.27 | 0.00073 | 30405 | Zinc finger protein 94 | 5 | 12 | 6 | 0.83 |
| AAAGTACACA | 1 | 0 | 1 | 0.27 | 0.00073 | 102799 | ESTs, weakly similar to zinc finger protein (M. musculus) | 1 | 1 | 1 | 1.00 |
| TTTAAGTTAA | 1 | 0 | 1 | 0.27 | 0.00073 | 21914 | ESTs, highly similar to ZINC FINGER PROTEIN 91 (H. sapiens) | 6 | 26 | 13 | 0.69 |
| GTACGTTTCT | 1 | 0 | 1 | 0.27 | 0.00073 | 155690 | M. musculus zinc finger protein Sp5 mRNA, complete cds | 2 | 2 | 1 | 1.00 |
| GTCCCTCTCT | 1 | 0 | 1 | 0.27 | 0.00073 | 41364 | ESTs, weakly similar to zinc finger protein s11-6 (M. musculus) | 7 | 13 | 4 | 0.77 |
| GTTCATACAA | 1 | 0 | 1 | 0.27 | 0.00073 | 132523 | ESTs, weakly similar to ZF90_MOUSE ZINC FINGER PROTEIN 90 (M. musculus) | 1 | 3 | 2 | 0.67 |
| AGACATCACA | 0 | 1 | 1 | 0.27 | 0.00073 | 20430 | zinc finger protein 105 | 5 | 7 | 3 | 1.00 |
| AGTCACCAAT | 0 | 1 | 1 | 0.27 | 0.00073 | 2927 | zinc finger protein 30 | 2 | 5 | 4 | 1.00 |
| CTTTCTGTTC | 0 | 1 | 1 | 0.27 | 0.00073 | 30405 | zinc finger protein 94 | 3 | 12 | 6 | 0.67 |
| CATTCCACAT | 0 | 1 | 1 | 0.27 | 0.00073 | 12891 | zinc finger protein 326 | 3 | 6 | 4 | 1.00 |
| GCGCACTCAG | 1 | 0 | 1 | 0.27 | 0.00073 | 24775 | ESTs, weakly similar to zinc finger protein 64 (M. musculus) | 4 | 8 | 5 | 1.00 |
| CTCACAAATC | 0 | 1 | 1 | 0.27 | 0.00073 | 117547 | ESTs, highly similar to zinc finger transcription factor REST protein (R. norvegicus) | 1 | 1 | 1 | 1.00 |
| AGAAGGTGGT | 0 | 1 | 1 | 0.27 | 0.00073 | 12930 | BcL6-associated zinc finger protein | 2 | 2 | 1 | 1.00 |
| LIM containing | |||||||||||
| GCCTGTGTAA | 10 | 13 | 23 | 6.3 | 0.017 | 21830 | reversion induced LIM gene | 28 | 34 | 4 | 0.91 |
| CACTTTTGCA | 2 | 4 | 6 | 1.6 | 0.0044 | 79380 | LIM homeobox protein 9 | 2 | 3 | 2 | 1.00 |
| TAAAGACACA | 1 | 1 | 2 | 0.55 | 0.0015 | 25785 | LIM domain binding 3 | 11 | 25 | 8 | 0.72 |
| GGATTTTACT | 0 | 1 | 1 | 0.27 | 0.00073 | 42242 | ISL1 transcription factor, LIM/homeodomain, (islet-1) | 3 | 6 | 2 | 0.67 |
| TATAGAGGAA | 0 | 1 | 1 | 0.27 | 0.00073 | 25785 | LIM domain binding 3 | 6 | 25 | 8 | 0.52 |
| ATCTCAGAAA | 0 | 1 | 1 | 0.27 | 0.00073 | 70551 | ESTs, weakly similar to LIM homeobox protein 9 (M. musculus) | 5 | 6 | 2 | 1.00 |
Of the 264 FGFs, transcription factors, related genes, and homologous ESTs identified in our limb SAGE libraries with the ehm-tag-mapping method, only 43 tags, listed here, were specific to the limb sibraries after a virtual subtraction of the non-limb pooled SAGE library.
Copies per cell was calculated assuming, on average, 500,000 transcripts in a cell (Hastie and Bishop 1976).
See Methods for a description of these values.
DISCUSSION
We performed a comprehensive analysis of gene expression between intact developing vertebrate forelimbs and hindlimbs. SAGE profiles were obtained and analyzed using a novel approach for extracting SAGE tags from high-quality, phred-analyzed sequence data. In addition, the ehm-tag-Mapping method was developed to provide additional information about the confidence of the tag-UniGene cluster match. Finally, by a direct comparison of the two limb SAGE libraries, as well as a comparison to additional, nonlimb SAGE libraries, we created a candidate set of differentially expressed genes between forelimbs and hindlimbs.
