

Abstract
Given that cardiovascular safety liabilities remain a major cause of drug attrition during preclinical and clinical development, adverse drug reactions, and post-approval withdrawal of medicines, the Medical Research Council Centre for Drug Safety Science hosted a workshop to discuss current challenges in determining, understanding and addressing ‘Cardiovascular Toxicity of Medicines’. This article summarizes the key discussions from the workshop that aimed to address three major questions: (i) what are the key cardiovascular safety liabilities in drug discovery, drug development and clinical practice? (ii) how good are preclinical and clinical strategies for detecting cardiovascular liabilities? and (iii) do we have a mechanistic understanding of these liabilities? It was concluded that in order to understand, address and ultimately reduce cardiovascular safety liabilities of new therapeutic agents there is an urgent need to:
Fully characterize the incidence, prevalence and impact of drug-induced cardiovascular issues at all stages of the drug development process.
Ascertain the predictive value of existing non-clinical models and assays towards the clinical outcome.
Understand the mechanistic basis of cardiovascular liabilities; by addressing areas where it is currently not possible to predict clinical outcome based on preclinical safety data.
Provide scientists in all disciplines with additional skills to enable them to better integrate preclinical and clinical data and to better understand the biological and clinical significance of observed changes.
Develop more appropriate, highly relevant and predictive tools and assays to identify and wherever feasible to eliminate cardiovascular safety liabilities from molecules and wherever appropriate to develop clinically relevant and reliable safety biomarkers.
Keywords: cardiovascular safety liabilities, medicines, adverse drug reaction, adverse event, patient safety, drug attrition
Introduction
Pharmaceutical industry surveys have revealed that over the last decade the number of new medicines being launched has fallen sharply despite significant investments in Research and Development (Munos, 2009). Over the same period, non-clinical and clinical safety has remained a major cause of drug attrition. Such attrition can occur during preclinical or clinical development and post-approval stage, resulting in withdrawal of marketed drugs accounting for approximately one-third of all drug discontinuation (Figure 1; Kennedy, 1997; Lasser et al., 2002; Kola and Landis, 2004; Shah, 2006; Redfern et al., 2010). A recent literature review of the reasons for drug attrition in non-clinical and clinical development, serious adverse drug reactions (ADRs) and withdrawal from the market place revealed that cardiovascular toxicity occurred more frequently than hepatotoxicity (Figure 2; Redfern et al., 2010). The data indicate that Phase I clinical trials are very safe at least from a cardiovascular point of view; this may reflect the effective preclinical testing and elimination of high-risk cardiovascular safety liabilities prior to entering clinical development. More worrying is the identification of more subtle, but high-risk, cardiovascular events either not detected in earlier clinical trials or not deemed to be biologically significant or clinically meaningful that emerge when drugs are administered for longer periods of time to larger patient populations (Figure 2; Lewington et al., 2002; Joy and Hegele, 2008; Paul et al., 2010; Redfern et al., 2010). Such high incidence and/or severity of cardiovascular ADRs in late-stage clinical development can lead to prescribing restrictions, additional pre- and/or post-approval monitoring, dose limiting toxicity, or ultimately drug discontinuation or withdrawal. Interestingly, the hepatic and cardiovascular toxicity profiles are somehow different. The incidence of hepatic ADRs is low and consistently lower than cardiovascular ADRs; this may reflect our ability to detect and discard drugs associated with hepatic more so than cardiovascular type A-ADRs during preclinical development. However, the high incidence of liver-related attrition observed in late clinical development or post-approval might be indicative of idiosyncratic reactions, not identified during preclinical or early clinical development. The difference in incidence of cardiovascular-related attrition post-approval noted between Fung et al. (2001) and Stevens and Baker (2009) may reflect the increased cardiovascular attrition related to arrhythmias over the last ∼15 years. Because of these unanticipated cardiovascular complications at the population scale, cardiovascular safety is of paramount importance in contemporary drug development. Cardiovascular safety liabilities are observed with both cardiovascular and non-cardiovascular pharmaceuticals and affect all components of the cardiovascular system, namely, the heart, blood vessels and blood constituents; in addition, the key role of the nervous and renal systems in modulating cardiovascular function should not be neglected. Cardiovascular side effects can occur after acute (i.e. single dose administration) or chronic treatment and can be functional and/or structural (i.e. histopathology) in nature. In an era marked by increased public scrutiny, escalating industry costs and finite resources at regulatory agencies, the need for efficient yet safe drug development is more important than ever.
Figure 1.
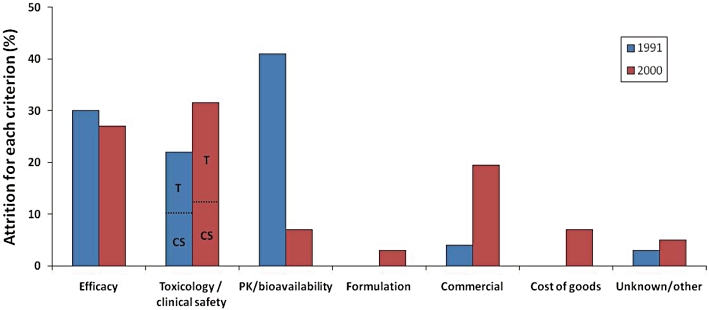
Reasons for drug attrition in 1991 and 2000. PK, pharmacokinetics; T, toxicology; CS, clinical safety. Modified from Kola and Landis (2004). Over the 10 year period safety, combining non-clinical toxicology and clinical, has remained a major cause of drug attrition accounting for approximately 30% of all drug discontinuation; during the same period attrition due to pharmacokinetics and bioavailability reasons has been reduced significantly, probably reflecting in part the effort placed on front-loading Drug Metabolism and Pharmacokinetics activities in the early discovery phases.
Figure 2.
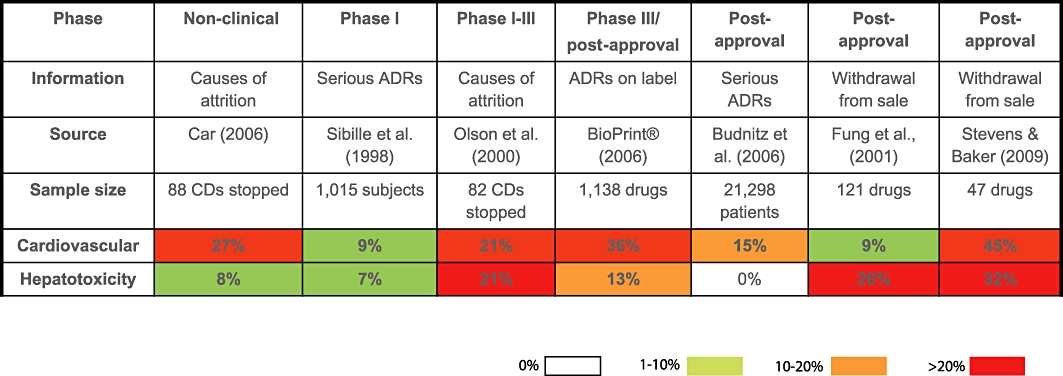
Evidence, prevalence and occurrence of safety liabilities relating to the cardiovascular and hepatic systems. Information was collated from published articles (Sibille et al., 1998; Olson et al., 2000; Fung et al., 2001; Budnitz et al., 2006; Car, 2006; Stevens and Baker, 2009) and from the commercially available database BioPrint® (Krejsa et al., 2003) to provide a guide to the prevalence of adverse drug reactions (ADRs) and their impact in terms of attrition, across the cardiovascular and hepatic systems. Some datasets relate to frequency (i.e. percentage) of candidate drugs or marketed drugs associated with the toxicity; others contain data on prevalence of the ADRs in volunteer subjects or patients. Cardiovascular data do not include haematological related attrition, withdrawal or ADRs. The data were collected over different time periods, so that there is no analysis of trends over time. Attrition has a greater financial impact than ADRs per se, and the further advanced a candidate drug (CD) or drug is in clinical development, the greater the financial impact. Therefore, cardiovascular toxicity has a greater impact than hepatotoxicity in terms of its contribution to drugs withdrawn from sale and CD attrition during clinical preclinical or clinical development. Some of the adverse events (AEs) contributing to the data are functional in nature, and so would be predictable from primary, secondary or safety pharmacology studies, whereas others are pathological in nature, so would be predictable from toxicology studies (histopathological end-points). Some AEs might be indicative of idiosyncratic reactions not identified during preclinical or clinical development (e.g. hepatotoxicity-related attrition observed in late clinical development or post-approval). The difference in incidence of cardiovascular-related attrition post-approval noted between Fung et al. (2001) and Stevens and Baker (2009) may reflect the increased cardiovascular-related attrition over the last ∼15 years particularly in relation to arrhythmias. Modify from Redfern et al. (2010).
Cardiovascular safety liability is seen by many to be an important area where non-competitive collaboration would bring benefits to all parties involved in terms of improving patient safety, increasing the numbers of new medicines registered and reducing drug development times and costs. On 28 January 2010, a workshop was hosted by the Medical Research Council (MRC) Centre for Drug Safety Science (CDSS; http://www.liv.ac.uk/drug-safety), University of Liverpool, in conjunction with the Association of the British Pharmaceutical Industry (ABPI) and the Medicines and Healthcare products Regulatory Agency (MHRA). It discussed current challenges in determining and understanding ‘Cardiovascular Toxicity of Medicines’. The MRC CDSS is a non-profit organization that brings together academic, industry and government scientists in the collaborative identification and resolution of emerging issues in drug safety (Park, 2008). The key aims of the workshop were to identify those areas of cardiovascular safety testing where our knowledge and understanding should be further strengthened and to recommend areas as to where collaborative efforts should be focused on. The workshop was attended by representatives from pharmaceutical companies, contract research organizations, regulatory agencies, world-leading cardiologists, oncologists and academics. Besides contributing to a range of stimulating presentations and discussion sessions, attendees were asked to provide information on our current understanding of cardiovascular toxicity (summarized in Tables 1–3) to allow a gap analysis to be performed. This analysis attempts to address three major areas:
Table 1.
