

Abstract
Gene expression studies bridge the gap between DNA information and trait information by dissecting biochemical pathways into intermediate components between genotype and phenotype. These studies open new avenues for identifying complex disease genes and biomarkers for disease diagnosis and for assessing drug efficacy and toxicity. However, the majority of analytical methods applied to gene expression data are not efficient for biomarker identification and disease diagnosis. In this paper, we propose a general framework to incorporate feature (gene) selection into pattern recognition in the process to identify biomarkers. Using this framework, we develop three feature wrappers that search through the space of feature subsets using the classification error as measure of goodness for a particular feature subset being “wrapped around”: linear discriminant analysis, logistic regression, and support vector machines. To effectively carry out this computationally intensive search process, we employ sequential forward search and sequential forward floating search algorithms. To evaluate the performance of feature selection for biomarker identification we have applied the proposed methods to three data sets. The preliminary results demonstrate that very high classification accuracy can be attained by identified composite classifiers with several biomarkers.
Over the past few years, the genomes of more than 39 organisms have been completely sequenced (Cummings and Relman 2000), with another 100 in progress (Lockhart and Winzeler 2000). With the human genome draft sequence in hand, the complete sequence of the entire genome will not be far behind. Availability of genetic sequence information in both public and private databases has gradually shifted genome-based research away from pure sequencing towards functional genomics and genotype–phenotype studies.
Among the most powerful and versatile tools for functional genomic studies are high-density DNA microarrays (Brown and Botstein 1999; Lipshulz et al. 1999). One of the most important applications of microarrays is to simultaneously monitor the expression of thousands or even tens of thousands of genes. A new discipline of gene expression profiling, which will play a fundamental role in biological research, pharmacology, and medicine, is emerging that allows the language of biology to be spoken in mathematical terms (Young 2000).
The practical applications of gene expression analyses are numerous and only beginning to be realized. One particularly powerful application of gene expression analyses is biomarker identification, which can be used for disease risk assessment, early detection, prognosis, prediction response to therapy, and preventative measures (Allgayer et al. 1997; Brien et al. 1998). Currently, the main strategy for disease diagnosis depends primarily on clinical evaluation and ultimately on clinical judgment that generally includes a careful medical history and physical examination (Growdon 1999). However, macro- and microscopic histology and morphology as the basis for disease diagnoses have some limitations, in particular, for early tumor detection (Mulshine 1999). Biomarkers can also be used to measure specific toxicity and efficacy profiles of a drug in preclinical trials or for assessing risk of environmental exposure (Bennett and Waters 2000; Rothberg et al. 2000; Steiner and Witzmann 2000).
Currently, the major tools for mapping disease genes are based on meiotic mapping within the paradigm of positional cloning (Collins 1995). A road toward identification of disease genes less traveled is functional analysis that studies mRNA and protein variations. Complementary to positional cloning, gene, including protein, expression analyses also may be employed to identify novel candidates for disease susceptibility loci (Niculescu et al. 2000). Functional analysis attempts to dissect disease processes and relevant biochemical pathways into component parts, which serve as intermediaries between genotypes and phenotype information and to bridge the gap between DNA information and trait information (Horvath and Baur 2000). We expected that the linkage studies and functional analysis would cross-validate the findings of each method, reducing the uncertainty inherent in the two approaches.
Biomarkers are expected to be highly accurate, efficient, and reliable for assessing disease risk and biological effect, simple to perform, and inexpensive. Microarrays provide rapid, efficient, and systematic approaches to searching biomarkers that are potential candidates with high accuracy for disease diagnosis and prognosis, putative targets of therapeutic agents, and understanding the basic biology of a disorder (Chow et al. 2001; Welsh et al. 2001). Although microarrays can generate a large amount of informative data, statistical and computational methods are required to reliably and efficiently discover biomarkers.
Most existing statistical and computational methods for gene expression data analysis have focused on differential gene expression, which is tested by simple calculation of fold changes, by t-test, F test, scoring methods (Hedenfalk et al. 2001; Welsh et al. 2001), or cluster analysis (Eisen et al. 1998; Tamayo et al. 1999; Tavazoie et al. 1999; Brazma and Vilo 2000; Butte et al. 2000; Getz et al. 2000). Although cluster analysis will continue to be a popular method for gene expression data analysis, it is an unsupervised learning method and cannot provide accurate prediction of diseases by itself. Supervised classification methods are available and offer a powerful alternative. The prediction strength (PS) method (Golub et al. 1999), support vector machine (SVM) (Furey et al. 2000; Moler et al. 2000), a naive Bayes Method (Moler et al. 2000) and Fisher's linear discriminant analysis (LDA) (Xiong et al. 2000) have been used for tumor classification. Chow et al. (2001) proposed to use some quantities that measure the ability of distinguishing tissue samples of genes and select subsets of genes with highest score as biomarkers.
However, the majority of current gene expression data analysis methods are not effective for biomarker identification and disease diagnosis for the following reasons. First, although the calculation of fold changes or t-test and F test can identify highly differentially expressed genes, the classification accuracy of identified biomarkers by these methods is, in general, not very high. Second, most scoring methods do not use classification accuracy to measure a gene's ability to discriminate tissue samples. Therefore, genes that are ranked according to these scores may not achieve the highest classification accuracy among genes in the experiments. Even if some scoring methods, which are based on classification methods, are able to identify biomarkers with high classification accuracy among all genes in the experiments, the classification accuracy of a single marker cannot achieve the required accuracy in clinical diagnosis. Third, to improve accuracy, several authors (Moler et al. 2000; Chow et al. 2001) used a combination of genes in the top of the list of ranked genes as a composite classifier. However, a simple combination of highly ranked markers according to their scores or discrimination ability may not be efficient for classification. Although two markers may carry good classification information when treated separately, there is little gain if they are combined together because of a high mutual correlation. Thus, complexity increases without much gain. Furthermore, using large number of biomarkers for diagnosis increases cost.
A fundamental problem in biomarker identification is how to efficiently sift through thousands or even tens of thousands of genes to select the ones related to disease pathophysiology. The goal of this research was to use feature (gene) selection incorporated into pattern recognition as a general framework for biomarker identification and optimal classifier generation. Using this framework, we attempted to systematically search optimal single biomarker classifier and composite classifiers that consist of a combination of biomarkers according to classification accuracy. To accomplish this goal, we developed three feature wrappers that are being “wrapped around” three learning algorithms: Fisher's LDA, logistic regression (LR), and SVMs. Because a learning algorithm is employed to evaluate each and every set of features considered, wrappers are prohibitively expensive to run. The computational time of searching algorithms is important to the success of feature selection. In this paper, we employ two search algorithms: sequential forward search (SFS) and sequential forward floating search (SFFS) algorithms. Therefore, feature selection is transformed into an optimization problem. This opens the way to use rich statistical and optimization methods and software for feature selection.