Determination of Tag Sequencing Error Rates
By analyzing most of our sequence data with phred and using eSAGE to prevent the extraction of SAGE tags with low-quality base calls, we were able to determine that ∼1 in 630 SAGE tags had sequencing errors. Even though 66% of the SAGE tags were observed once, factoring in this low tag sequencing error rate decreases the total number of unique transcripts by only 38. These high-quality mouse SAGE libraries are an ideal resource for identifying and mapping genes to finished genomic DNA sequence.
This error estimate does not account for mutations that may have occurred from polymerase errors in the numerous enzymatic and cloning steps of the SAGE protocol. The use of high-fidelity polymerases in future SAGE experiments could minimize these errors. Nevertheless, doubling our error estimate, a conservative approach considering the average polymerase error rate (Cha and Thilly 1993) and the amount of sequence data we generated, still results in a relatively low tag sequencing error rate, which would correspond to ∼440 out of 136,856 tags arising because of sequencing errors or cloning mutations.
Why So Many Unique Transcripts in a Limb?
Unique transcripts totaling 36,300 were identified from a very narrow time frame of the developing mouse limb. In addition, we were identifying new tags at a rate of 16% toward the end of our tag sequencing effort (Fig. 2), indicating that we had not yet sampled all of the unique, rare transcripts in the limb. This may partially reflect the fact that our in vivo limb bud population is a heterogenous population of cells. Nevertheless, maintaining the spatiotemporal relationships of these cells has the advantage of taking an accurate snapshot of gene expression in an in vivo context.
Including the six additional nonlimb SAGE libraries (Table 5), a total of 99,473 unique transcripts was identified in these mouse tissues. This number is three times the current gene estimate of ∼30,000 genes from cross-species comparisons (Crollius et al. 2000), EST database information (Ewing and Green 2000), or analyses of the draft human genome sequences (International Human Genome Sequencing Consortium 2001; Venter et al. 2001). This is remarkable, considering the relatively small proportion of tissue types these SAGE libraries represent.
This discrepancy has been observed before and arises for several reasons. First, current gene estimates count variant forms of the same transcript as one gene. Because many genes have more than one unique transcript, arising from multiple polyadenylation sites or alternative splicing events, there may be multiple, authentic SAGE tags representing the same “gene.” Second, there could be an inflated unique tag count because of sequencing or cloning errors. At least for our SAGE libraries, we know this is not the case. Third, it is possible that current gene estimates are too low. Indeed, there are reports of gene estimates in the 100,000 to 150,000 range (Liang et al. 2000), which may be more consistent with the SAGE data. It is also interesting to note that SAGE tags were used to identify novel open reading frames in yeast studies, after the complete yeast genome was sequenced (Velculescu et al. 1997). Fourth, there could be spurious transcription from nonspecific sites, creating nonfunctional transcripts at a basal level. Finally, gene estimates from the analysis of genome sequence data do not include the numerous nonprotein-encoding, polyadenylated transcripts (see http://www.ensembl.org/Genesweep for the definition of a “gene”) that are likely included in the count of unique transcripts for SAGE libraries. cDNA sequencing efforts and other experimental annotation strategies capable of mapping SAGE tags to specific gene regions may help to resolve this issue.
Known Differentially Expressed Genes are Detected
By comparing the forelimb and hindlimb SAGE libraries, we were able to detect the differential expression of the three currently known major regulators of limb identity: Pitx1, Tbx4, and Tbx5. Furthermore, we also detected the differential expression of several Hox genes identified previously by whole-mount in situ hybridization experiments as being differentially expressed in chick and mouse. Because Tbx5 and many of the Hox genes were expressed at low levels in our SAGE libraries, their differential expression was not statistically significant at this level of sequencing. Nevertheless, the appropriate detection of previously known differentially expressed genes has validated this approach to detect novel, in vivo differences.
Pitx1 was the most abundant limb-specific transcription factor in our combined limb SAGE libraries and the most differentially expressed SAGE tag. We did not expect the other known differentially expressed genes to be in the low-abundance classes. Nevertheless, the fact that Tbx5 and the limb-specific Hox genes are in low-abundance classes supports our hypothesis that other SAGE tags at this expression level are valid candidates for novel regulators of limb identity and morphology.
A Candidate Set of Differentially Expressed Genes
Our forelimb and hindlimb SAGE libraries were very similar: 93.4% of the genes were twofold different or less, and 99.8% of the SAGE tags were not statistically different at a significance level of 1%. However, statistical tests and fold difference calculations cannot accurately assess the low-abundance transcripts. In these cases, the virtual subtraction approach may be better suited. Resources would be better spent verifying this candidate subset of differentially expressed genes with other methods, rather than to sequence an additional one million SAGE tags to identify tags that will become statistically significant.
Pitx1, Tbx4, and Tbx5 are expressed throughout the entire limb structure during a time that starts before and continues after the specific stage of limb development assayed here. Nevertheless, our experiment may not have detected potential upstream regulators in the Pitx1-Tbx hierarchy that are no longer expressed at this stage of development. This system may also have difficulties detecting genes expressed in a particular subset of limb cells such that their representation in the entire population is too low to be observed at this depth of sequencing. With the improvement of methods to perform SAGE on 1000-fold less RNA (Datson et al. 1999; Peters et al. 1999; Virlon et al. 1999), it will be possible to investigate gene expression at earlier time points of limb development, potentially identifying genes upstream of the Pitx1-Tbx hierarchy.