What are the key cardiac liabilities?
| Tissue | Toxicity | Drug discovery | Drug development | Clinical practice | Drug class/disease specific |
|---|---|---|---|---|---|
| Heart | Proarrhythmic potential | hERG screening strategies are in place in most pharmaceutical companies leading to a reduced number of QT prolonging drugs in development pipelines. Primary focus on hERG/QT. Screening other ion channels (e.g. NaV1.5 and KCNQ1/KCNE1) and profiling in cardiac tissue is also being undertaken (Gallacher et al., 2007; Pugsley et al., 2009; Pollard et al., 2010). | QT prolongation is a developability issue as opposed to a reliable predictor of TdP risk. On occasion other arrhythmias mediated through non-hERG mechanisms, for example, conduction slowing. hERG trafficking effects or non-hERG-related issues are found in telemetry studies; this can be a big issue when detected late, for example, trafficking effects that show up only after prolonged exposure. QT shortening associated type arrhythmia is an emerging issue (Pollard et al., 2010; Shah, 2010; Valentin et al., 2010). | QT determination and prediction on likely outcome of TQT study. If interval signals have been detected this may translate to rhythm changes in patient groups. Better understanding of this translation is required (Darpo, 2010; Wallis, 2010). In addition to QT/TdP ADRs the ECG provides valuable parameters reporting other ECG/cardiac disorders. (Gussak et al., 2004). | Class specific tends to be dealt with in drug discovery as in some cases it is chemically related (hERG) or target related (e.g. ion channel projects for disease modulation). The problem is exacerbated by certain diseases, for example, diabetes, pre-existing heart conditions. |
| Myocardial ischaemia | Rare event. Currently detected via ECG morphology changes (Detweiler, 1983) and sometimes radionuclide scanning. | Rare clinical event. Detected through clinical signs | The consequences of ischaemia (angina pectoris and myocardial infarction) may only be detected through post-marketing observations. | Exacerbated in ‘at-risk patients’ with pre-existing disease. | |
| Myocardial necrosis | Quite frequent finding. Identified in standard histological examination as part of early repeat dose tox studies. There are currently few predictive in vitro methods (Partridge et al., 2005). Standard and non-standard histological examination (LM, EM). | Can be monitored via troponin and other biomarkers. Potential for imaging (e.g. echocardiography) to follow functional consequence of CV damage. Radionuclide markers can also be used. This method is translatable (Hanton et al., 2008). | Unlikely to be detected in clinical practice – dependent on post-marketing observations (Borer et al., 2007). Require biomarkers that predict rather than report damage (e.g. troponin). Imaging provides translational bridge from preclinical observations. | Some examples with characteristic haemodynamic profiles (e.g. increased heart work). Some non-specific mechanisms that are diffuse throughout heart and more likely due to direct cell tox as opposed to a response to functional/haemodynamic change. | |
| Heart failure | Rare event. Heart weight is used as a crude index during pathology. There may be functional end-points (e.g. contractility) that flag risk of heart failure. | Rare clinical event. Usually detected through clinical signs. Potential to include imaging (e.g. ultrasound and echocardiography) to assess heart failure markers – however, this is currently a non-standard assessment. | Unlikely to be detected in clinical practice – dependent on post-approval observations Imaging can be used if flagged from non-clinical work. | nda | |
| Coronary artery disorders | Rare event. | Rare clinical event. Detected through clinical signs. | Clinical consequences are currently only detected at the post-approval stage. | Dependent on ‘at-risk patients’ with pre-existing disease. | |
| Cardiac valve disorders | Rare event. 5-HT2B agonism has been implicated in valvulopathy (proliferative type) seen with norfenfluramine (metabolite of fenfluramine ‘Fen-Phen’), pergolide and others (Roth, 2007). Alerts may come from receptor binding data (e.g. pergolide-like pattern). | Imaging can be used to follow progression and impact of valve damage on valve function. Potential to use echocardiography but rarely included in preclinical studies (Hanton et al., 2008). Preclinical assessment relies on careful histopathological examination of cardiac valves. | Imaging and echocardiography can be used. | 5-HT2B link and associated QSAR. Migraine prophylaxis, Parkinson (dopamine agonists) appetite suppressant and, hyperlipidaemic disorders have all been linked. | |
| Endocardial disorders | Although rare, examples do exist (Belhani et al., 2006). Identification of endocardial disorders is not part of standard assessment. Detected during pathology if present (Chekmarev et al., 2008). | May be detected during histopathological assessment at end of repeat dose tox studies. | nda | nda | |
| Vessel | Hypo/hypertension | Common findings in non-clinical studies. Receptors involved in vascular control routinely included in secondary pharmacology screens (Whitebread et al., 2005). BP screening in telemetered rats is a frequently used approach. Isolated vessel assessment can be performed. Often lack an understanding on whether BP effect is due to change in cardiac output or peripheral vascular resistance (Taylor et al., 2007). One issue for studies is that in early phases there is seldom enough test material to conduct experiments in larger animals. | Quite common and detectable in first in human studies. Question of assay sensitivity to detect small changes that may have a long-term consequence. Require repeat dose assessment of BP risk and to understand relationship to clinical stopping criteria and clinical symptoms (e.g. syncope for hypotension or increased CV events. BP measurements are a standard part of the ICH S7 compliant safety pharmacology studies in non-rodents. New technology may soon allow measurement of BP also in toxicity studies (JET technology, i.e. implanting a monitor in the femoral artery). Some regulators see even small increases of BP as problematic, which will make accurate BP measurements an important part of preclinical safety studies. | Postural hypotension can be detected symptomatically (and investigated with tilt-table testing clinically), but hypertension may not. Powering of clinical studies important to detect BP signal. Single dose and repeated dose Phase I studies tend to identify trends. Combining preclinical and clinical BP data in PKPD models can be beneficial. The key challenge faced is the translation from non-clinical studies to man and also defining the magnitude of effect that is a concern. | Impact in patients with pre-existing disease, for example, drug-induced increased in BP in patients with high BP and CV risk factors. Class effects are known, although for novel targets some upfront work understand potential involvement of target in CV function may be necessary (requires tool compounds). Impact of BP effect in patient populations is currently not well incorporated in risk management plans. |
| Vascular disorders | nda | nda | nda | nda | |
| Arteriosclerosis/stenosis | Overt drug-induced changes are very rare. However, potential for indirect risk, for example, change in plasma lipid levels. Platelet function can be assessed for cause. | Potential to use imaging techniques | nda | nda | |
| Vascular insufficiency | nda | nda | nda | nda | |
| Necrosis | Identified in standard histological examination as part of early repeat dose tox studies. | Identified in standard histological examination as part of early repeat dose tox studies. | nda | nda | |
| Vascular inflammation | Vasculitis is a relatively common finding preclinically but difficult to manage going forward. The issue is clinical relevance (Brott et al., 2005; Louden et al., 2006). | Rare clinical event that probably has a distinct pathology from drug-induced injury in non-clinical species. | nda | nda | |
| Blood | Embolism and thrombosis | Quite rare findings non-clinically | May be detected in the clinic via coagulation times, blood/platelet aggregation, etc. | nda | nda |
| Vascular haemorrhagic disorders | nda | nda | nda | nda | |
| Coagulation | nda | nda | nda | nda | |
| Aggregation | nda | nda | nda | nda |
hERG, human ether-a-go-go-related gene; TdP, Torsades de Pointes; ADR, adverse drug reaction; ECG, electrocardiogram; nda, no data available; BP, blood pressure; PK, pharmacokinetic; PD, pharmacodynamic; ICH, International Conference on Harmonization; CV, cardiovascular; QSAR, quantitative structure-activity relationship.
Table 3.
Do we have a mechanistic understanding of the liabilities?
| Tissue | Toxicity | Do we have a mechanistic understanding of the toxicity? |
|---|---|---|
| Heart | Proarrhythmic potential | For some but not all arrhythmias our knowledge is limited to QT and not to how arrhythmias translate to TdP. |
| Broad profiling of drugs against cardiac ion channels, action potential morphology on isolated tissues (e.g. Purkinje fibre, isolated hearts) and also ECG morphology are all available methods. | ||
| Have not been successful at converting mechanistic understanding in to new, regulatory accepted, biomarkers. | ||
| The contribution of genetics to the problem must also be considered. | ||
| Often when compound development is stopped due to proarrhythmic potential the exact mechanism is not completely known. | ||
| Myocardial ischaemia | Ischaemia can occur from increased oxygen demand and/or reduced supply and both of these have been studied non-clinically, for example, changes in vascular tone/coronary blood flow as well as cardiac function (heart rate and contractility). | |
| Myocardial necrosis | Knowledge of mechanisms is limited. | |
| Heart failure | Generally no, however, effects on cardiac contractility can be measured, but understanding the mechanisms that can change contractility is limited at the cellular level. | |
| Coronary artery disorders | Pharmacological mechanisms that can cause coronary artery constriction are well defined although the effects on endothelial function are less clear. | |
| Cardiac valve disorders | Only in some rare cases do we have an understanding of mechanisms but in general we have a poor understanding of drug effects on valve function. | |
| Pericardial disorders | Some inflammatory mechanisms can cause pericardial damage. | |
| Endocardial disorders | Some inflammatory mechanisms can cause endocardial damage. | |
| Vessel | Hypo/hypertension | Classical pharmacological mechanisms that affect vascular tone are well defined. However, others are not well defined. |
| Complex multifactorial control of BP often confounds identification of a specific mechanism. Some examples where site of action understood (e.g. vascular resistance and/or cardiac output change) centrally versus peripherally mediated and the role of some receptors is understood. | ||
| Vascular disorders | Causes are not well defined. | |
| Arteriosclerosis/stenosis | Changes in lipids and other factors are defined, but impact of inflammatory mechanisms is less clear. | |
| Vascular insufficiency | Causes are not well defined. | |
| Necrosis | Causes are not well defined. | |
| Vascular inflammation | Causes are not well defined. | |
| Blood | Embolism and thrombosis | Causes are not well defined. |
| Vascular haemorrhagic disorders | Causes are not well defined. | |
| Coagulation | Causes are not well defined. | |
| Aggregation | Causes are well defined. |
TdP, Torsades de Pointes; ECG, electrocardiogram; BP, blood pressure.