RESULTS
To evaluate the performance of feature wrappers for biomarker identification, we analyzed three data sets. One data set consists of expression profiles for 2000 genes using an Affymetrix oligonucleotide array in 22 normal and 40 cancer colon tissues, which were originally downloaded from the Web site at http://www.molbio.princeton.edu/colondata (Alon et al. 1999) and can now be retrieved from the Web site at http://www.sph.uth.tmc.edu/hgc. The second data set is expression profiles for 3226 genes using a cDNA microarray in seven BRCA1 mutation-positive, eight BRCA2 mutation-positive, and seven sporadic breast tumor samples (Hedenfalk et al. 2001). The third data set is expression profiles for 8102 genes in 40 tissue samples from 20 patients, 20 of which were obtained before treatment and 20 of which were obtained after an average of 16-week treatment of doxorubicin (Perou et al. 2000).
Before presenting the results, we first describe two ways of measuring classification accuracy. When the collection of total samples is used as both training and test data sets, the classification accuracy is referred to as the within-sample prediction accuracy. When the training and test samples are separate data sets, the classification accuracy is referred to as the out-of-sample prediction accuracy because test samples are used for the calculation of accuracy. Tables 1 and 2 compare the within-sample prediction accuracy of the single markers selected by LDA and the class-prediction method (Hedenfalk et al. 2001) for classifying BRCA1 mutation-positive and BRCA2 mutation-positive tumors, respectively. We have ordered the genes in the data set according to their classification accuracy or P values. In Tables 1 and 2 we selected the genes at the top of the list (those with higher accuracy or smaller P value). Hedenfalk et al. (2001) used a total of 9 clones (γ = 0.0001) in Table 1 and 11 clones in Table 2 and a class-prediction method to classify BRCA1 mutation-positive and BRCA2 mutation-positive tumors. The achieved accuracy rates for classifying BRCA1 mutation-positive and BRCA2 mutation-positive were 95.4% and 81.82%, respectively. The accuracy for classifying BRCA2 mutation-positive tumors by the class-prediction method is not very high, because the set of biomarkers for class prediction includes the clones 784830 and 366824. Although these two clones are highly differentially expressed (as measured by a t-test, γ = 0.0001) between BRCA2 mutation-positive and BRCA2 mutation-negative tumors, both of them have only 77.23% classification accuracy according to LDA. Tables 1 and 2 clearly demonstrate that genes at the top of the list (those with smaller P values) may not have the highest classification accuracy. Hence, ranking genes according to their t or F statistic values may not be the best strategy to select biomarkers for classification.
Table 1.
Top Accuracy Genes Selected by LDA and Class Prediction Method for Classifying BRCA1 Mutation Positive Tissue Samples
| Colon | Description | Accuracy |
|---|---|---|
| LDA | ||
| 212198 | tumor protein p53-binding protein, 2 | 0.954545 |
| 897646 | splicing factor, arginine/serine-rich 4 | 0.954545 |
| 344352 | ESTs | 0.954545 |
| 42888 | interleukin enhancer binding factor 2, 45kD | 0.954545 |
| 366647 | butyrate response factor 1 (EGF-response factor 1) | 0.954545 |
| 242037 | Human putative cyclic G1 interacting protein mRNA, partial sequence | 0.909091 |
| 248531 | guanine-monophosphate synthetase | 0.909091 |
| 46182 | CTP synthase | 0.909091 |
| 840702 | selenophosphate synthase; Human selenium donor protein | 0.909091 |
| 811930 | KIAA0020 gene product | 0.909091 |
| 687397 | Ras suppressor protein 1 | 0.909091 |
| 566887 | chromobox homolog 3 (Drosophila HP1 gamma) | 0.909091 |
| 81331 | fatty acid binding protein 5 (psoriasis-associated) | 0.909091 |
| 202034 | ESTs, highly similar to 45kDa splicing factor [Heme sapiens] | 0.909091 |
| 307843 | ESTs | 0.909091 |
| 247818 | ESTs | 0.909091 |
| 46019 | minichromosome maintenance deficient (S. cerevisiae) 7 | 0.909091 |
| 32790 | mutS (E. coli) homolog 2 (colon cancer, nonpolyposis type 1) | 0.909091 |
| Class Prediction Method (Hedenfalk et al. 2001) | ||
| 212198 | tumor protein p53-binding protein, 2 | 0.954545 |
| 366647 | butyrate response factor 1 (EGF-response factor 1) | 0.954545 |
| 840702 | selenophosphate synthetase; Human selenium donor protein | 0.909091 |
| 566887 | chromobox homolog 3 (Drosophila HP1 gamma) | 0.909091 |
| 307843 | ESTs | 0.909091 |
| 247818 | ESTs | 0.909091 |
| 46019 | minichromosome maintenance deficient (S. cerevisiae) 7 | 0.909091 |
| 26082 | very low density lipoprotein receptor | 0.863636 |
| 897781 | keratin 8 | 0.818182 |
Table 2.