Current studies in our laboratory are focused on confirming the differential expression of these candidate genes by other methods. The results of these studies will be reported separately. We have also been developing novel methods (that rely on other molecular sequence databases instead of UniGene) for identifying SAGE tags that currently have no match to a UniGene cluster.
One of the advantages of SAGE is that our data will become more informative with time. As methods to match unidentified SAGE tags become more reliable, and genome sequencing efforts are completed, the number of SAGE tags matching genes will continually increase. Furthermore, as the number of SAGE libraries available for comparative analyses keeps growing, we will be able to further refine our limb-specific set of genes.
Candidates for Human Limb Malformation Syndromes
There are numerous inherited limb malformation syndromes, with or without additional organ system involvement, for which the genetic basis has not been identified. A more sophisticated approach for identifying candidate genes can be envisioned that uses the limb-specific set of genes presented here, the rapidly expanding mouse genomic sequence data, and methods to map expressed genes to physical chromosomal locations (Caron et al. 2001). Further refinement of a candidate gene set could come from the ability to rapidly compare additional, independently generated SAGE libraries (representing an expanding variety of tissue sources) with the phenotypic knowledge of a particular syndrome.
Finding correlations between the tissue expression distribution of specific genes and affected organ systems, combined with linkage data and chromosomal mapping information, will be a powerful integrated approach for the study of inherited diseases. Furthermore, understanding the complex regulation that results in the divergence of a single structure within the same organism may provide new insights into the evolutionary complexity observed among species (Capdevila and Izpisúa-Belmonte 2000; Ruvinsky and Gibson-Brown 2000).
METHODS
Tissue Collection and RNA Isolation
Embryonic day 11.5 embryos were harvested from timed-pregnant B6C3Fe mice (Jackson Labs) maintained in a temperature-regulated room on a 12-h light/dark cycle. Noon on the day at which vaginal plugs were detected was designated embryonic day 0.5. Embryos were dissected in ice-cold, RNase-free PBS, and limb buds were harvested under a dissecting microscope and staged according to Wanek et al. (1989). To minimize differences in gene expression due to developmental timing, only stage 4–5 hindlimbs and stage 5–6 forelimbs were collected for RNA isolation.
Limb buds were placed immediately in Trizol as they were harvested from the embryo. Total RNA was purified from forelimbs and hindlimbs separately and stored under 70% ethanol. Approximately 1.6 μg of total RNA was obtained from each limb bud. Poly(A) mRNA was purified from pooled total RNA pellets using the OligoTex Midi kit (Qiagen).
SAGE Library Synthesis
SAGE libraries were constructed following the SAGE protocol v1.0c or e (available at http://www.sagenet.org) essentially as described (Velculescu et al. 1995) using 4 μg of poly(A) mRNA. DiTag amplifications were performed with 96 × 50 μL PCRs, each containing 1 μL of a 1 : 200 dilution of the DiTag ligation. The reaction buffer and primer concentrations recommended in the SAGE protocol were used with 5 U of Taq polymerase (Engelke et al. 1990) per reaction. The DiTag ligation was amplified with 26 cycles of 30 sec (95°), 30 sec (55°), and 1 min (72°), followed by 5 min (72°) in an MJ Research 96-well thermocycler using a heated lid. To increase the stability of a solution containing free DiTags, the purified NlaIII digest of the amplified linker-DiTags was resuspended in TE supplemented with 50 mM NaCl (Michiels et al. 1999) before loading on a 12% polyacrylamide gel resolved in 2× TAE. DiTag concatemers were cut with SphI and purified twice from an 8% polyacrylamide gel before cloning them into SphI-digested pZERO-1 (Invitrogen Corp.).
SAGE Tag Sequencing
Bacterial colonies harboring plasmid inserts with DiTag concatemers were picked with toothpicks and dissolved in 50 μL of 1× GIBCO BRL PCR buffer supplemented with 1.5 mM MgCl2, 200 μM dNTPs (Amersham), 0.8 ng/μL forward (5′-TGTGCTGCAAGGCGATTAAGTTGG-3′) and reverse (5′-CCAGGCTTTACACTTTATGCTTCC-3′) primers that flank the cloning site (Bies et al. 1992), and 2.5 U of Taq polymerase. Plasmid templates were released from the bacteria by an initial incubation for 2 min at 94° and were then amplified with 30 cycles of 30 sec (94°), 30 sec (60°), and 1.5 min (72°) followed by a final extension for 5 min at 72°. Amplified PCR products were purified in 96-well format using the Qiagen PCR cleanup kit, the Millipore MultiScreenFB plates, or the Millipore MultiScreen-PCR plates and eluted with 10 mM Tris (pH 8.0). Templates were sequenced with the T7 primer using either dye-primer cycle sequencing on a Pharmacia ALFexpress system or with big-dye terminators on an ABI PRISM 3700 DNA Analyzer.