What are the key cardiovascular safety liabilities in drug discovery, drug development and clinical practice?
How good are preclinical and clinical strategies for detecting cardiovascular liabilities?
Do we have a mechanistic understanding of these liabilities?
Information was gathered using the headings defined by the Food and Drug Administration's (FDA) Adverse Event Reporting System (AERS) for cardiovascular-related adverse events (AEs; Anon, 2010a; Figure 3). The data represent the cumulative number of cardiac (top panel) or vascular (bottom panel) AEs reported over the last 40 years; over that period there has been a large number of cardiac and vascular AEs reported. Within each category, the AEs are ranked by decreasing order of incidence. As shown in Figure 3, there are six main AE categories of cardiac and vascular side effects for which over 10 000 reports are available. The very large number of AERS reports related to arrhythmias is likely to reflect the increased scrutiny around drug-induced QT prolongation and associated arrhythmias over the last decade. The highest incidence of AEs in each category (i.e. the top six categories) have been considered as part of the questionnaire and analysis conducted prior to and during the workshop. Although such AEs data should be taken with caution, as a causal relationship between an AE and a medicine is not always demonstrated, they do provide some indication on where to focus our efforts in order to reduce cardiovascular-related safety liabilities. This publication incorporates the key issues highlighted during the workshop along with the gap analysis and identifies key areas where a concerted effort could make a real difference by reducing cardiovascular liabilities of new medicines.
Figure 3.
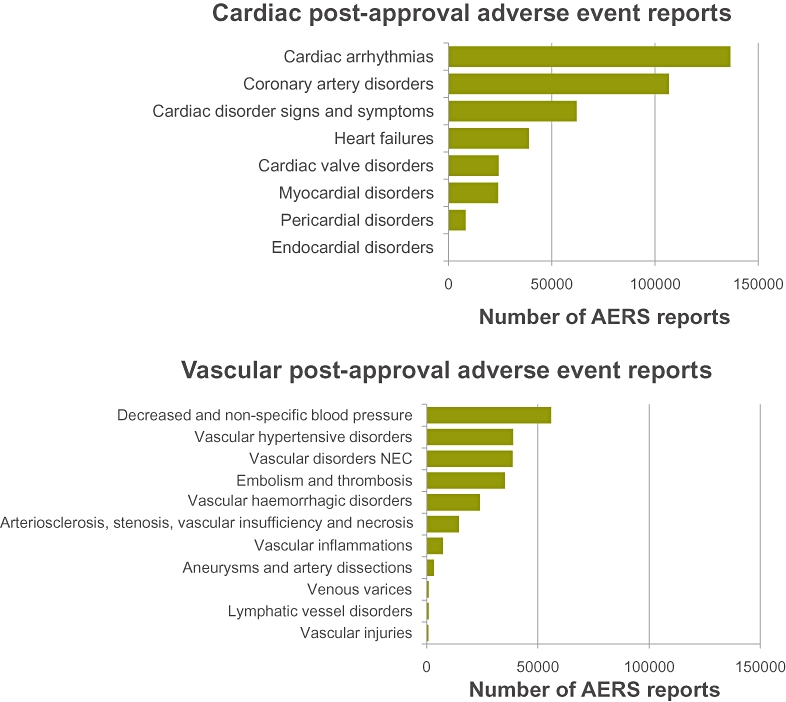
Cumulative cardiac and vascular adverse events (AEs) reported to the US Food and Drug Administration Adverse Event Reporting System (AERS) since 1969; the documents compiled in PharmaPendium (https://www.pharmapendium.com) refer to the Spontaneous Reporting System prior to 2000 and to the AERS from 2000 onwards, so the reports prior to that are more sporadic and less detailed. The annual number of AERS reports that exist in PharmaPendium has more than doubled over the last decade from a value of ∼200 000 in 2000. The data represent the cumulative number of cardiac (top panel) or vascular (bottom panel) AEs reported over that period of time. Within each category, the AEs are ranked by decreasing order of incidence. Such AEs data should be taken with caution, as the linkages with drugs are not always demonstrated. Overall there are six main AE categories of cardiac and vascular side effects, for which over 10 000 reports are available. Note the very large number of AERS reports related to arrhythmias probably reflects the increased scrutiny around drug-induced QT prolongation and Torsades de Pointes over the last decade. NEC, not elsewhere classified.
What are the key cardiovascular safety liabilities in drug discovery, drug development and clinical practice? Table 1
Using cardiovascular safety-related terms as outlined in the FDA's AERS, workshop attendees were invited to provide information on the key cardiovascular liabilities that pharmaceutical companies faced (Table 1). Information was segregated into data for the different stages of the drug development process, from discovery through to reaching market and clinical practice; information on whether drug classes had specific problems associated with them or specific diseases impacted on cardiovascular safety was also requested. The gap analysis revealed that the majority of AEs reported in the FDA's AERS are often not well described and the relationship between an AE and a drug is not necessarily established. It is worth highlighting that the frequency of serious ADRs leading eventually to discontinuation or withdrawal of a drug can be extremely low [e.g. less than 1 in 100 000 patients experienced Torsades de Pointes (TdP) with terfenadine].
Using the data summarized by Shah (2006), drug-induced TdP, a potentially fatal arrhythmia, was the reason for around one-third of all drug withdrawals between 1990 and 2006. Indeed, even though TdP is extremely rare, for all the agents withdrawn there was evidence of TdP; these drugs were associated with prolongation of the QT interval on the electrocardiogram (ECG) and block of the human ether-a-go-go-related gene (hERG) channel (Redfern et al., 2003). Shah's data in 2006 are further supported by the high number of cardiac arrhythmias-related AEs reported in the FDA's AERS (Figure 3; Anon, 2010a); although predominantly populated by QT-related arrhythmias (i.e. TdP), such dataset also includes non-QT-related arrhythmias (data not shown). Furthermore, Stevens and Baker (2009) reported a higher incidence of cardiovascular-related drug withdrawal compared with Fung et al. (2001) (45% vs. 9%, respectively); this may reflect the increased withdrawal related to arrhythmias over the last ∼15 years. Myocardial ischaemia, myocardial necrosis, heart failure and coronary artery disorders do not always appear to be highlighted during drug development, rather they are reported at the post-approval stage indicating that current preclinical assays and clinical development studies are failing to capture all these liabilities. Whereas myocardial necrosis is sometimes observed in development and is relatively well understood, for some drugs myocardial necrosis is not detected during development, only for it to be reported at the post-approval stage. The detection of myocardial necrosis in development would be improved by employing biomarkers that ideally would predict the onset of myocardial necrosis, as the currently accepted marker reports damage that has already occurred (Baubuin and Jaffee, 2005; Reinhold et al., 2010; Thygesen et al., 2010; Tijsen et al., 2010). Analysis of the data collected suggests that cardiac valve, pericardial and endocardial disorders, as well as disorders affecting blood components (e.g. embolism and thrombosis, vascular haemorrhagic disorders, coagulation and aggregation; see Tables 1–3) are rare events with few instances reported during drug development; however, these serious AEs are reported post-approval.
The information gathered indicates that at all stages of the drug development pipeline compounds that cause hypo- or hypertension and tachy- or bradycardia, can be detected and these data used to manage risk during subsequent development. It is becoming increasingly recognized that small cardiovascular changes may be very relevant to longer-term clinical outcomes. For example, it has been suggested that increases in blood pressure (BP) as small as 2 mmHg may be associated with increased morbidity and mortality (Lewington et al., 2002), although this view is not shared by all especially as convincing clinical studies are not readily available. However, more recent studies suggest that patients experiencing small, chronic drug-induced increases in BP (5 mmHg) or heart rate (5 beats·min−1) have a 6–23% greater risk of suffering heart failure, cardiac ischaemia or cerebral stroke events (Paul et al., 2010). Interestingly, preclinical studies conducted prior to first in human trials may only be powered to identify relatively large changes in a given parameter (Guth et al., 2009). The true meaning of any observed change is often only understood when a sufficiently large number of patients have been exposed to a given drug often for a prolonged length of time. It is possible that even small changes in these parameters result in the impairment of cardiovascular function and/or loss of cardiovascular homeostasis with the potential to damage cardiovascular organ systems as well as other organs (e.g. kidney), precipitating increases in cardiovascular-related mortality and morbidity (Lewington et al., 2002; Valentin et al., 2009b; Chalmers and Arima, 2010). Likewise, vascular inflammation and injury is often reported in preclinical models (Brott et al., 2005; Louden et al., 2006), while the clinical relevance and consequence of these observations remains unclear, as compounds with a drug-induced vascular injury liability have been successfully developed (e.g. potassium channel openers; phosphodiesterase inhibitors; endothelin receptor antagonists). Surprisingly for many vascular disorders reported preclinically (e.g. arteriosclerosis, vascular insufficiency and vascular necrosis); there remains a lack of data to confidently translate from preclinical observations to man (Valentin et al., 2009a,b).
How good are preclinical and clinical strategies for detecting cardiovascular liabilities? Table 2
Table 2.