Top Accuracy Genes Selected by LDA and Class Prediction Method for Classifying BRCA2 Mutation Positive Tissue Samples
| Colon | Description | Accuracy |
|---|---|---|
| LDA | ||
| 175123 | mitogen-activated protein kinase 7 | 1.000000 |
| 714106 | plasminogen activator, urokinase | 0.954545 |
| 210887 | suppression of tumorigenicity 13 (colon carcinoma) | 0.954545 |
| 29054 | ARP1 (actin-related protein 1, yeast) homolog A | 0.954545 |
| 36775 | hydroxyacyl-Coenzyme A dehydrogenase | 0.954545 |
| 21652 | catenin (cadherin-associated protein), alpha 1 (102kD) | 0.909091 |
| 233721 | insulin-like growth factor binding protein 2 (36kD) | 0.909091 |
| 666377 | zinc finger protein 161 | 0.909091 |
| 50413 | armadillo repeat gene deletes in velocardiofacial syndrome | 0.909091 |
| 179804 | PWP2 (periodic tryptophan protein, yeast) homolog | 0.909091 |
| 563444 | forkhead (Drosophila)-like 5 | 0.909091 |
| 345423 | DKFZP564M112 protein | 0.909091 |
| 246194 | ESTs | 0.909091 |
| 23014 | mitogen-activated protein kinase 1 | 0.909091 |
| 51209 | protein phosphatase 1, catalytic subunit, beta isoform | 0.909091 |
| 341130 | retinoblastoma-like 2 (p130) | 0.909091 |
| 345645 | plate-derived growth factor beta polypeptide | 0.909091 |
| Class Prediction Method (Hedenfalk et al. 2001) | ||
| 36775 | hydroxyacyl-Coenzyme A dehydrogenase | 0.954545 |
| 29054 | ARP1 (actin-related protein 1, yeast) homolog A (centractin alpha) | 0.954545 |
| 666377 | Zinc finger protein 161 | 0.909091 |
| 50413 | armadillo repeat gene deletes in velocardiofacial syndrome | 0.909091 |
| 31842 | UDP-galactose transporter related | 0.863636 |
| 51209 | protein phosphatase 1, catalytic subunit, beta isoform | 0.909091 |
| 345645 | plate-derived growth factor beta polypeptide | 0.909091 |
| 340644 | integrin, beta 8 | 0.863636 |
| 344109 | proliferating cell nuclear antigen | 0.863636 |
| 784830 | D123 gene product | 0.772727 |
| 366824 | cyclin-dependent kinase 4 | 0.727273 |
Among the 18 genes with the highest accuracy for classifying BRCA1 mutation-positive tumors in Table 1 are the p53-binding protein (212198), Ras suppressor protein (687397), psoriasis-associated protein (81331), and DNA repair gene MSH2 (32790), which are related to the development of tumors. Among the 17 genes with the highest accuracy for classifying BRCA2 mutation-positive tumors in Table 2 are MAPK1 (23014), MAPK7 (175123), suppression of tumorogenicity (210887), and semia sarcoma viral oncogene homolog (345645), which are all involved in tumurogenesis.
Tables 1 and 2 show that the use of even a single marker can achieve very high accuracy. This may be due to small sample size in the experiment. In general, using a single marker for classification cannot achieve high accuracy, which is demonstrated in Table 3. Table 3 shows that the highest accuracy for classifying breast tumor tissue samples before and after treatment using a single marker as a classifier selected by LDA is 77.5%. To improve the accuracy, we combined several markers together to generate a composite classifier and used the SFFS algorithm to search subsets of optimal composite classifiers with the highest accuracy among all possible composite classifiers with the same number of genes in the composite classifier. As shown in Table 3, the accuracy of the selected optimal composite classifier with three genes can reach 100%.
Table 3.
Accuracy of Single Classifier and Composite Classifier for Classifying Breast Tumor Tissue Sample Before and After Treatment
| Gene access number | Gene name | Gene access number | Gene name | Gene access number | Gene name | Accuracy |
|---|---|---|---|---|---|---|
| T62179 | FOSB | 0.775 | ||||
| AA598794 | CTGF | 0.775 | ||||
| W96134 | JUN | 0.775 | ||||
| R12840 | AA005202 | ESTs | 0.925 | |||
| R12840 | AA027875 | HBA2 | 0.925 | |||
| AA343173 | SPN | AA040944 | AA114864 | ESTs | 1.00 | |
| R12840 | FOS | AA027875 | HBA2 | AA045342 | ESTs | 1.00 |
| R12841 | FOS | AA027875 | HBA2 | T95903 | ESTs | 1.00 |
| AA700604 | SORD | R12841 | FOS | AA027875 | HBA2 | 100 |
| H62594 | GW128 | R12842 | FOS | AA027875 | HBA2 | 1.00 |
| AA402766 | SMP1 | R12843 | FOS | AA027875 | HBA2 | 1.00 |
| AA460599 | COPS5 | R12844 | FOS | AA027875 | HBA2 | 1.00 |
| H15707 | TRAM | R12845 | FOS | AA027875 | HBA2 | 1.00 |
| AA045587 | TAF2J | R12846 | FOS | AA027875 | HBA2 | 1.00 |
Several authors (Chow et al. 2001; Hedenfalk et al. 2001) proposed to use a combination of genes in the top of the list in which genes were ranked according to some discrimination quantity. To examine whether this is a good strategy for producing a composite classifier we provide Table 4, which shows combination of two genes with within-sample prediction accuracy >92%. Two remarkable features from Table 4 are evident. First, at least one gene in the composite classifier has low classification accuracy. Second, although the accuracies of both genes in the composite classifier are low, their combination may have high accuracy.
Table 4.
Top 15 Combinations of Two Genes for Classifying Colon Tumor Samples
| Access number | Gene name | Accuracy single marker | Access number | Gene name | Accuracy single marker | Accuracy combination |
|---|---|---|---|---|---|---|
| Z50753 | GUCA2B | 0.75806 | H22579 | 0.59677 | 0.93548 | |
| Z50753 | GUCA2B | 0.75806 | X67155 | KNSL5 | 0.56452 | 0.93548 |
| Z50753 | GUCA2B | 0.75806 | H22579 | 0.53226 | 0.93548 | |
| R87126 | 0.82258 | U31215 | GRM1 | 0.64516 | 0.91935 | |
| H20709 | MYL6 | 0.66129 | T63484 | 0.53226 | 0.91935 | |
| H20709 | MYL6 | 0.66129 | L39874 | DCTD | 0.51613 | 0.91935 |
| R88740 | ATP5J | 0.51613 | T90350 | SFPQ | 0.6129 | 0.91935 |
| Z50753 | GUCA2B | 0.75806 | X70326 | MACMARCKS | 0.69355 | 0.91935 |
| H08393 | 0.70968 | M84490 | MAPK3 | 0.6129 | 0.91935 | |
| D26018 | POLD3 | 0.46774 | R44301 | NR3C2 | 0.72581 | 0.91935 |
| Z50753 | GUCA2B | 0.75806 | H06061 | PRO0082 | 0.54839 | 0.91935 |
| Z50753 | GUCA2B | 0.75806 | R72374 | ACTN4 | 0.54839 | 0.91935 |
| M36634 | VIP | 0.77419 | J05032 | DARS | 0.62903 | 0.91935 |
| R87126 | 0.82258 | Z15009 | LAMC2 | 0.6129 | 0.91935 | |
| R87126 | 0.82258 | T65938 | TPT1 | 0.6129 | 0.91935 |
To further demonstrate the potential power of a combination of several genes for distinguishing different types of tissues and to compare the performance of SFS and SFFS search algorithms, we calculated the maximum accuracy for classifying 22 normal and 40 tumor colon tissue samples as a function of number of genes used for classification. The results are shown in Figure 1, which includes the classification accuracy for the total collection of tissue samples. SFFS (combination) and SFFS (forward) denote the SFFS algorithms, which started with two genes obtained by searching all possible combinations of two genes and by an SFS algorithm, respectively. Several interesting features emerge from Figure 1. First, the classification accuracy of the optimal subsets of genes searched by SFFS algorithm is greater than or equal to that obtained by SFS algorithm. Second, the accuracy increased when sizes of subsets of selected genes increased and quickly reached 100% accuracy for the SFFS algorithm, but suddenly dropped to 50% when the size of selected subsets of genes was >60 (which is close to total sample size of 62). It is well known that when the number of features used for classification is greater than the number of samples to be classified, the sample covariance matrix will become singular, and Fisher's LDA cannot be applied to such case. Third, it is interesting to note that the classification accuracy of optimal subsets of genes with size 4 searched by SFFS algorithm is 100%. This example demonstrates that using a small number of genes can achieve a high accuracy of classification. To visualize such a possibility, we plot Figure 2, using expression levels of three genes (accession numbers H22579, Z50753, and R67343; http://www.molbio.princeton.edu/colondata). From Figure 2 we can see that most normal and tumor tissue samples were separated. To compare maximum classification accuracy, which can be achieved by the three learning algorithms, we plot Figure 3. In Figure 3, SVMs used two kernel functions: linear and polynomial of degree P = 3, and γ is set to γ = 10. Figure 3 demonstrates that the LR performs better than LDA and SVMs, but the difference in accuracy between LDA and LR is very small.