SAGE Data Acquisition
eSAGE v1.10c (Margulies and Innis 2000b) was used to extract and analyze the SAGE data. To assure SAGE tags were extracted only from high-quality sequences, data from the ALFexpress were edited manually with ALFwin v2.10 (AP-Biotech) to exclude low-quality regions. Sequence trace files generated on the ABI PRISM 3700 DNA Analyzer were analyzed with the phred base-calling algorithm (Ewing et al. 1998). PHD-formatted output files (*.phd.1) generated from phred-analyzed sequence trace data were read by eSAGE, which was programmed to automatically exclude sequences with phred quality values <20. Average phred scores were calculated with several Perl scripts kindly provided by R.H. Lyons (University of Michigan, Ann Arbor).
SAGE Tag Matching
We developed an algorithm to reliably match SAGE tags to UniGene clusters that is similar to that developed by Lash et al. (2000) and also incorporates ideas presented by Caron et al. (2001). The ehm-tag-Mapping method is implemented through the use of several Perl scripts designed to extract tag-to-UniGene cluster information from the UniGene flatfiles available at NCBI. Briefly, the ehm-tag-Mapping method extracts a SAGE tag from each sequence in a UniGene cluster, only if the orientation and 3′ end of the sequence can be confirmed by identifying poly(A) signals and/or tails. To minimize the extraction of SAGE tags from entries with potential sequencing errors, SAGE tags not representing at least 20% of all tags extracted from a given UniGene cluster are removed from the final ehm-tag-Mapping flatfile.
In addition to the SAGE tag sequence, UniGene ID number, and description, three additional parameters are reported for a SAGE tag that provide additional criteria on which to base the confidence of the tag-to-UniGene cluster match: (1) the number of times this particular SAGE tag was extracted from sequence files in the UniGene cluster (Num), (2) the total number of times any SAGE tag was extracted from sequence files in the UniGene cluster (Sum), and (3) the number of unique SAGE tag sequences extracted from the UniGene cluster (Count). As stated in the last paragraph, Num/Sum must be ≥0.2 for a SAGE tag to be listed in the ehm-tag-Mapping file. For clarity, the ehm-tag-Mapping log file output for UniGene clusters Mm.1 and Mm.5 (the first two clusters in the UniGene database) are reported in Figure 6. As an example, the Num, Sum, and Count values for UniGene cluster Mm.1 are 59, 77, and 18, respectively.
Figure 6.

Representative output from the ehm-tag-Mapping log file for the first two analyzed UniGene clusters. The first entry of the log file has been annotated for explanation purposes after the ###.
To keep the false-positive rate of this method to a minimum, the algorithm was specifically designed to prevent the extraction of SAGE tags from sequence entries for which there was no obvious poly(A) signal and/or tail. Because (1) poly(A) tails are sometimes removed from sequences before they are added to the public databases, and (2) a small proportion of genes will contain noncanonical poly(A) signals, there may be a number of “true” positive Tag-to-UniGene cluster matches that are missed by this method.
A fourth Fit value (Equation 1) was calculated to summarize the Num, Sum, and Count values into a single informative number that is correlated with the quality of the tag-UniGene cluster match.
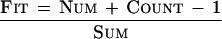 |
1 |
The Fit value represents the proportion of tag-UniGene cluster matches that come from the given tag sequence plus the Count of all other tag matches in a given UniGene cluster. Therefore, Fit values of 1 indicate that all other tags matching the UniGene cluster were observed once, and are therefore likely due to sequencing errors. Values <1 indicate an increasing proportion of other tags matching the UniGene cluster. As a representative example, the Fit values for UniGene clusters Mm.1 and Mm.5 are 0.99 and 1, respectively. In our experience, Fit values >0.9 generally result from UniGene clusters with numerous EST sequencing errors and usually are indicative of only one SAGE tag matching a UniGene cluster. Values <0.8 may indicate that an additional SAGE tag correctly matches the UniGene cluster.
The method presented by Caron et al. (2001) is more complex. Among other additions, they adjust for multiple types of sequencing errors, which results in matching a slightly higher number of experimental SAGE tags to UniGene clusters. However, their algorithms are not easily portable (because of several complex interactions with multiple in-house databases and programming modules), and a tag-to-gene matching file is presently available for human data only. As with their method, ours also significantly reduces the false-positive rate relative to those of earlier reliable tag Mapping builds available at NCBI (http://ncbi.nlm.nih.gov/SAGE).
A current ehm-tag-Mapping file was generated in ∼8 h on an 800 MHz PIII PC, running RedHat Linux 6.2. Because of the versatile portability of Perl and relatively stable format of the UniGene databases, in theory, these scripts can be implemented on a variety of platforms and for all species of UniGene clusters, which currently include human, mouse, rat, zebrafish, and cow. All Perl scripts used to implement the ehm-tag-Mapping method are available on request.