How good are preclinical and clinical strategies for detecting cardiovascular liabilities?
| How effective are current strategies at detecting and dealing with identified toxicities? | How effective are current biomarkers (e.g. diagnosis, prognosis, etc.) | ||||
|---|---|---|---|---|---|
| Tissue | Toxicity | Preclinically | Clinically | Preclinically | Clinically |
| Heart | Proarrhythmic potential | The QT focus has improved ECG monitoring in non-clinical safety studies and so we have a higher chance of detecting rhythm disturbance non-clinically. In addition, ECG intervals are better quantified and therefore any changes can be used as an early marker of potential arrhythmias. Non-hERG – improving understanding and development of appropriate strategies. | With improved ECG monitoring for QT reasons, there is a higher chance of detecting rhythm disturbance in healthy volunteers. Non-hERG – improving understanding and development of appropriate strategies. However, the TQT study may be too restrictive. It may be important to monitor factors other than repolarization prolongation (Bass et al., 2007; Gallacher et al., 2007). | ECG interval changes are essentially biomarkers for arrhythmias. However, the magnitudes of ECG intervals changes that are a cause for clinical concern are not fully defined. hERG and QT are good markers for flagging development risk, but not so effective in defining TdP risk. New biomarkers predicting TdP risk are required as are new tools to integrate preclinical and clinical data to address need for TQT. The importance of the contribution of genetics to drug-induced QT/TdP problems should be considered. | ECG interval changes are essentially biomarkers for arrhythmias. However, the magnitude of change that would be a concern and is predictive for arrhythmias is not defined. hERG/QT – good for flagging development risk, not so effective for TdP risk. Not ideal but the best tool we currently have for assessment. |
| Myocardial ischaemia | Unlikely that ischaemic events would be detected unless such changes cause T-wave changes and overt histological damage. | Improved ECG monitoring would detect T-wave changes. However, more subtle effects, not relevant to healthy volunteers may be missed, but these may be relevant to certain patient populations, for example, tegaserod. | ECG morphology change would be a biomarker in addition to troponin. ST segment is a potential translatable biomarker but currently has limited preclinical validation. | ECG morphology change could be a biomarker in addition to troponin. | |
| Myocardial necrosis | Can be detected in toxicology studies and also through use of biomarkers, for example, troponin (current gold standard). Histopathology does not predict risk, it simply reports outcome. Need progressive approach from in vitro models. Molecular imaging of necrosis and apoptosis should be considered (Chun et al., 2008). | Can be monitored via troponin and other biomarkers. | Troponin is used as a biomarker, but cTn reports damage, need predictive biomarker. Imaging could be used but not routinely used preclinically. | Troponin is used as a biomarker. Although not routinely used molecular imaging of necrosis and apotosis is available in expert clinical centres. | |
| Heart failure | Can be detected via monitoring LV function and cardiac contractility in animals (routine in some companies, but not all). Otherwise dependent on monitoring the impact of any changes in cardiac output via effects on BP. | In the absence of non-clinical signals detection would be dependent on clinical signs. | Measurement of left ventricular function and ejection fraction as well as using echocardiography and MRI. | Left ventricular function and ejection fraction as well as echocardiography. | |
| Coronary artery disorders | Unlikely that ischaemic events would be detected unless such changes cause T-wave changes and overt histological damage. Techniques are available to investigate drug effects on coronary blood flow, but these are not routinely used unless other signals are detected. | Improved ECG monitoring would detect T-wave changes. However, more subtly effects, not relevant to healthy volunteers may be missed, but these may be relevant to certain patient populations, for example, tegaserod. | No obvious biomarkers. Coronary diameter and flow can be measured in animals. | No obvious biomarkers. Measure coronary artery diameter. | |
| Cardiac valve disorders | Structural changes can be detected in toxicology studies. However, the experience with Fen-Phen demonstrates that this can be difficult. Histopathology and 5HT2B screening detects problematic compounds. | nda | Echocardiography can be used to detect functional changes in valve function. However, it is a rare event normally detected by histopathology. | Echocardiography can be used to detect functional changes in valve function. But this may require repetitive echos at the time of treatment to determine the precise cause of the problem. | |
| Pericardial disorders | Structural changes can be detected in toxicology studies. | nda | nda | nda | |
| Endocardial disorders | Structural changes can be detected in toxicology studies. | nda | nda | nda | |
| Vessel | Hypo/hypertension | Good systems are in place to detect effects that may impact the safety of first in human studies. However, there are questions related to the sensitivity of the assays to detect ‘small effects’ that may be relevant to longer-term toleration and risk. Tends to detect BP risk, but not always in the right direction as clinical. Acute assessment routinely performed, repeat dosing to determine cause – may be missing chronic BP effects. | BP and HR can be easily monitored through clinical development. However, the ability to detect and respond to small changes that may be clinically relevant is variable. | BP is translatable biomarker. Uncertainty exists in terms of sensitivity and magnitude of change related to clinical concern (e.g. hypertension and CV events). | nda |
| Vascular disorders | nda | nda | nda | nda | |
| Arteriosclerosis/stenosis | Risk factors can be detected in animals, for example, lipid changes as well as any pathology changes. | Risk factors can be detected and monitored in the clinic. | nda | Blood vessel imaging. | |
| Vascular insufficiency | nda | nda | nda | nda | |
| Necrosis | Histopathology can detect necrosis. | nda | nda | nda | |
| Vascular inflammation | Identified in standard histological examination as part of early repeat dose tox studies. | Can be detected though clinical signs. | A lot of work is being done in this area to validate biomarkers, although none has been universally accepted. | A lot of work is being done in this area to validate biomarkers, although none has been universally accepted. | |
| Blood | Embolism and thrombosis | Potential to detect effects in pathology. | Can be detected though clinical signs. | Investigate effects on clotting times and coagulation pathway. | Investigate effects on clotting times and coagulation pathway. |
| Vascular haemorrhagic disorders | Potential to detect effects in pathology. | Can be detected though clinical signs. | nda | nda | |
| Coagulation | Can study drug effects on clotting times and coagulation pathway. | Can study drug effects on clotting times and coagulation pathway. | Can study drug effects on clotting times and coagulation pathway. | Can study drug effects on clotting times and coagulation pathway. | |
| Aggregation | Drug effects on platelet function can be studied non-clinically. | Drug effects on platelet function can be studied clinically. | Drug effects on platelet function can be studied non-clinically. | Drug effects on platelet function can be studied clinically. | |
ECG, electrocardiogram; hERG, human ether-a-go-go-related gene; TdP, Torsades de Pointes; BP, blood pressure; nda, no data available; MRI, magnetic resonance imaging; HR, heart rate.
Information was gathered on our understanding of the issues that had been highlighted in Table 1 and whether we have the tools to be able to identify all cardiovascular liabilities (summarized in Table 2). Data requested addressed the effectiveness of current strategies at detecting and dealing with identified adverse effects clinically and/or preclinically, and whether we have effective preclinical and clinical biomarkers to allow for diagnosis and prognosis of cardiovascular liabilities (Valentin et al., 2009a,b). It should be noted that preclinical safety evaluation is almost always conducted in young healthy animals that do not carry the pathophysiological background underpinning disease conditions, and a key question is whether these preclinical models accurately reflect the patient population that will be exposed to a drug.
Although the mechanisms underpinning drug-induced TdP are far from being fully understood, over the last 15 years a significant scientific understanding has been gained into the molecular mechanisms and predisposing factors of drug-induced QT prolongation. So despite an imperfect biomarker of drug-induced TdP, QT prolongation is accepted as a surrogate marker of TdP. Indeed all the drugs that do induced TdP are associated with prolongation of the QT interval (Redfern et al., 2003). Industry, academia and regulatory agencies have worked closely together in developing a rigorous QT testing paradigm (Pugsley et al., 2008; 2009; Vik et al., 2008; Pollard et al., 2010; Valentin, 2010; Valentin et al., 2010; Wallis, 2010). Genetic and pharmacological evidence highlighting the pivotal role of hERG was a critical step in understanding how to start addressing this issue. It led to the development of hERG assays with the rapid throughput needed for the short timescales required in early drug discovery. The resulting volume of hERG data has fostered in silico models to help chemists design compounds with reduced hERG potency. In early drug discovery, a pragmatic approach based on exceeding a given potency value has been required to decide when a compound is likely to carry a low QT risk, to support its progression to late-stage discovery. At this point, the in vivo efficacy and metabolism characteristics of the potential drug are generally defined, as well as its safety profile, which includes usually a dog study to assess QT interval prolongation risk. The hERG and in vivo QT data, combined with the likely indication and the estimated free drug level for efficacy, are put together to assess the risk that the potential drug will prolong QT in man. Further data may be required to refine the risk assessment before making the major investment decisions for full development (e.g. assessment of the proarrhythmic potential per se). The non-clinical data are essential to inform decisions about compound progression and to optimize the design of clinical QT studies; a negative clinical ‘thorough QT study’ provides reassurance regarding the torsadogenic potential of a drug. Emerging data indicate that preclinical QT-related assays are overall good predictors of the clinical outcome (Wallis, 2010). However, even with this well-defined system, there are substantial gaps in our knowledge in terms of understanding the genesis of TdP and non-TdP arrhythmia in preclinical and clinical setting and their relationship to changes in the QT interval and other ECG changes (Hoffmann and Warner, 2006; Pollard et al., 2010; Shah, 2010; Valentin et al., 2010).
The picture is much less clear with regard to other AEs such as myocardial ischaemia and myocardial necrosis. In the case of ischaemia it is unlikely to be detected unless there are changes to the ST wave of the ECG or overt histological damage. In the case of myocardial necrosis, troponin release into the serum can be used as a biomarker of damage, but this current gold standard of histopathological assessment does not predict risk, it simply reports cardiac myocytes damage. The risk of heart failure, cardiomyopathy and coronary artery disorders can be detected by monitoring left ventricular function and coronary blood flow. Sensitive and predictive imaging technologies such as ultrasound, echocardiography and magnetic resonance imaging can be used to measure changes in heart function that might precede myocardial damage. These technologies and parameters are directly translatable to humans; however, they are not available routinely in all laboratories and therefore not consistently included in drug development programmes unless triggered by other signals (Hanton et al., 2008). Drug effects on the cardiac inotropic and lusitropic state can be monitored routinely via the invasive measurement of left ventricular function and derived parameters (i.e. LV dP/dt max & min; Pugsley et al., 2008), although such end-points are not routinely accessible in the clinic, they are assumed to be translatable to humans. The role of calcium handling in myocytes, endothelial and smooth muscle cells is still little understood but could prove to be a rich avenue for investigation (Bers, 2002). Other disorders such as cardiac valve, pericardial and endocardial disorders can be detected in toxicology studies via histopathological examination. However, the current inability to translate between species means it is often difficult to predict how these preclinical observations may manifest in patients. As has been noted previously, the acute absence of predictive biomarkers of cardiovascular side effects at all stages of drug development is again emphasized by the data collected here. Many different initiatives and much research is being devoted to the identification of novel biomarkers; however, to date none has been universally accepted either clinically or preclinically (e.g. troponin, Baubuin and Jaffee, 2005; Omland et al., 2009; Thygesen et al., 2010; miRNA, Tijsen et al., 2010; brain natriuretic peptides, Reinhold et al., 2010). An additional challenge is to determine the sensitivity and specificity of these clinical biomarkers when applied in the non-clinical setting.