Figure 1.

Maximum within-sample prediction accuracy as a function of number of genes for classifying colon tumors that can be achieved by LDA using SFS and SFFS search algorithms.
Figure 2.

Expression levels of three genes with accession numbers H22579, Z50573, and R67343 in 62 colon tissue samples.
Figure 3.

Maximum within-sample prediction accuracy which was evaluated from the total collection of 62 colon tissue samples and by LDA, LR, and SVM with two kernel functions: linear and polynomial of degree P = 3 learning methods using SFFS search algorithm.
Because it is not reliable to use the total sample for evaluating the accuracy of classification methods, to get a realistic estimate of classification accuracy one procedure is to split the total sample into a training sample and a validation sample. The training sample is used to construct the classification function and the validation sample is used to evaluate it. We used leave-one-out cross-validation procedure (i.e., each time hold out one sample as a validation set and develop a classification function based on the remaining samples and then classify the “held-out” sample using the function constructed from the training data) to calculate the average classification accuracy. The procedure was repeated for each training sample in turn. Figure 4 plots maximum average classification accuracy over the cross-validation trials, which can be achieved by using SFFS searching algorithms and the three learning methods LDA, LR, and SVM with a linear and a polynominal kernel function of degree of p = 3. It is clear from Figure 4 that when the number of genes is 3 and 4, SVM with the polynomial kernel function has the highest classification accuracy 93.5%, but in other cases LR has higher accuracy than that of LDA and SVM methods. It was reported that Furey et al. (2000) used SVM and all 2000 or top 1000 genes achieved only 90% accuracy. Figure 4 demonstrates the important point that using a much smaller number of genes can achieve higher accuracy than that of using thousands of genes.
Figure 4.

Maximum average out-of-sample prediction accuracy over the leave-one-out cross-validation set of colon tissue samples, which was achieved by LDA, LR and SVM with two kernel functions: linear and polynomial of degree P = 3 function learning methods using SFFS search algorithm.
Table 5 lists the 15 genes with the highest within-sample and out-of-sample prediction accuracies for classifying colon tumors that were estimated by LR from the total collection of samples and leave-one-out cross-validation data set. Table 5 shows that the top 15 genes that were inferred from the total collection of samples and leave-one-out validation data set are the same, but their rank in the list differs somewhat. Table 5 demonstrates that to search a list of genes with high accuracy, we can use the total collection of sample, which will save a lot of computational time.
Table 5.
Top 15 Genes for Classifying Colon Tumor Samples Searched from Total Collection of Samples and Cross-Validation Set
| Access number | Gene name | Accuracy | |
|---|---|---|---|
| total sample | cross-validation | ||
| M63391 | DES | 0.854839 | 0.854839 |
| M76378 | EST | 0.83871 | 0.822581 |
| J05032 | DARS | 0.83871 | 0.790323 |
| M76378 | EST | 0.822581 | 0.822581 |
| R87126 | EST | 0.822581 | 0.822581 |
| M22382 | HSPD1 | 0.822581 | 0.822581 |
| M76378 | EST | 0.822581 | 0.822581 |
| J02854 | MYRL2 | 0.806452 | 0.806452 |
| M26383 | IL8 | 0.806452 | 0.790323 |
| T60155 | ACTA2 | 0.790323 | 0.774194 |
| H40095 | MIF | 0.790323 | 0.758065 |
| T92451 | TPM2 | 0.790323 | 0.790323 |
| R36977 | GTF3A | 0.790323 | 0.774194 |
| R64115 | EST | 0.790323 | 0.758065 |
| X63629 | CDH3 | 0.790323 | 0.790323 |
To examine how the selected optimal subsets of genes depend on the learning algorithms, we provide Table 6. It summarizes the results of 10 selected genes with the highest classification accuracy, which is evaluated using total collection of 62 colon tissue samples, by three learning algorithms. Table 6 demonstrates that 7 out of 10 genes are common to three learning algorithms although their orders in the table for the three learning algorithms have some differences. However, the classification accuracies of the gene DARS evaluated by the three learning algorithms are quite different. Table 6 shows that the majority of the selected genes by feature selection are less dependent on the learning algorithms.
Table 6.