To summarize our SAGE data, we have found it useful to classify SAGE tags as representing known genes, ESTs, or no match. However, the algorithm used to build UniGene clusters sometimes assigns the sequences of one gene to multiple UniGene clusters, resulting in the same SAGE tag matching multiple UniGene clusters. To adjust for this, an extended set of Perl scripts and Access 97 (Microsoft Corp.) queries was designed to analyze the description line of each UniGene cluster and classify each SAGE tag as matching a gene or EST. SAGE tags matching both a gene and an EST were classified as matching only a gene.
Determination of Unknown SAGE Tags for Selected Genes
To validate the ability of this experimental system to detect molecules important in the establishment of limb identity and morphology, we determined the sequence of several SAGE tags corresponding to genes, notably Tbx4, Tbx5, Hoxa13, and Hoxd13, which could not be determined with available sequence data in the genomic databases. To identify these SAGE tags and determine their corresponding abundance in our SAGE libraries, the 3′ ends of these genes were cloned and sequenced.
For Tbx4 and Tbx5, we sequenced whole-mount in situ hybridization clones (kindly provided by V.E. Papaioannou, Columbia University, NY) and used this information to design gene-specific primers for 3′ RACE experiments (Frohman et al. 1988). 3′ RACE was performed with the GIBCO BRL 3′ RACE kit following the manufacturer’s protocol. First-strand cDNA was generated from limb bud total RNA isolated from embryonic day 11.5 mouse embryos. Cloned RACE products were found to include the 3′ ends for both genes. Interestingly, Tbx4 contains a noncanonical poly(A) signal, GATAAA (Beaudoing et al. 2000).
For Hoxd13, the sequence of a clone we have used to prepare whole-mount in situ hybridization probes (kindly provided by D. Duboule, University of Geneva) was obtained by several rounds of primer walking and found to include the 3′ end of the transcript. Finally, for Hoxa13, a genomic fragment extending beyond the 3′ end of the coding region was cloned and sequenced.
Test for an Overall Difference between Two SAGE Populations
Before determining if specific pair-wise differences exist between the two populations of SAGE tags, a formal statistical test was used to validate an overall difference between forelimbs and hindlimbs. We also used this overall test to determine the reproducibility of subpopulations of data from a single SAGE library (Margulies et al. 2001). Because the χ2 test of independence performs poorly when tag counts are less than five (Sokal and Rohlf 1995), a Monte Carlo simulation approach was used to determine the overall difference between two populations of SAGE data.
In this approach, data sets were generated randomly, keeping the row and column totals of the observed data set fixed. χ2 values were then calculated from each randomly generated data set. This process was repeated 200 times to obtain a distribution of χ2 values under the null hypothesis of no difference between two populations of SAGE data. An empirical P value was calculated by comparing the observed χ2 value with the values generated under the null hypothesis. A program was written in S-Plus 2000 (Insightful Corp.) to perform this Monte Carlo simulation and is available on request.
Acknowledgments
We thank D.T. Burke, M.R. Hughes, and M.D. Hagen for assistance with the large-scale sequencing efforts, D.P. Mortlock for cloning and sequencing the 3′ end of Hoxa13, H.V. Jagadish for helpful discussions regarding database design for improved tag-gene matching, M.E. Williams for sequencing the Hoxd13 whole-mount in situ hybridization clone and, along with T.M. Williams and B.E. Koester, assistance with preparing SAGE concatemers for sequencing. We also thank J.L. Curtis, M.W. Glynn, and J.V. Moran for helpful suggestions with the manuscript. E.H.M. is supported by the Institutional Training Program in Genomic Science (T32 HG00040). This work was supported by an NIH grant (HD34059) and by a University of Michigan Bioinformatics Program pilot grant.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL innis@umieh.edu; FAX (734) 763-3784.
Article published on-line before print: Genome Res., 10.1101/gr.192601.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.192601.