In relation to BP changes, there are good preclinical methods in place that detect effects that may be useful in the design of first in human studies. BP measurements are important in terms of monitoring vital physiological function and relatively straightforward, and therefore they are routinely used clinically, providing a good example of a translatable biomarker. However, there are questions as to the sensitivity and specificity of preclinical and clinical measures of BP in current use and whether the small changes in BP can be accurately detected, and if so whether they translate into a clinically meaningful risk especially following chronic drug treatment (Lewington et al., 2002; Valentin et al., 2009a,b; Chalmers and Arima, 2010). It is also likely that this risk depends on the patient population, for example a higher risk may be anticipated in elderly patients with cardiovascular risk factors compared with young healthy individuals. Moreover, certain aspects of drug-induced cardiovascular disturbances (e.g. orthostatic hypotension, baro-reflex dys-regulation) are not routinely addressed during preclinical development. Other vascular disorders captured by preclinical testing and observations are translatable to the clinic, such as, arteriosclerosis where plasma changes in lipids can be measured in animals and changes detected in the clinic; likewise blood coagulation and aggregation are amenable to both preclinical and clinical assessment. However, some disorders can only be confirmed by histopathology such as necrosis, while others although observed preclinically, are of disputed relevance to the clinic (e.g. vascular inflammation and injury; Brott et al., 2005; Louden et al., 2006).
Do we have a mechanistic understanding of the liabilities? Table 3
Chemical liabilities
Finally, data were gathered on whether we have a mechanistic understanding of the toxicities described (Table 3). As we have discussed, the most studied effects are those that are associated with QT interval prolongation and associated proarrhythmic potential and much has been learnt regarding some of the underlying mechanisms. It has been shown that a vast majority of compounds prolong the QT interval through the inhibition of a single molecular target – the hERG-encoded potassium ion channel. This has enabled medicinal chemists to develop structure–activity relationships such that few compounds are synthesized that have high potency as hERG channel inhibitors (Gavaghan et al., 2007; Pugsley et al., 2008; Pollard et al., 2010; Valentin et al., 2010; Wallis, 2010). This was achieved by concerted efforts involving industry and academic scientists and regulators, often working closely together (Valentin et al., 2010). However, cardiovascular safety is responsible for a large number of drug withdrawals only a small proportion of which are due to arrhythmias. Much is understood regarding changes in BP and classical pharmacological mechanisms that affect vascular tone are well defined. But even when parameters such as BP are well understood the often complex multifactorial control of BP frequently confounds the identification of a specific mechanism of a drug-induced effect. This situation is exacerbated by variation in the patient population and underlying cardiovascular risk, making prediction of injury to a patient due to drug-induced BP changes extremely difficult.
From the biological information collected for the safety liabilities listed, it is clear that in most cases we understand the processes that are affected but we currently lack the understanding of why certain compounds produce a given ADR. When the chemical structures of compounds that have been withdrawn have been analysed it is clear that they show structural diversity; that is, they cover most of the ‘drug-like’ space outlined by the medicinal chemist. Only a few structural motifs have been reported that are associated with cardiovascular safety liabilities (Gavaghan et al., 2007; Aronov, 2008; Chekmarev et al., 2008; Frid and Matthews, 2010), making it very difficult for medicinal chemists to develop structure–activity relationships, although there are emerging data that suggest an association between physicochemical drug properties and in vivo or in vitro toxicological profiles (Krejsa et al., 2003; Leeson and Springthorpe, 2007; Price et al., 2009). However, in most instances, we do not understand the mechanistic basis that underlies why only some compounds produce cardiovascular safety liabilities. This combination of a lack of a chemical motif and an incomplete understanding of the biology results in compounds with a cardiovascular liability being able to progress through the drug development process before ultimately failing late in clinical development, at considerable costs to both companies and patient safety.
Emerging evidence suggests that cardiac drug disposition and/or local cardiac drug concentration might play a role in drug-induced cardiac side effects. For instance, many transporters and metabolizing enzymes are expressed in the heart; several of these transporters and P450s are associated with evidence of drug transport or metabolism in the heart. Furthermore, several drugs transported and/or metabolized by these transporters or enzymes are associated with cardiovascular toxicities (McBride et al., 2009; Reiss et al., 2009). Moreover, drug cardiac tissue concentration appeared to be a key factor in exacerbating the arrhythmogenic proclivity of drugs (Titier et al., 2004). Although current technology exists to perform tissue distribution studies, they are not routinely conducted during the early phases of drug discovery due to the need for radioactive compound but tend to be performed late in the drug discovery phases; clearly compounds that do accumulate in the heart or vasculature may deserve a closer look. To this effect, the use of novel technology such as Positron Emission Tomography might be valuable. Further research is required to understand the full impact of such observations.
Target liabilities
One emerging problem in cardiovascular safety is the increasing prevalence of toxicity from targeted cancer therapies, notably protein kinase inhibitors (Cheng and Force, 2010; Zuppinger and Suter, 2010). These synthetic small molecular weight compounds inhibit the activity of several protein kinases, either associated with neoplastic transformation of cells within the tumour, or present in the associated vascular endothelial cells of blood vessels supplying the tumour leading to angiogenesis inhibition (Holmes et al., 2007). However, these kinases are also expressed in cardiac myocytes and cardiac endothelial cells and play a crucial role in normal homeostasis within the cardiac tissue. Consequently, inhibition of these kinases within cardiac tissue due to ‘on-target toxicity’ has the potential to result in cardiovascular AEs. Furthermore, such liability can be compounded by the fact that kinase inhibitors such as sunitinib (Sutent®) and sorafenib (Nexavar®) often display broad selectivity increasing the potential for ‘off-target toxicity’ (Zuppinger and Suter, 2010). A major challenge is to understand how we predict and manage the ADRs associated with kinase inhibitors. Novel in vitro screens that can detect cardiovascular adverse effect liability with these agents at the preclinical stage are needed.
In some instances it might not be possible to remove the cardiovascular risk associated with a compound, for example for some anti-cancer agents, as the pathways that a given drug targets in the tumour are also intrinsic to the proper functioning of cardiac tissue (Cheng and Force, 2010; Zuppinger and Suter, 2010). It is a paradox of current therapy that as it has become more effective, with better survival rates with some drugs, such as anthracyclines, higher rates of heart failure are being observed 6–7 years post treatment. As healthy heart tissue can defend against stress by several different mechanisms (e.g. NO, GF, gp130, Neu/HER), the risk of cardiovascular AEs is also heightened by newer drugs that target some of these defence mechanisms (e.g. anti-HER2: trastuzumab – Herceptin®). With improved understanding of the mechanism by which drugs work and the mechanism by which toxicities arise, it is possible to manage and wherever feasible to mitigate the risk of cardiovascular side effects. For example, when concomitant treatment of chemotherapy and trastuzumab is used then high incidence (i.e. 25%) of cardiac dysfunction are observed. However if trastuzumab is used after completion of the chemotherapy course then the level of cardiac dysfunction falls (3–18%). In this instance the risk is manageable by changing clinical treatment design with a reduction in the level of AEs observed (De Keulenaer et al., 2010; Peng et al., 2010). Other treatments target cardiac tissue due to their intrinsic mechanism of action (e.g. bevacizumab – Avastin®), which binds the VEGF ligand and inhibits angiogenesis; therefore, it is difficult to envisage how risk can be avoided. A clear understanding of the benefit to risk ratio is needed for these promising new medicines and a realization that the naturally risk averse approach of our society may be denying potentially beneficial compounds from reaching the market; all those involved in development medicines may need to adopt a new approach to cardiovascular risk assessment (Borer et al., 2007).
Discussion
This document represents an important step towards the identification of the problems surrounding drug-induced cardiovascular side effects at all stages of drug discovery, clinical development and post-approval. It is clear that in some areas we have a good understanding of potential liability (e.g. drug-induced QT prolongation and associated proarrhythmic risk). However, as highlighted in this manuscript, there are other areas that we do not understand fully (e.g. tissue exposure, pharmacokinetic and pharmacodynamic relationship, metabolism, transporters, structural changes and the inter-relationship between vascular biology and cardiac structural changes due to drug treatment). The authors acknowledge that the gap analysis outlined in this manuscript is obtained from information collected from one meeting and that other data may be available from other sources to complement and expand the analysis. We recognize that this is the beginning of an ongoing process and welcome correspondence with other interested parties regarding data available for cardiovascular safety and further discussions on how to progress the field. However, it is beyond dispute that cardiovascular AEs is an area of significant need both in terms of patient safety and its impact upon drug attrition, drug withdrawal and ADRs. For the field to progress several areas were identified where action must be taken to:
Fully characterize the incidence, the prevalence and the impact of drug-induced cardiovascular issues at all stages of drug discovery, development and post-approval. This could be achieved by retrieving, sharing, analysing and interrogating cardiovascular safety-related data hold in privately own or publicly available repositories and databases.
Ascertain the predictive value of existing non-clinical models and assays towards the clinical outcome. This could be achieved by building and expanding on existing initiatives; by sharing, interrogating, analysing and interpreting complex preclinical and clinical pharmacodynamic and pharmacokinetic cardiovascular datasets.
Understand the mechanistic basis of cardiovascular liabilities; by addressing those areas where it is currently not possible to predict clinical outcome based on preclinical safety data and that are most likely to succeed and to have the highest impact on patient safety and drug attrition. Research collaborations, cooperations and consortia efforts between scientists of pharmaceutical companies, contract research organizations, non-profit organizations and academic institutions are required.
Provide scientists in all disciplines (e.g. clinician, pharmacologist, physiologist, toxicologist and pathologist) with additional skills to enable them to better integrate preclinical and clinical data and, to better understand the biological and clinical significance of any changes observed. Significant investments in cross-functional, pan disciplines training and educational programmes at national and international levels are required.