Ten Selected Genes with Highest Classification Accuracy Using Linear Discriminant Analysis (LDA), Logistic Egression (LR), and Support Vector Machine (SVM) for Classifying Colon Tumor
| LDR | LR | SVM | |||
|---|---|---|---|---|---|
| gene access number | accuracy | gene access number | accuracy | gene access number | accuracy |
| M63391(DES) | 0.8548 | M63391(DES) | 0.8548 | M6391(DES) | 0.8387 |
| M76378(EST,245) | 0.8226 | M76378(EST,245) | 0.8387 | M76378(EST,245) | 0.8226 |
| M76378(EST,267) | 0.8226 | M76378(EST,267) | 0.8226 | M76378(EST,267) | 0.8387 |
| J05032(DARS) | 0.8387 | ||||
| R87126(EST) | 0.8226 | R87126(EST) | 0.8226 | ||
| M76378(EST,765) | 0.8226 | M76378(EST,765) | 0.8226 | M76378(EST,765) | 0.8387 |
| J02854(MYRL2) | 0.7903 | J02854(MYRL2) | 0.8065 | J02854(MYRL2) | 0.8387 |
| U25138(KCNMB1) | 0.7742 | U25138(KCNMB1) | 0.7903 | U25138(KCNMB1) | 0.8065 |
| T92451(TPM2) | 0.7903 | T92451(TPM2) | 0.7903 | T92451(TPM2) | 0.7742 |
| H08393(EST) | 0.7903 | ||||
| X86693(SPACL1) | 0.7742 | ||||
| M36634(VIP) | 0.7742 | ||||
| M26383(IL8) | 0.8065 | ||||
| T60155(ACTA2) | 0.7903 | T60155(ACTA2) | 0.7742 | ||
| T61629(LAMR1) | 0.7742 | ||||
DISCUSSION
Emerging advances in microarray “chip” technology allow the simultaneous analysis of expression patterns for thousands of gene sequences (i.e., chip features) and will serve as precursors to genome-wide functional analyses. These studies open new avenues for identifying complex disease genes and biomarkers for disease diagnosis and for assessing drug efficacy and toxicity. To achieve this goal, it is fundamental to develop a sound framework for biomarker discovery. In this paper, we formulated the problem of biomarker identification as feature selection incorporated into pattern recognition (i.e., we formulated it into an optimization problem). This general framework has two parts. One part comes from pattern recognition theory that provides an objective function. Classification accuracy, a quantity used to measure the discriminating ability, was taken as the objective function in this paper. The second part comes from search algorithms or optimization methods that provide algorithms to search global optimal solutions. This general framework allows us to systematically and efficiently search biomarkers from large volumes of expression data by using rich statistical and computational methods and software in pattern recognition and data mining.
Feature selection serves two purposes: (1) to reduce dimensionality of the data and improve classification accuracy, and (2) to identify genes that are relevant to the cause and consequences of disease or can be used as biomarkers for diagnosis of disease, measuring drug toxicology and efficacy. The first practical application area of gene expression data analysis is disease diagnosis. Classification accuracy and cost are two important indices for disease diagnosis. The great advantage of microarrays is that they are able to simultaneously monitor the expression of thousands or even ten thousands of genes, which provides extremely useful information. However, if whole-genome expression profiles are used for disease diagnosis, the prediction accuracy will be low and the cost of diagnosis will be high. Theoretically, having more genes should give more discriminating power. But, as shown in this paper, using a large number of genes for classification can dramatically reduce the classification accuracy.
It is well recognized that improved accuracy results from reducing the dimensionality of the data. Now the question is how many genes are required and which genes are selected to ensure the required classification accuracy. To address these problems, we have analyzed three available expression data sets. In this paper, we showed that when the sample size is small, using one selected biomarker reached very high accuracy and when the sample size is moderate (<100), a combination of three or four markers, which we called a composite classifier, achieved >90% accuracy. Here we must point out that the results from small sample sizes are not reliable. To further investigate the feasibility of biomarkers for disease diagnosis, we probably need to have ∼1000 samples. In this situation, more than five biomarkers are expected to be required. It bodes well for the following scenario. Initial basic research and clinical trials will monitor the expressions of thousands or even tens of thousands of genes in several hundred or a thousand samples using microarrays to identify subsets of genes providing optimal classification accuracy. Clinical applications will then monitor only this small subset of genes, avoiding the cost and complexity of large-scale gene expression array.
Recently, several authors (Moler et al. 2000; Chow et al. 2001) have proposed to simply combine genes that were highly ranked according to some quantity to measure discrimination ability as a composite classifier. Intuitively, this strategy to select a combination of biomarkers for improving classification accuracy is appealing. However, our preliminary results showed that not all genes in the composite classifier have high classification accuracy and that in some cases although the accuracy of each gene is quite low, their combination may lead to high accuracy. The optimal combination of genes with high accuracy should be systematically searched by a feature selection procedure.
Furthermore, we have demonstrated that feature selection is a powerful tool to determine the number of genes and what genes should be used for classification. Both accuracy and computational time depend on the learning and search algorithms. Classification function is determined by learning algorithms and has a large impact on the classification accuracy.
It has been argued that because feature selection is typically done in an off-line manner, the execution time of a particular algorithm is not as critical as the optimality of the feature subset it generates. Although this may be true for feature sets of moderate size, for sets involving thousands or even ten thousands of features, the computational requirement of feature selection is extremely important. Because SVMs involves quadratic programming that is computationally expensive we used a least square version of SVMs, which can reduce the computational time. Even if we used faster versions of SVMs, LDA and LR run much faster than SVMs.
Although an exhaustive search is sufficient to guarantee optimality of selected composite classifier, it is computationally prohibitive as the number of feature subsets increases. To solve this problem a number of suboptimal selection techniques have been proposed, which essentially trade off the optimality of the selected subset for computational efficiency. It has been recognized that no unique optimal approach to the feature selection exists (Pudil and Novovicova 1998). In this paper, two heuristic algorithms, SFS and SFFS, were employed. The results showed that SFFS algorithm can search composite classifiers with higher accuracy than SFS algorithm. This may be due to the fact that for the SFS algorithm the nesting of biomarker subsets might rapidly cause deteriorating performance. The computational time of SFFS algorithm is only slightly more than that of SFS algorithm.
Genome-wide gene expression data analyses open a new avenue for biomarker identification. Although the results presented here are encouraging, they are limited. Some important factors such as sample size, which may have a large impact on the biomarker identification, and whole-genome functional analysis have not been discussed and should be investigated in the future.
METHODS
Classification Task and Data Representation
In a typical tissue classification task, data is represented as a table of examples. Each example is described by a fixed number of measurements, or features along with a label that denotes its class (type of tissues). Features are typically gene expression levels, sex, age, and environmental variables such as drug dosages. The label variable and the features are denoted by a vector.
Tissue classification begins with a set of training examples, denoted by
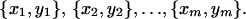 |
Learning a classifier involves inducing a model from the training data set that can be used to classify a new feature vector into one of the existing classes. This new data is often referred to as the testing data set.
Problem Formulation of Feature Selection
Let X be the original set of features with size k, that is, the number of features in the set. Let Z be the selected subset, Z ⊆ X. To evaluate the worth of features for classification, we introduce a feature selection criterion function for the set that is denoted by C(X). In feature wrappers, we use classification accuracy, which is defined as the percentage of the correctly classified tissue samples and hence is directly related to the performance of classification, as the criterion function. The selected subset of features Z is used to construct the classification model. Formally, the problem of feature selection is to find a subset Z ⊆ X such that
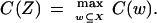 |
There are two ways to estimate classification accuracy. One procedure is to use the total collection of tissue samples to estimate the parameters in the classification model and the classification accuracy. Because of the possibility of over-fitting the data that arises from using the same data to both build and judge the classification model, the generalization performance of the model induced from the total collection of tissue samples may not be good for future samples. This will affect the quality of the selected features for classifying new tissue samples. To overcome this problem, we use a leave-one cross-validation strategy to estimate the classification accuracy. A collection of n tissue samples is split into n − 1 training samples and 1 test sample. The n − 1 training samples are used to construct the classification model. We use the constructed classification model to classify the test sample. The label assigned by a trained classification model can be true or false. This procedure is repeated n times to produce the training and test samples from the total collection of samples in turn. The classification accuracy is estimated to be the ratio of the total number of correctly classified samples by the trained models in all generated test samples by the leave-one-out procedure divided by the total number of samples.