REFERENCES
- Audic S, Claverie JM. The significance of digital gene expression profiles. Genome Res. 1997;7:986–995. doi: 10.1101/gr.7.10.986. [DOI] [PubMed] [Google Scholar]
- Basson CT, Bachinsky DR, Lin RC, Levi T, Elkins JA, Soults J, Grayzel D, Kroumpouzou E, Traill TA, Leblanc-Straceski J, et al. Mutations in human TBX5 cause limb and cardiac malformation in Holt-Oram syndrome. Nat Genet. 1997;15:30–35. doi: 10.1038/ng0197-30. [DOI] [PubMed] [Google Scholar]
- Beaudoing E, Freier S, Wyatt JR, Claverie JM, Gautheret D. Patterns of variant polyadenylation signal usage in human genes. Genome Res. 2000;10:1001–1010. doi: 10.1101/gr.10.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bies RD, Phelps SF, Cortez MD, Roberts R, Caskey CT, Chamberlain JS. Human and murine dystrophin mRNA transcripts are differentially expressed during skeletal muscle, heart, and brain development. Nucleic Acids Res. 1992;20:1725–1731. doi: 10.1093/nar/20.7.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capdevila J, Izpisúa-Belmonte JC. Perspectives on the evolutionary origin of tetrapod limbs. J Exp Zool. 2000;288:287–303. doi: 10.1002/1097-010X(20001215)288:4<287::AID-JEZ2>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Caron H, van Schaik B, van der Mee M, Baas F, Riggins G, van Sluis P, Hermus MC, van Asperen R, Boon K, Voute PA, et al. The human transcriptome map: Clustering of highly expressed genes in chromosomal domains. Science. 2001;291:1289–1292. doi: 10.1126/science.1056794. [DOI] [PubMed] [Google Scholar]
- Cha RS, Thilly WG. Specificity, efficiency, and fidelity of PCR. PCR Meth Applic. 1993;3:S18–S29. doi: 10.1101/gr.3.3.s18. [DOI] [PubMed] [Google Scholar]
- Chrast R, Scott HS, Papasavvas MP, Rossier C, Antonarakis ES, Barras C, Davisson MT, Schmidt C, Estivill X, Dierssen M, et al. The mouse brain transcriptome by SAGE: Differences in gene expression between P30 brains of the partial trisomy 16 mouse model of down syndrome (Ts65Dn) and normals. Genome Res. 2000;10:2006–2021. doi: 10.1101/gr.10.12.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn MJ, Tickle C. Limbs: A model for pattern formation within the vertebrate body plan. Trends Genet. 1996;12:253–257. doi: 10.1016/0168-9525(96)10030-5. [DOI] [PubMed] [Google Scholar]
- Cohn MJ, Patel K, Krumlauf R, Wilkinson DG, Clarke JDW, Tickle C. Hox9 genes and vertebrate limb specification. Nature. 1997;387:97–101. doi: 10.1038/387097a0. [DOI] [PubMed] [Google Scholar]
- Crollius HR, Jaillon O, Bernot A, Dasilva C, Bouneau L, Fischer C, Fizames C, Wincker P, Brottier P, Qu'etier F, et al. Estimate of human gene number provided by genome-wide analysis using Tetraodon nigroviridis DNA sequence. Nat Genet. 2000;25:235–238. doi: 10.1038/76118. [DOI] [PubMed] [Google Scholar]
- Datson NA, van der Perk-de Jong J, van den Berg MP, de Kloet ER, Vreugden-hil E. MicroSAGE: A modified procedure for serial analysis of gene expression in limited amounts of tissue. Nucleic Acids Res. 1999;27:1300–1307. doi: 10.1093/nar/27.5.1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelke DR, Krikos A, Bruck ME, Ginsburg D. Purification of Thermus aquaticus DNA polymerase expressed in Escherichia coli. Analyt Biochem. 1990;191:396–400. doi: 10.1016/0003-2697(90)90238-5. [DOI] [PubMed] [Google Scholar]
- Ewing B, Green P. Base-calling of automated sequencer traces using Phred. II. Error probabilities. Genome Res. 1998;8:186–194. [PubMed] [Google Scholar]
- ————— Analysis of expressed sequence tags indicates 35,000 human genes. Nat Genet. 2000;25:232–234. doi: 10.1038/76115. [DOI] [PubMed] [Google Scholar]
- Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using Phred. I. Accuracy assessment. Genome Res. 1998;8:175–185. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- Frohman MA, Dush MK, Martin GR. Rapid production of full-length cDNAs from rare transcripts: Amplification using a single gene-specific oligonucleotide primer. Proc Natl Acad Sci. 1988;85:8998–9002. doi: 10.1073/pnas.85.23.8998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson-Brown JJ, Agulnik SI, Chapman DL, Alexiou M, Garvey N, Silver LM, Papaioannou VE. Evidence of a role for T-box genes in the evolution of limb morphogenesis and the specification of forelimb/hindlimb identity. Mech Dev. 1996;56:93–101. doi: 10.1016/0925-4773(96)00514-x. [DOI] [PubMed] [Google Scholar]