Develop more appropriate, highly relevant and predictive tools and assays to identify and wherever feasible to eliminate cardiovascular safety liabilities from molecules and wherever appropriate to develop clinically relevant and reliable safety biomarkers. This includes a better understanding of the pharmacology of drugs, their targets, pharmacokinetic and pharmacodynamic relationship, metabolism and disposition. This would require concerted efforts and investments from academia, service and technology providers as well as biopharmaceutical companies.
Currently there are several initiatives in the cardiovascular safety area as outlined in Table 4 (Bass et al., 2008; Piccini et al., 2009; Stummann et al., 2009; Cavero, 2010; Pettit et al., 2010); most of these tackle some of the issues that have been described in this manuscript but tend to focus on known liabilities that have been well described and mechanisms proposed. Other initiatives have produced positions or white papers describing aspects of cardiovascular safety that would benefit from being followed up with concrete research projects. Some offer a ‘pragmatic approach’ that will inform regulatory processes; however, what is required are approaches that discover the fundamental mechanisms of cardiovascular liability that will allow a step change in the detection and assessment of cardiovascular liability in drug discovery and development. Few if any of the current initiatives are addressing the identification and characterization of cardiovascular liabilities and the impact that they have on late-stage clinical development. Several of the initiatives are also under pressure financially or have not been progressed due to the fiscal pressures on industry and governments. So although it is clear that those in industry are aware of the issue, not enough is being done and that is why it is believed that a new initiative is required. By presenting and recognizing existing initiatives, we clearly highlight the importance of ensuring complementarity and wherever feasible additivity if not synergistic effects with existing initiatives.
Table 4.
Background information and objectives of key initiatives focusing on cardiovascular safety liabilities
| Identification | Background | Objectives | References |
|---|---|---|---|
| Animal Model Framework – Association of the British Pharmaceutical Industry (ABPI) | Cross pharmaceutical companies initiative under the auspice of the ABPI aimed at developing a conceptual framework to define and classify key models used in safety pharmacology with respect to specific end-points that are evaluated in animals and humans. | To investigate the translation of non-clinical safety pharmacology assays and models (including cardiovascular models) to the phase I clinical outcome. | Valentin JP et al., 2009a. J Pharmacol Toxicol Methods 60: 152–158. |
| Top Institute (TI) Pharma | Public–private partnership aimed at improving development of socially valuable medicines. Formed by Dutch Government, Universities and Pharmaceutical Industry. | To investigate the translation of non-clinical assays of QT prolongation to the clinic and patient outcome. Use mechanistic PK/PD modelling to predict risk from non-clinical data. Examine relationship between QT prolongation and sudden death. | Anon (2010b)http://www.tipharma.com/about-our-institute/about-ti-pharma.html |
| European Centre for the Validation of Alternative Methods (ECVAM) | To promote acceptance of alternative methods to reduce, refine or replace the use of laboratory animals. | ‘Alternative Methods for Drug-induced Cardiotoxicity’ workshop. Recommendations published. However, ECVAM no longer focusing on pharmaceuticals. | Stummann TC et al., 2009. Cardiovasc Toxicol 9: 107–125. |
| Cardiac Safety Research Consortium (CSRC) | To advance scientific knowledge on cardiac safety for new and existing medical products by building a collaborative environment based upon the principles of the FDA's Critical Path Initiative as well as other public health priorities. | Intent is to develop evaluative tools, standards, validated tests and cardiovascular biomarkers related to broader aspects of cardiac safety including, but not limited to arrhythmia, thrombosis, myocardial infarction and heart failure. | Anon (2010c)https://www.cardiac-safety.org/Piccini JP et al., 2009. Am Heart J 158: 317–326. |
| Safety Pharmacology Society (SPS) | Best practice initiative led under the auspice of Safety Pharmacology Society involving Pharmaceutical companies, CROs and Regulatory agencies | ‘Cardiovascular Best Practices Workshop’ aimed at improving the quality and consistency of cardiovascular in vivo Safety Pharmacology studies and cardiovascular end-point measurement in toxicology studies. | Cavero I, 2010. Expert Opin 10: 319–333. |
| Stem Cell for Safer Medicines (SC4SM) | Public–private collaboration, established through the vehicle of a not-for-profit company, whose mission is to enable the creation of a bank of stem cells lines. | The aim of the SC4SM programme is to develop stem cell technology for use at an early stage in safety testing, to eliminate new drug candidates, which may have side effects in humans, before animal or human trials are conducted. Stem cells may provide a route to deriving sufficient quantities of hESC-derived cardiomyocytes that can be used in high-throughput in vitro cardiac safety screens. | Anon (2010g)http://findarticles.com/p/articles/mi_hb5255/is_16/ai_n35675936/ |
| HESI – Cardiac Technical Committee | Global scientific, non-profit, organization since 1989. Focus on human and environmental risk assessment and toxicology. The Cardiac Technical Committee encompasses four sub-committees. | Cardiac Biomarkers. Focus on cardiac troponin as a marker of cardiac injury. Evaluate current experimental practice in assessment of haemostasis. Predictive Cardiovascular Safety. To identify and pursue opportunities to develop a more predictive/translational approach to cardiovascular safety evaluation. Functional Cardiovascular Toxicity Evaluation. Facilitate discussion around the use of functional measures of cardiovascular effects for safety evaluation. Proarrhythmia Models. Understand of the value of non-clinical repolarization assays for predicting drug-induced QT prolongation. | Anon (2010f)http://www.hesiglobal.org/i4a/pages/index.cfm?pageid=1Pettit SD et al., 2010. J Pharmacol Toxicol Methods 61: 1–2. |
| Critical Path institute's Predictive Safety Testing Consortium (PSTC) | PSTC is a public–private partnership established in 2006 and directed by the non-profit Critical Path Institute. | Consortium of pharmaceutical companies who share and validate each other's safety testing methods. Current working groups include vascular injury and cardiac hypertrophy. | Anon (2010d)http://www.c-path.org/pstc.cfm |
| Innovative Medicine Initiative (IMI) | Public–private partnership between the pharmaceutical industry represented by the European Federation of Pharmaceutical Industries and Associations (EFPIA) and the European Union represented by the European Commission. | Cardiovascular liabilities were originally included in the IMI JU Scientific Priorities 2010 (published on IMI website 16/03/2010): Safety Pillar: General Safety, Scientific Priority A: ‘Assessment of drug induced toxicity in relevant organs – surrogates for early drug failure’. However, it was subsequently not included in the final ‘2010 Call for proposals’ (published online 04/10/2010) that specified DILI. | Anon (2010e)http://www.imi.europa.eu/ |
Having reviewed the field and other initiatives in this area it is believed that what is currently missing is a true pharmacological approach to define the issue. We simply do not understand enough about the mechanisms that result in cardiovascular dysfunction and key to being able to address this are well-defined chemical reagents that are highly selective for the pathways important for cardiovascular liability. To achieve this the definition of a training set of compounds that will allow us to link the chemistry of the compounds to biological perturbation of the pathways is proposed. This new initiative will need to collect information from the pharmaceutical industry on compounds with associated liability and build training sets of compounds that can then be tested in current and novel models, assays and screens. It is believed that it will be too time-consuming a task to collect this information for every cardiovascular liability so it is proposed that in the first instance a small number of liabilities are selected (e.g. cardiac contractility), and the relevant information and compounds are collated. These compounds can then be tested in the same relevant test systems that have been selected by the stakeholders and the signatures of cardiovascular liabilities can be looked for.
Clearly the issue of cardiovascular safety liability in drug development is an area that would benefit from Innovative Medicines Initiative (IMI) support. Cardiovascular toxicity was announced as being one of the IMI indicative call topics earlier in 2010; however, it has not progressed to the proposal stage. The reasons for this are unclear; however, the area is ideally suited to an IMI approach as it is an emerging problem for the development of safe medicines in which both academia and industry scientists have the opportunity to synergize and innovate in tackling the problem. The authors ask that European Federation of Pharmaceutical Industries and Associations members and IMI look again at this issue. By better coordinating our activities and focusing on key drug development challenges there is an opportunity to advance our understanding of the causes of cardiovascular safety, improve the likelihood of success in drug development and ultimately improve patient safety. Organization such as the MRC CDSS and the IMI have a key role to play in fostering this non-competitive collaborative approach involving pharmaceutical companies, academic institutions and the regulators. It is proposed to host a follow-up workshop to define the liability to be selected and put in place a mechanism for the collection of the data from companies, its safe storage and interrogation followed by compound selection and testing. We would welcome all interested stakeholders to join this venture and their contribution to moving the field forward.
Acknowledgments
The authors wish to acknowledge the contribution of all participants to the ‘Cardiovascular Toxicity of Medicines’ workshop held under the auspice of MRC CDSS, the ABPI and the MHRA that took place in Liverpool on 28 January 2010. The authors wish to express their thanks to: Dr Will Redfern from AstraZeneca for collecting the data on safety-related attrition presented in Figure 2 and for creating Figure 2 and Dr Alex Harmer from AstraZeneca for extracting the cardiovascular AEs data from the AERS that are presented in Figure 3 and for creating Figure 3.
Glossary
Abbreviations
- ABPI
Association of the British Pharmaceutical Industry
- ADR
adverse drug reaction
- AE
adverse event
- AERS
Adverse Events Reporting System
- BP
blood pressure
- CD
candidate drug
- CDSS
Centre for Drug Safety Science
- FDA
Food and Drug Administration
- hERG
human ether-a-go-go-related gene
- ICH
International Conference on Harmonization
- IMI
Innovative Medicines Initiative
- MHRA
Medicines and Healthcare products Regulatory Agency
- MRC
Medical Research Council
- nda
no data available
- PK/PD
pharmacokinetic and pharmacodynamic
- TdP
Torsades de Pointes
Conflict of interest
Some authors of this paper are employed in the pharmaceutical industry or serve as consultants to the pharmaceutical industry. However, the subjects presented in the paper do not advocate or support purchase of any of the products offered by the respective organizations.