Learning Algorithm
The use of classification accuracy as a criterion function makes feature selection dependent on the learning algorithms. Throughout this paper, three learning algorithms are used as a basis for the development of feature wrappers for biomarker identification: Fisher's LDA, LR, SVMs.
Fisher's LDA
Fisher's LDA has been a widely used tool for classification in machine learning. Because of its simplicity and high computational speed, LDA was our first choice for classification and gene selection and was applied to gene expression-based tumor classification. Fisher's approach does not assume that the observations are normally distributed. But, it does implicitly assume that the population covariance matrices are equal (Johnson and Wichern 1982). Tissues are classified on the basis of k selected feature variables. Suppose that nN normal and nT tumor tissue samples are examined. For tissue sample i, we have the vector Yi‘ = (Yi1, Yi2, ..., Yik). The Yi’s for normal (N) and tumor (T) samples constitute the following data matrix,
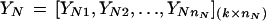 |
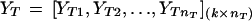 |
From these data matrices, the sample mean vectors and covariance matrices are determined by
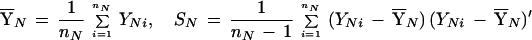 |
 |
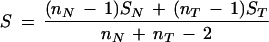 |
Fisher's idea was to transform the multivariate observations YNi and YTi into univariate observations ZNi and ZTi such that Z's were separated as much as possible. Fisher suggested taking linear combinations of the Y's to generate Z's, which can be easily maipulated mathematically. The midpoint, m̂, between the two univariate sample means, 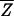 N = (
N = ( N −
N −  T)‘ S−1
T)‘ S−1 N and
N and 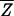 T = (
T = (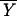 N −
N −  T)′ S−1
T)′ S−1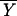 T, is given by
T, is given by
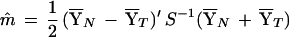 |
The classification rule based on Fisher’s linear discrimination function for an unknown sample, Y0, is as follows
 |
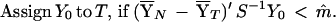 |
LR Model
Some environments, such as smoking, with exposure to carcinogens will cause changes in patterns of gene expression. Suppose that we collect tissue samples from patients who are divided into two groups: smoking and nonsmoking. The pattern of gene expression profiles for tumor and normal lung tissue samples collected from smokers may be different from that of nonsmokers. Sex, ethnicity, genotypes at oncogenes, tumor suppressor genes, and drug metabolism enzymes may also affect the pattern of gene expression. These variables are qualitative. The LDA is a linear statistical method for classification. Although it can still simultaneously deal with both quantitative and qualitative variables, in this case, its discriminatory power will be reduced. A practical alternative method that includes both continuous and discrete variables is Cox's LR method (1970). The LR model is also a simple nonlinear method for classification.
Suppose that there are n tissue samples. For each of the n tissue samples, there are k independent variables xji, j = 1, ..., k. These variables can be either qualitative variables, such as sex, age, and race, or quantitative variables, such as gene expression levels.
In the LR model, the dependence of the probability of being disease on independent variables including gene expression levels and other discrete variables is assumed to be
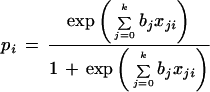 |
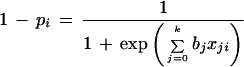 |
where pi = P(yi = 1|x1i,…,xki), x0i = 1 and bj are unknown coefficients, and yi = 1, abnormal tissue; yi = 0, normal. The logarithm of the ratio of pi and 1 − pi is a simple linear function of the xji. We define log odds as
 |
The maximum likelihood method can be used to estimate the coefficients bj's. Let y1,y2,…,yn be the observed class label on the n individuals. Thus, the likelihood for n tissue samples
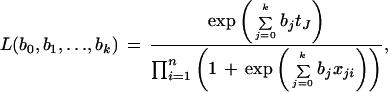 |
where tj = Σ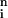 =1 xjiyi. The log-likelihood function is
=1 xjiyi. The log-likelihood function is
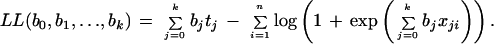 |
By maximizing the log-likelihood function we can obtain the maximum likelihood estimates of bj's. Then, for a given new sample x1,x2,…,xk, we determine its identity by p = P(y = 1|x1,…,xk).
SVMs
The past few years have seen the rise of SVMs as powerful tools for solving classification problems (Burges 1998; Christianini and Shawe-Taylor 2000). The basic idea that drove the initial development of SVMs is that for a given learning task, with a given finite amount of training data, the best generalization performance will be achieved by the balance between the accuracy attained on that particular training set and the ability of the machine to learn any training set without error. The SVM classifier typically follows from the solution to a quadratic programming (QP) problem. However, the QP requires expensive computation. This will create serious problems for the selection of thousands of features. To avoid heavy computation, in this paper, we use least square SVM (Suykens and Vandewalle 1999).