- Gibson-Brown JJ, Agulnik SI, Silver LM, Niswander L, Papaioannou VE. Involvement of T-box genes Tbx2-Tbx5 in vertebrate limb specification and development. Development. 1998;125:2499–2509. doi: 10.1242/dev.125.13.2499. [DOI] [PubMed] [Google Scholar]
- Hastie ND, Bishop JO. The expression of three abundance classes of messenger RNA in mouse tissues. Cell. 1976;9:761–774. doi: 10.1016/0092-8674(76)90139-2. [DOI] [PubMed] [Google Scholar]
- Hostikka SL, Capecchi MR. The mouse Hoxc11 gene: Genomic structure and expression pattern. Mech Dev. 1998;70:133–145. doi: 10.1016/s0925-4773(97)00182-2. [DOI] [PubMed] [Google Scholar]
- Hsieh-Li HM, Witte DP, Weinstein M, Branford W, Li H, Small K, Potter SS. Hoxa11 structure, extensive antisense transcription, and function in male and female fertility. Development. 1995;121:1373–1385. doi: 10.1242/dev.121.5.1373. [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Isaac A, Rodriguez-Esteban C, Ryan A, Altabef M, Tsukui T, Patel K, Tickle C, Izpisúa-Belmonte JC. Tbx genes and limb identity in chick embryo development. Development. 1998;125:1867–1875. doi: 10.1242/dev.125.10.1867. [DOI] [PubMed] [Google Scholar]
- Johnson RL, Tabin CJ. Molecular models for vertebrate limb development. Cell. 1997;90:979–990. doi: 10.1016/s0092-8674(00)80364-5. [DOI] [PubMed] [Google Scholar]
- Lanctôt C, Moreau A, Chamberland M, Tremblay ML, Drouin J. Hindlimb patterning and mandible development require the Pitx1 gene. Development. 1999;126:1805–1810. doi: 10.1242/dev.126.9.1805. [DOI] [PubMed] [Google Scholar]
- Lash AE, Tolstoshev CM, Wagner L, Schuler GD, Strausberg RL, Riggins GJ, Altschul SF. SAGEmap: A public gene expression resource. Genome Res. 2000;10:1051–1060. doi: 10.1101/gr.10.7.1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li QY, Newbury-Ecob RA, Terrett JA, Wilson DI, Curtis AR, Yi CH, Gebuhr T, Bullen PJ, Robson SC, Strachan T, et al. Holt-Oram syndromes caused by mutations in TBX5, a member of the Brachyury (T) gene family. Nat Genet. 1997;15:21–29. doi: 10.1038/ng0197-21. [DOI] [PubMed] [Google Scholar]
- Liang F, Holt I, Pertea G, Karamycheva S, Salzberg SL, Quackenbush J. Gene index analysis of the human genome estimates approximately 120,000 genes. Nat Genet. 2000;25:239–240. doi: 10.1038/76126. [DOI] [PubMed] [Google Scholar]
- Logan M, Tabin C J. Role of Pitx1 upstream of Tbx4 in specification of hindlimb identity. Science. 1999;283:1736–1739. doi: 10.1126/science.283.5408.1736. [DOI] [PubMed] [Google Scholar]
- Maichele AJ, Farwell N J, Chamberlain JS. A B2 repeat insertion generates alternate structures of the mouse muscle γ-phosphorylase kinase gene. Genomics. 1993;16:139–149. doi: 10.1006/geno.1993.1151. [DOI] [PubMed] [Google Scholar]
- Man MZ, Wang X, Wang Y. POWER_SAGE: Comparing statistical tests for SAGE experiments. Bioinformatics. 2000;16:953–959. doi: 10.1093/bioinformatics/16.11.953. [DOI] [PubMed] [Google Scholar]
- Margulies EH, Innis JW. Building arms or legs with molecular models. Pediatr Res. 2000a;47:2–3. doi: 10.1203/00006450-200001000-00002. [DOI] [PubMed] [Google Scholar]
- ————— eSAGE : Managing and analysing data generated with serial analysis of gene expression (SAGE) Bioinformatics. 2000b;16:650–651. doi: 10.1093/bioinformatics/16.7.650. [DOI] [PubMed] [Google Scholar]
- Margulies EH, Kardia SLR, Innis JW. Identification and prevention of a GC content bias in SAGE libraries. Nucleic Acids Res. 2001;29:e60. doi: 10.1093/nar/29.12.e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michiels EMC, Oussoren E, Van Groenigen M, Pauws E, Bossuyt PMM, Voute PA, Baas F. Genes differentially expressed in medulloblastoma and fetal brain. Physiol Genomics. 1999;1:83–91. doi: 10.1152/physiolgenomics.1999.1.2.83. [DOI] [PubMed] [Google Scholar]
- Nelson CE, Morgan BA, Burke AC, Laufer E, DiMambro E, Murtaugh LC, Gonzales E, Tessarollo L, Parada LF, Tabin C. Analysis of Hox gene expression in the chick limb bud. Development. 1996;122:1449–1466. doi: 10.1242/dev.122.5.1449. [DOI] [PubMed] [Google Scholar]
- Niswander L. Developmental biology. Legs to wings and back again. Nature. 1999;398:751–752. doi: 10.1038/19625. [DOI] [PubMed] [Google Scholar]
- Papenbrock T, Visconti RP, Awgulewitsch A. Loss of fibula in mice over-expressing Hoxc11. Mech Dev. 2000;92:113–123. doi: 10.1016/s0925-4773(99)00344-5. [DOI] [PubMed] [Google Scholar]