References
- Anon. Adverse Event Reporting System (AERS) – U.S. Food and Drug Administration – U.S. Department of Health & Human Services. 2010a. http://www.fda.gov/drugs/guidancecomplianceregulatoryinformation/surveillance/adversedrugeffects/default.htm (accessed on 1st July 2010)
- Anon. Top Institute Pharma. 2010b. http://www.tipharma.com/about-our-institute/about-ti-pharma.html (accessed on 22nd November 2010)
- Anon. Cardiac Safety Research Consortium. 2010c. https://www.cardiac-safety.org/ (accessed on 22nd November 2010)
- Anon. Critical Path Institute's Predictive Safety Testing Consortium. 2010d. http://www.c-path.org/pstc.cfm (accessed on 22nd November 2010)
- Anon. Innovative Medicines Initiative. 2010e. http://www.imi.europa.eu/ (accessed on 22nd November 2010)
- Anon. The Health and Environmental Sciences Institute. 2010f. http://www.hesiglobal.org/i4a/pages/index.cfm?pageid=1 (accessed on 22nd November 2010)
- Anon. Stem Cells for Safer Medicines (SC4SM) 2010g. http://findarticles.com/p/articles/mi_hb5255/is_16/ai_n35675936/ (accessed on 31th March 2011)
- Aronov AM. Ligand structural aspects of hERG channel blockade. Curr Top Med Chem. 2008;8:1113–1127. doi: 10.2174/156802608785700061. [DOI] [PubMed] [Google Scholar]
- Bass A, Valentin JP, Fossa AA, Volders PG. Points to consider emerging from a mini-workshop on cardiac safety: assessing torsades de pointes liability. J Pharmacol Toxicol Methods. 2007;56:91–94. doi: 10.1016/j.vascn.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Bass AS, Darpo B, Breidenbach A, Bruse K, Feldman HS, Garnes D, et al. International Life Sciences Institute (Health and Environmental Sciences Institute, HESI) initiative on moving towards better predictors of drug-induced torsades de pointes. Br J Pharmacol. 2008;154:1491–1501. doi: 10.1038/bjp.2008.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baubuin L, Jaffee AS. Troponin; the biomarker of choice for the detection of cardiac injury. CMAJ. 2005;173:1191–1202. doi: 10.1503/cmaj.050141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belhani D, Frassati D, Mégard R, Tsibiribi P, Bui-Xuan B, Tabib A, et al. Cardiac lesions induced by neuroleptic drugs in the rabbit. Exp Toxicol Pathol. 2006;57:207–212. doi: 10.1016/j.etp.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- Borer JS, Pouleur H, Abadie E, Follath F, Wittes J, Pfeffer MA, et al. Cardiovascular safety of drugs not intended for cardiovascular use: need for a new conceptual basis for assessment and approval. Eur Heart J. 2007;28:1904–1909. doi: 10.1093/eurheartj/ehm209. [DOI] [PubMed] [Google Scholar]
- Brott D, Jones H, Gould S, Valentin JP, Evans G, Richardson RJ, et al. Current status and future directions for diagnostic markers of drug-induced vascular injury. Cancer Biomark. 2005;1:15–28. doi: 10.3233/cbm-2005-1104. [DOI] [PubMed] [Google Scholar]
- Budnitz DS, Pollock DA, Weidenbach KN, Mendelsohn AB, Schroeder TJ, Annest JL. National surveillance of emergency department visits for outpatient adverse drug effects. JAMA. 2006;296:1858–1866. doi: 10.1001/jama.296.15.1858. [DOI] [PubMed] [Google Scholar]
- Car B. Enabling technologies in reducing drug attrition due to safety failures. Am Drug Discov. 2006;1:53–56. [Google Scholar]
- Cavero I. Cardiovascular system assessment best practices: a Safety Pharmacology Society meeting. Expert Opin Drug Saf. 2010;9:855–866. [Google Scholar]
- Chalmers J, Arima H. Importance of blood pressure lowering in type 2 diabetes: focus on ADVANCE. J Cardiovasc Pharmacol. 2010;55:340–347. doi: 10.1097/fjc.0b013e3181d26469. [DOI] [PubMed] [Google Scholar]
- Chekmarev DS, Kholodovych V, Balakin KV, Ivanenkov Y, Ekins S, Welsh WJ. Shape signatures: new descriptors for predicting cardiotoxicity in silico. Chem Res Toxicol. 2008;21:1304–1314. doi: 10.1021/tx800063r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Force T. Molecular mechanisms of cardiovascular toxicity of targeted cancer Therapeutics. Circ Res. 2010;106:21–34. doi: 10.1161/CIRCRESAHA.109.206920. [DOI] [PubMed] [Google Scholar]
- Chun HJ, Narula J, Hofstra L, Wu JC. Intracellular and extracellular targets of molecular imaging in the myocardium. Nat Clin Pract Cardiovasc Med. 2008;5(Suppl 2):S33–S41. doi: 10.1038/ncpcardio1161. [DOI] [PubMed] [Google Scholar]
- Darpo B. The thorough QT study four years after the implementation of the ICH E14 guidance. Br J Pharmacol. 2010;159:49–57. doi: 10.1111/j.1476-5381.2009.00487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Keulenaer GW, Doggen K, Lemmens K. The vulnerability of the heart as a pluricellular paracrine organ lessons from unexpected triggers of heart failure in targeted ErbB2 anticancer therapy. Circ Res. 2010;106:35–46. doi: 10.1161/CIRCRESAHA.109.205906. [DOI] [PubMed] [Google Scholar]
- Detweiler DK. Electrocardiographic monitoring in toxicological studies: principles and interpretations. Adv Exp Med Biol. 1983;161:579–607. doi: 10.1007/978-1-4684-4472-8_35. [DOI] [PubMed] [Google Scholar]
- Frid AA, Matthews EJ. Prediction of drug-related cardiac adverse effects in humans – B: use of QSAR programs for early detection of drug-induced cardiac toxicities. Regul Toxicol Pharmacol. 2010;56:276–289. doi: 10.1016/j.yrtph.2009.11.005. [DOI] [PubMed] [Google Scholar]
- Fung M, Thornton A, Mybeck K, Wu JH, Hornbuckle K, Muniz E. Evaluation of the characteristics of safety withdrawal of prescription drugs from worldwide pharmaceuticals markets – 1960 to 1999. Drug Inf J. 2001;35:293–317. [Google Scholar]
- Gallacher DJ, Van de Water A, van der Linde H, Hermans AN, Lu HR, Towart R, et al. In vivo mechanisms precipitating torsades de pointes in a canine model of drug-induced long-QT1 syndrome. Cardiovasc Res. 2007;76:247–256. doi: 10.1016/j.cardiores.2007.06.019. [DOI] [PubMed] [Google Scholar]
- Gavaghan CL, Arnby CH, Blomberg N, Strandlund G, Boyer S. Development, interpretation and temporal evaluation of a global QSAR of hERG electrophysiology screening data. J Comput Aided Mol Des. 2007;21:189–206. doi: 10.1007/s10822-006-9095-6. [DOI] [PubMed] [Google Scholar]
- Gussak I, Litwin J, Kleiman R, Grisanti S, Morganroth J. Drug-induced cardiac toxicity: emphasizing the role of electrocardiography in clinical research and drug development. J Electrocardiol. 2004;37:19–24. doi: 10.1016/j.jelectrocard.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Guth BD, Bass AS, Briscoe R, Chivers S, Markert M, Siegl PKS, et al. Comparison of electrocardiographic analysis for risk of QT interval prolongation using safety pharmacology and toxicological studies. J Pharmacol Toxicol Methods. 2009;60:107–116. doi: 10.1016/j.vascn.2009.05.006. [DOI] [PubMed] [Google Scholar]
- Hanton G, Eder V, Rochefort G, Bonnet P, Hyvelin JM. Echocardiography, a non-invasive method for the assessment of cardiac function and morphology in preclinical drug toxicology and safety pharmacology. Expert Opin Drug Metab Toxicol. 2008;4:681–696. doi: 10.1517/17425255.4.6.681. [DOI] [PubMed] [Google Scholar]
- Hoffmann P, Warner B. Are hERG channel inhibition and QT interval prolongation all there is in drug-induced torsadogenesis? A review of emerging trends. J Pharmacol Toxicol Methods. 2006;53:87–105. doi: 10.1016/j.vascn.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Holmes K, Roberts OL, Thomas AM, Cross MJ. Vascular endothelial growth factor receptor-2: structure, function, intracellular signalling and therapeutic inhibition. Cell Signal. 2007;19:2003–2012. doi: 10.1016/j.cellsig.2007.05.013. [DOI] [PubMed] [Google Scholar]
- Joy TR, Hegele RA. The failure of torcetrapib: what have we learned? Br J Pharmacol. 2008;154:1379–1381. doi: 10.1038/bjp.2008.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy T. Managing the drug discovery/development interface. Drug Discov Devel. 1997;2:436–444. [Google Scholar]
- Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov. 2004;3:711–715. doi: 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- Krejsa CM, Horvath D, Rogalski SL, Penzotti JE, Mao B, Barbosa F, et al. Predicting ADME properties and side effects: the bioprint approach. Curr Opin Drug Discov Devel. 2003;6:470–480. [PubMed] [Google Scholar]
- Lasser KE, Allen PD, Woolhandler SJ, Himmelstein DU, Wolfe SM, Bor DH. Timing of new black box warnings and withdrawals for prescription medications. JAMA. 2002;287:2215–2220. doi: 10.1001/jama.287.17.2215. [DOI] [PubMed] [Google Scholar]
- Leeson D, Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat Rev. 2007;6:881–890. doi: 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- Lewington S, Clarke R, Qizilbash N, Peto R, Collins R, Prospective Studies Collaboration Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet. 2002;360:1903–1913. doi: 10.1016/s0140-6736(02)11911-8. [DOI] [PubMed] [Google Scholar]
- Louden C, Brott D, Katein A, Kelly T, Gould S, Jones H, et al. Biomarkers and mechanisms of drug-induced vascular injury in non-rodents. Toxicol Pathol. 2006;34:19–26. doi: 10.1080/01926230500512076. [DOI] [PubMed] [Google Scholar]