Given a training set  indicating the class (type of tissue), SVM formulations start from the assumption that all the training data satisfy the following constraints:
indicating the class (type of tissue), SVM formulations start from the assumption that all the training data satisfy the following constraints:
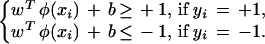 |
Here the nonlinear mapping φ(·) maps the input data into a higher dimensional space and w is a normal to the hyperplane. Note that the dimension of w is not specified (it can be infinite dimensional). Suppose we have some hyperplane that separates the positive from the negative examples (a “separating hyperplane”). Define the “margin” of a separating hyperplane to be the summation of shortest distance from the separating hyperplane to the closest positive and negative examples. It can be shown that the margin is simply 2/√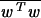 . Our goal is to find the pair of hyperplanes that gives the maximum margin. This can be accomplished by minimizing wTw, subject to the above constraints. In least squares SVMs, the above optimization problem is formulated as
. Our goal is to find the pair of hyperplanes that gives the maximum margin. This can be accomplished by minimizing wTw, subject to the above constraints. In least squares SVMs, the above optimization problem is formulated as
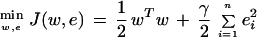 |
which is subject to the equality constraints
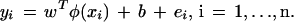 |
where γ is a penalty parameter. The Lagrangian multiplier method can be used to solve this equality constrained optimization problem. The Lagrangian is given by
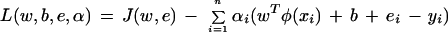 |
with Lagrange multipliers αi. The conditions for optimality
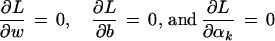 |
give
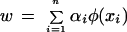 |
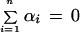 |
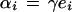 |
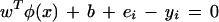 |
Some algebra yields the following set of linear equations
 |
where yT = [y1,…,yn], τT = [1,…,1], αT = [α1,…,αn] and the Mercer condition
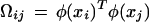 |
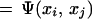 |
have been applied, where Ψ(xi,xj) is a kernel function. Once we have trained a SVM, we determine on which side of the decision boundary given test pattern x lies and assign the corresponding class label, i.e., we take the class of x to be sgn(f(x)) where f(x) is given by
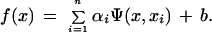 |
The following four functions:  (linear SVM), Ψ(x,xi) = (x
(linear SVM), Ψ(x,xi) = (x x + 1)p (polynomial SVM of degree p), Ψ(x,xi) = exp{−∥x − xi∥2/ς2} (Radial Basis Function SVM) and Ψ(x,xi) = tanh(κx
x + 1)p (polynomial SVM of degree p), Ψ(x,xi) = exp{−∥x − xi∥2/ς2} (Radial Basis Function SVM) and Ψ(x,xi) = tanh(κx x + θ) (two-layer sigmoidal neural network) can be used as kernel functions.
x + θ) (two-layer sigmoidal neural network) can be used as kernel functions.
Search Algorithms
Because a learning algorithm is employed to evaluate each and every set of features considered, feature wrappers are very expensive to run. The search algorithms are fundamental to the success of the biomarker identification. Although an exhaustive search can find optimal solutions, it requires an extremely large number of computations. To overcome this difficulty, we adopt two heuristic searching algorithms: SFS and SFFS (Sahiner et al. 2000).
SFS
The procedures for sequential forward selection are as follows:
Compute the criterion value (classification accuracy) for each of the features. Select the feature with the best value.
Form all possible two-dimensional vectors that contain the winner from the previous step. Compute the criterion value for each of them and select the best one.
Form all three-dimensional vectors expanded from the two-dimensional winners, and select the best one. Continue this process until reaching the prespecified dimension of the feature vector say, l.
SFFS
The SFS algorithm suffers from the so-called nesting effect. That is, once a feature is chosen, there is no way for it to be discarded later on. To overcome this problem, the sequential floating algorithm was proposed (Pudil et al. 1994).
Suppose m variables have already been selected from the complete set B = {xj, j = 1,…,k}, so that the selected variables form the set Am (and the criterion value C(Am) is known). The values C(Ai),i = 1,2,…,m − 1 are also known and stored for further usage.
Step 1 (inclusion)
Using SFS, select a variable xm+1 from the set of unselected variables B − Am and form the set Am+1 so that the most significant variable with respect to Am is added to Am, i.e., Am+1 = Am + xm+1.
Step 2 (conditional exclusion)
Find the least significant variable in the set Am+1. If xm+1 is the least significant variable in the set Am+1, i.e.,
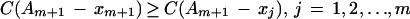 |
then set m = m + 1 and return to step 1. If the least significant variable in the set Am+1 is xr, r = 1,2,…,m, i.e., C(Am+1 − xr) > C(Am) then exclude xr from the set Am+1, i.e., A = Am+1 − xr. If m = 2, then set Am = A
= Am+1 − xr. If m = 2, then set Am = A , C(Am) = C(A
, C(Am) = C(A ) and return to step 1, otherwise go to step 3.
) and return to step 1, otherwise go to step 3.
Step 3 (continuation of conditional exclusion)
Find the least significant variable xs in the set A . If C(A
. If C(A − xs) ≤ C(Am−1), then set Am = A
− xs) ≤ C(Am−1), then set Am = A , C(Am) = C(A
, C(Am) = C(A ) and return to step 1. If C(A
) and return to step 1. If C(A − xs) > C(Am−1), then exclude xs from the set A
− xs) > C(Am−1), then exclude xs from the set A and form a new reduced set A
and form a new reduced set A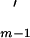 , i.e., A
, i.e., A = Am − xs. Set m = m − 1. If m = 2, then set Am = A
= Am − xs. Set m = m − 1. If m = 2, then set Am = A , C(Am) = C(A
, C(Am) = C(A ) and return to step 1, otherwise return to step 3.
) and return to step 1, otherwise return to step 3.
Initialization
The algorithm is initialized by value m = 0. A set A0 is empty. SFS algorithm or an exhaustive search of all possible combinations of two features is used for finding an initial set with two feature variables. Start with a step 1. The resulting set is Am.
Acknowledgments
M.M.X., X.Z.F., and J.Y.Z. are supported by NIH grants GM56515 and HL 5448. We thank Joshua M. Akey for his helpful comments on this paper, which helped to improve its presentation. We also thank Dr. Yidong Chen for providing an Excel form of gene expression data in hereditary breast cancer.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL mxiong@utsph.sph.uth.tmc.edu; FAX (713) 500-0900.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.190001.