- Peters DG, Kassam AB, Yonas H, O'Hare EH, Ferrell RE, Brufsky AM. Comprehensive transcript analysis in small quantities of mRNA by SAGELite. Nucleic Acids Res. 1999;15:e39. doi: 10.1093/nar/27.24.e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson RL, Jacobs DF, Awgulewitsch A. Hox-3.6: Isolation and characterization of a new murine homeobox gene located in the 50 region of the Hox-3 cluster. Mech Dev. 1992;37:151–166. doi: 10.1016/0925-4773(92)90077-w. [DOI] [PubMed] [Google Scholar]
- Peterson RL, Papenbrock T, Davda MM, Awgulewitsch A. The murine Hoxc cluster contains five neighboring AbdB-related Hox genes that show unique spatially coordinated expression in posterior embryonic subregions. Mech Dev. 1994;47:253–260. doi: 10.1016/0925-4773(94)90043-4. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Esteban C, Tsukui T, Yonei S, Magallon J, Tamura K, Izpisúa-Belmonte JC. The T-box genes Tbx4 and Tbx5 regulate limb outgrowth and identity. Nature. 1999;398:814–818. doi: 10.1038/19769. [DOI] [PubMed] [Google Scholar]
- Ruvinsky I, Gibson-Brown JJ. Genetic and developmental basis for serial homology in vertebrate limb evolution. Development. 2000;127:5233–5244. doi: 10.1242/dev.127.24.5233. [DOI] [PubMed] [Google Scholar]
- Sokal RR, Rohlf FJ. Biometry: The principles and practice of statistics in biological research. 3rd edition. New York: Freeman; 1995. [Google Scholar]
- Szeto DP, Ryan AK, O'Connell SM, Rosenfeld MG. P-OTX: A PIT-1-interacting homeodomain factor expressed during anterior pituitary gland development. Proc Natl Acad Sci. 1996;93:7706–7710. doi: 10.1073/pnas.93.15.7706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szeto DP, Rodriguez-Esteban C, Ryan AK, O'Connell SM, Liu F, Kioussi C, Gleiberman AS, Izpisúa-Belmonte JC, Rosenfeld MG. Role of the Bicoid-related homeodomain factor Pitx1 in specifying hindlimb morphogenesis and pituitary development. Genes & Dev. 1999;13:484–494. doi: 10.1101/gad.13.4.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takabatake Y, Takabatake T, Takeshima K. Conserved and divergent expression of T-box genes Tbx2-Tbx5 in Xenopus. Mech Dev. 2000;91:433–437. doi: 10.1016/s0925-4773(99)00329-9. [DOI] [PubMed] [Google Scholar]
- Takeuchi JK, Koshiba-Takeuchi K, Matsumoto K, Vogel-Hopker A, Naitoh-Matsuo M, Ogura K, Takahashi N, Yasuda K, Ogura T. Tbx5 and Tbx4 genes determine the wing/leg identity of limb buds. Nature. 1999;398:810–814. doi: 10.1038/19762. [DOI] [PubMed] [Google Scholar]
- Tamura K, Yonei-Tamura S, Belmonte JC. Differential expression of Tbx4 and Tbx5 in zebrafish fin buds. Mech Dev. 1999;87:181–184. doi: 10.1016/s0925-4773(99)00126-4. [DOI] [PubMed] [Google Scholar]
- Taylor KD, Piko L. Patterns of mRNA prevalence and expression of B1 and B2 transcripts in early mouse embryos. Development. 1987;101:877–892. doi: 10.1242/dev.101.4.877. [DOI] [PubMed] [Google Scholar]
- Velculescu VE, Zhang L, Vogelstein B, Kinzler KW. Serial analysis of gene-expression. Science. 1995;270:484–487. doi: 10.1126/science.270.5235.484. [DOI] [PubMed] [Google Scholar]
- Velculescu VE, Zhang L, Zhou W, Vogelstein J, Basrai MA, Bassett DE, Hieter P, Vogelstein B, Kinzler KW. Characterization of the yeast transcriptome. Cell. 1997;88:243–251. doi: 10.1016/s0092-8674(00)81845-0. [DOI] [PubMed] [Google Scholar]
- Velculescu VE, Madden SL, Zhang L, Lash AE, Yu J, Rago C, Lal A, Wang CJ, Beaudry GA, Ciriello KM, et al. Analysis of human transcriptomes. Nat Genet. 1999;23:387–388. doi: 10.1038/70487. [DOI] [PubMed] [Google Scholar]
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- Virlon B, Cheval L, Buhler JM, Billon E, Doucet A, Elalouf JM. Serial microanalysis of renal transcriptomes. Proc Natl Acad Sci. 1999;96:15286–15291. doi: 10.1073/pnas.96.26.15286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanek N, Muneoka K, Holler-Dinsmore G, Burton R, Bryant SV. A staging system for mouse limb development. J Exp Zool. 1989;249:41–49. doi: 10.1002/jez.1402490109. [DOI] [PubMed] [Google Scholar]
- Weatherbee SD, Carroll SB. Selector genes and limb identity in arthropods and vertebrates. Cell. 1999;97:283–286. doi: 10.1016/s0092-8674(00)80737-0. [DOI] [PubMed] [Google Scholar]
- Zhang L, Zhou W, Velculascu VE, Kern SE, Hruban RH, Hamilton SR, Vogelstein B, Kinzler KW. Gene expression profiles in normal and cancer cells. Science. 1997;276:1268–1272. doi: 10.1126/science.276.5316.1268. [DOI] [PubMed] [Google Scholar]