- McBride BF, Yang T, Liu K, Urban TJ, Giacomini KM, Kim RB, et al. The organic cation transporter, OCTN1, expressed in the human heart, potentiates antagonism of the HERG potassium channel. J Cardiovasc Pharmacol. 2009;54:63–71. doi: 10.1097/FJC.0b013e3181abc288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munos B. Lessons from 60 years of pharmaceutical innovation. Nat Rev Drug Dis. 2009;8:959–968. doi: 10.1038/nrd2961. [DOI] [PubMed] [Google Scholar]
- Olson H, Betton G, Robinson D, Thomas K, Monro A, Kolaja G, et al. Concordance of the toxicity of pharmaceuticals in humans and in animals. Regul Toxicol Pharmacol. 2000;32:56–67. doi: 10.1006/rtph.2000.1399. [DOI] [PubMed] [Google Scholar]
- Omland T, de Lemos JA, Sabatine MS, Christophi CA, Rice MM, Jablonski KA, et al. for the Prevention of Events with Angiotensin Converting Enzyme Inhibition (PEACE) Trial Investigators A sensitive cardiac troponin T assay in stable coronary artery disease. N Engl J Med. 2009;361:2538–2547. doi: 10.1056/NEJMoa0805299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park BK. Medical Research Council (MRC) Centre for Drug Safety science. 2008. http://www.liv.ac.uk/research/environment/Drug_Safety_Centre.htm (accessed 9th May 2010)
- Partridge CR, Johnson CD, Ramos KS. In vitro models to evaluate acute and chronic injury to the heart and vascular systems. Toxicol In Vitro. 2005;19:631–644. doi: 10.1016/j.tiv.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Paul L, Hastie CE, Li WS, Harrow C, Muir S, Connell JMC, et al. Resting heart rate pattern during follow-up and mortality in hypertensive patients. Hypertension. 2010;55:567–574. doi: 10.1161/HYPERTENSIONAHA.109.144808. [DOI] [PubMed] [Google Scholar]
- Peng X, Pentassuglia L, Sawyer DB. Emerging anticancer therapeutic targets and the cardiovascular system. Is there cause for concern? Circ Res. 2010;106:1022–1034. doi: 10.1161/CIRCRESAHA.109.211276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettit SD, Berridge B, Sarazan RD. A call for more integrated cardiovascular safety assessment. J Pharmacol Toxicol Methods. 2010;61:1–2. doi: 10.1016/j.vascn.2009.08.001. [DOI] [PubMed] [Google Scholar]
- Piccini JP, Whellan DJ, Berridge BR, Finkle JK, Pettit SD, Stockbridge N, et al. CSRC/HESI Writing Group Current challenges in the evaluation of cardiac safety during drug development: translational medicine meets the Critical Path Initiative. Am Heart J. 2009;158:317–326. doi: 10.1016/j.ahj.2009.06.007. [DOI] [PubMed] [Google Scholar]
- Pollard CE, Abi-Gerges N, Bridgland-Taylor MH, Easter A, Harmer A, Hammond TG, et al. An introduction to QT interval prolongation and non-clinical approaches to assessing and reducing risk. Br J Pharmacol. 2010;159:12–21. doi: 10.1111/j.1476-5381.2009.00207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price DA, Blagg J, Jones L, Greene N, Wager T. Physicochemical drug properties associated with in vivo toxicological outcomes: a review. Expert Opin Drug Metab Toxicol. 2009;5:921–931. doi: 10.1517/17425250903042318. [DOI] [PubMed] [Google Scholar]
- Pugsley MK, Authier S, Curtis MJ. Principles of safety pharmacology. Br J Pharmacol. 2008;154:1382–1399. doi: 10.1038/bjp.2008.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugsley MK, Authier S, Towart R, Gallacher DJ, Curtis MJ. Beyond the safety assessment of drug-mediated changes in the QT interval … what's next? J Pharmacol Toxicol Methods. 2009;60:24–27. doi: 10.1016/j.vascn.2009.07.001. [DOI] [PubMed] [Google Scholar]
- Redfern WS, Carlsson L, Davis AS, Lynch WG, MacKenzie I, Palethorpe S, et al. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc Res. 2003;58:32–45. doi: 10.1016/s0008-6363(02)00846-5. [DOI] [PubMed] [Google Scholar]
- Redfern WS, Ewart L, Hammond TG, Bialecki R, Kinter L, Lindgren S, et al. Impact and frequency of different toxicities throughout the pharmaceutical life cycle. The Toxicologist. 2010;114:1081. [Google Scholar]
- Reinhold T, Berghöfer A, Willich SN. Is the determination of biomarkers worth its price? Review of the literature taking brain natriuretic peptides (BNP) as an example. Herz. 2010;35:1–10. doi: 10.1007/s00059-010-3312-8. [DOI] [PubMed] [Google Scholar]
- Reiss AB, Anwar F, Chan ES, Anwar K. Disruption of cholesterol efflux by coxib medications and inflammatory processes: link to increased cardiovascular risk. J Investig Med. 2009;57:695–702. doi: 10.2310/JIM.0b013e31819ec3c7. [DOI] [PubMed] [Google Scholar]
- Roth BL. Drugs and valvular heart disease. N Engl J Med. 2007;356:6–9. doi: 10.1056/NEJMp068265. [DOI] [PubMed] [Google Scholar]
- Shah RR. Can pharmacogenetics help rescue drugs withdrawn from the market? Pharmacogenomics. 2006;7:889–908. doi: 10.2217/14622416.7.6.889. [DOI] [PubMed] [Google Scholar]
- Shah RR. Drug-induced QT interval shortening: potential harbinger of proarrhythmia and regulatory perspective. Br J Pharmacol. 2010;159:58–69. doi: 10.1111/j.1476-5381.2009.00191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibille M, Deigat N, Janin A, Kirkesseli S, Durand DV. Adverse events in phase-I studies: a report in 1015 healthy volunteers. Eur J Clin Pharmacol. 1998;54:13–20. doi: 10.1007/s002280050413. [DOI] [PubMed] [Google Scholar]
- Stevens JL, Baker TK. The future of drug safety testing: expanding the view and narrowing the focus. Drug Discov Today. 2009;14:162–167. doi: 10.1016/j.drudis.2008.11.009. [DOI] [PubMed] [Google Scholar]
- Stummann TC, Beilmann M, Duker G, Dumotier B, Fredriksson JM, Jones RL, et al. Report and recommendations of the workshop of the European Centre for the Validation of Alternative Methods for drug-induced cardiotoxicity. Cardiovasc Toxicol. 2009;9:107–125. doi: 10.1007/s12012-009-9045-3. [DOI] [PubMed] [Google Scholar]
- Taylor GL, Patel B, Sullivan AT. Evaluation of blood flow parameters in addition to blood pressure and electrocardiogram in the conscious telemetered beagle dog. J Pharmacol Toxicol Methods. 2007;56:212–217. doi: 10.1016/j.vascn.2007.04.003. [DOI] [PubMed] [Google Scholar]
- Thygesen K, Mair J, Katus H, Plebani M, Venge P, Collinson P, et al. the Study Group on Biomarkers in Cardiology of the ESC Working Group on Acute Cardiac Care Recommendations for the use of cardiac troponin measurement in acute cardiac care. Eur Heart J. 2010;31:2197–2204. doi: 10.1093/eurheartj/ehq251. [DOI] [PubMed] [Google Scholar]
- Tijsen AJ, Creemers EE, Moerland PD, de Windt LJ, van der Wal AC, Kok WE, et al. MiR423-5p as a circulating biomarker for heart failure. Circ Res. 2010;106:1035–1039. doi: 10.1161/CIRCRESAHA.110.218297. [DOI] [PubMed] [Google Scholar]
- Titier K, Canal M, Deridet E, Abouelfath A, Gromb S, Molimard M, et al. Determination of myocardium to plasma concentration ratios of five antipsychotic drugs: comparison with their ability to induce arrhythmia and sudden death in clinical practice. Toxicol Appl Pharmacol. 2004;199:52–60. doi: 10.1016/j.taap.2004.03.016. [DOI] [PubMed] [Google Scholar]
- Valentin JP. Reducing QT liability and proarrhythmic risk in drug discovery and development. Br J Pharmacol. 2010;159:5–11. doi: 10.1111/j.1476-5381.2009.00547.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentin JP, Bialecki R, Ewart L, Hammond TG, Leishman D, Lindgren S, et al. A framework to assess the translation of safety pharmacology data to humans. J Pharmacol Toxicol Methods. 2009a;60:152–158. doi: 10.1016/j.vascn.2009.05.011. [DOI] [PubMed] [Google Scholar]
- Valentin JP, Keisu M, Hammond TG. Predicting human adverse drug reactions from non-clinical safety studies. In: Gad SC, editor. Clinical Trials Handbook. Hoboken, NJ: John Wiley & Sons, Inc; 2009b. pp. 87–113. [Google Scholar]
- Valentin JP, Pollard CE, Lainee P, Hammond T. Value of nonclinical repolarization assays in supporting the discovery and development of safer medicines. Br J Pharmacol. 2010;159:25–33. doi: 10.1111/j.1476-5381.2009.00530.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vik T, Pollard C, Sager P. Early clinical development: evaluation of drug-induced torsades de pointes risk. Pharmacol Ther. 2008;119:210–214. doi: 10.1016/j.pharmthera.2008.05.006. [DOI] [PubMed] [Google Scholar]
- Wallis RM. Integrated risk assessment and predictive value to humans of non-clinical repolarisation assays. Br J Pharmacol. 2010;159:115–121. doi: 10.1111/j.1476-5381.2009.00395.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitebread S, Hamon J, Bojanic D, Urban L. Keynote review: in vitro safety pharmacology profiling: an essential tool for successful drug development. Drug Discov Today. 2005;10:1421–1433. doi: 10.1016/S1359-6446(05)03632-9. [DOI] [PubMed] [Google Scholar]
- Zuppinger C, Suter TM. Cancer therapy-associated cardiotoxicity and signaling in the myocardium. J Cardiovasc Pharmacol. 2010;56:141–146. doi: 10.1097/FJC.0b013e3181e0f89a. [DOI] [PubMed] [Google Scholar]