REFERENCES
- Allgayer H, Heiss MM, Schildberg FW. Prognostic factors in gastric cancer. Br J Surg. 1997;84:1651–1664. [PubMed] [Google Scholar]
- Alon U, Brakai N, Notterman DA, Gish K, Ybarra S, Mack D, Levine AJ. Broad patterns of gene expression revealed by clustering analysis of tumor and normal colon tissues probed by oligonucleotide arrays. Proc Natl Acad Sci. 1999;96:6745–6750. doi: 10.1073/pnas.96.12.6745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DA, Waters MD. Applying biomarker research. Environ Health Perspect. 2000;108:907–910. doi: 10.1289/ehp.00108907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazma A, Vilo J. Gene expression data analysis. FEBS Lett. 2000;480:17–24. doi: 10.1016/s0014-5793(00)01772-5. [DOI] [PubMed] [Google Scholar]
- Brien TP, Depowski PL, Sheeehan CE, Ross JS, McKenna BJ. Prognostic factors in gastric cancer. Mol Pathol. 1998;11:870–877. [PubMed] [Google Scholar]
- Brown PO, Botstein D. Exploring the new world of the genome with DNA microarrays. Nat Genet. 1999;21:33–37. doi: 10.1038/4462. [DOI] [PubMed] [Google Scholar]
- Burges CJC. A tutorial on support vector machines for pattern recognition. Data Mining and KnowlDiscov. 1998;2:121–167. [Google Scholar]
- Butte AJ, Tamayo P, Slonim D, Golub TR, Kohane IS. Discovering functional relationships between RNA expression and chemotherapeutic susceptibility using relevance networks. Proc Natl Acad Sci. 2000;97:12182–12186. doi: 10.1073/pnas.220392197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow ML, Moler EJ, Mian IS. Identifying marker genes in transcription profiles data using a mixture of feature relevance experts. Physiol Genomics. 2001;5:99–111. doi: 10.1152/physiolgenomics.2001.5.2.99. [DOI] [PubMed] [Google Scholar]
- Christianini N, Shawe-Taylor J. An introduction to support vector machines and other kernel-based learning methods. London: Cambridge University Press; 2000. [Google Scholar]
- Collins FS. Positional cloning moves from perditional to traditional. Nat Genet. 1995;9:347–350. doi: 10.1038/ng0495-347. [DOI] [PubMed] [Google Scholar]
- Cox DR. The analysis of binary data. 1st ed. London: Methuen; 1970. [Google Scholar]
- Cummings CA, Relman DA. Using DNA microarrays to study host-microbe interactions. Emerg Infect Dis. 2000;6:513–525. doi: 10.3201/eid0605.000511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furey T, Cristianini N, Duffy N, Bednarski D, Schummer M, Haussler D. Support vector machines classification and validation of cancer tissue samples using microarray expression data. Bioinformatics. 2000;16:906–914. doi: 10.1093/bioinformatics/16.10.906. [DOI] [PubMed] [Google Scholar]
- Getz G, Levine E, Domany E. Coupled two-way clustering analysis of gene microarray data. Proc Natl Acad Sci. 2000;97:12079–12084. doi: 10.1073/pnas.210134797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, Coller H, Loh ML, Downing JR, Caligiuri MA, Bloomfield CD, Lander ES. Molecular classification of cancer class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- Growdon JH. Biomarkers of Alzheimer disease. Arch Neurol. 1999;56:281–283. doi: 10.1001/archneur.56.3.281. [DOI] [PubMed] [Google Scholar]
- Hedenfalk I, Duggan D, Chen Y, Radmacher M, Bittner M, Simon R, Meltzer P, Gusterson B, Esteller M, Mark R, et al. Gene-expression profiles in hereditary breast cancer. N Engl J Med. 2001;344:539–548. doi: 10.1056/NEJM200102223440801. [DOI] [PubMed] [Google Scholar]
- Horvath S, Baur MP. Future directions of research in statistical genetics. Stat Med. 2000;19:3337–3343. doi: 10.1002/1097-0258(20001230)19:24<3337::aid-sim828>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Johnson RA, Wichern DW. Applied multivariate statistical analysis. Englewood Cliffs, NJ: Prentice-Hall, Inc.; 1982. [Google Scholar]
- Lipshulz RJ, Fodor SP, Gingeras TR, Lockhart DJ. High density synthetic oligonucleotide arrays. Nat Genet. 1999;21:20–24. doi: 10.1038/4447. [DOI] [PubMed] [Google Scholar]
- Lockhart DJ, Winzeler EA. Genomics, gene expression and DNA arrays. Nature. 2000;405:827–836. doi: 10.1038/35015701. [DOI] [PubMed] [Google Scholar]
- Moler EJ, Chow ML, Mian IS. Analysis of molecular profile data using generative and discriminative methods. Physiol Genomics. 2000;4:109–126. doi: 10.1152/physiolgenomics.2000.4.2.109. [DOI] [PubMed] [Google Scholar]
- Mulshine JL. Reducing lung cancer risk: Early detection. Chest. 1999;116(Suppl):493S–496S. doi: 10.1378/chest.116.suppl_3.493s. [DOI] [PubMed] [Google Scholar]
- Niculescu AB, III, Segal DS, Kuczenski R, Barrett T, Hauger RL, Kelsoe JR. Identifying a series of candidate genes for mania and psychosis: A convergent functional genomics approach. Physiol Genomics. 2000;4:83–91. doi: 10.1152/physiolgenomics.2000.4.1.83. [DOI] [PubMed] [Google Scholar]
- Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, et al. Molecular portraits of human breast tumors. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- Pudil P, Novovicova J. Novel methods for subset selection with respect to problem knowledge. IEEE Intelligent Systems. 1998;10:66–74. [Google Scholar]
- Pudil P, Novovicova J, Kittler J. Floating search methods in feature selection. Patt Recogn Lett. 1994;15:1119–1125. [Google Scholar]
- Rothberg BEG, Ramesh TM, Burgess CE. Integrating expression-based drug response and SNP-based pharmacogenetic strategies into a single comprehensive pharmacogenomics program. Drug Develop Res. 2000;49:54–64. [Google Scholar]
- Sahiner B, Chan HP, Petrick N, Wagner RF, Hadjiiski L. Feature selection and classifier performance in computer-aided diagnosis: The effect of finite sample size. Med Phys. 2000;27:1509–1522. doi: 10.1118/1.599017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner S, Witzmann FA. Proteomics: Applications and opportunities in preclinical drug development. Electrophoresis. 2000;21:2099–2104. doi: 10.1002/1522-2683(20000601)21:11<2099::AID-ELPS2099>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Suykens JAK, Vandewalle J. Least squares support vector machine classifiers. Neural Processing Lett. 1999;9:293–300. [Google Scholar]
- Tamayo P, Slonim D, Mesirov J, Zhu Q, Kitareewan S, Dmitrovsky E, Lander ES, Golub TR. Interpreting patterns of gene expressions with self-organizing maps: Methods and application to hematopoietic differentiation. Proc Natl Acad Sci. 1999;96:2907–2912. doi: 10.1073/pnas.96.6.2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavazoie S, Hughes JD, Campbell MJ, Cho RJ, Church GM. Systematic determination of genetic network architecture. Nat Genet. 1999;22:281–285. doi: 10.1038/10343. [DOI] [PubMed] [Google Scholar]
- Welsh JB, Zarrinkar PP, Sapinoso LM, Kern SG, Behling CA, Monk BJ, Lockhart DJ, Burger RA, Hampton GM. Analysis of gene expression profiles in normal and neoplastic ovarian tissue samples identifies candidate molecular markers of epithelial ovarian cancer. Proc Natl Acad Sci. 2001;98:1176–1181. doi: 10.1073/pnas.98.3.1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong MM, Jin L, Li W, Boerwinkle E. Tumor classification using gene expression profiles. Biotechniques. 2000;29:1264–1270. doi: 10.2144/00296bc02. [DOI] [PubMed] [Google Scholar]
- Young RA. Biomedical discovery with DNA arrays. Cell. 2000;102:9–15. doi: 10.1016/s0092-8674(00)00005-2. [DOI] [PubMed] [Google Scholar]