

Abstract
Sphingolipids represent a class of diverse bioactive lipid molecules that are increasingly appreciated as key modulators of diverse physiologic and pathophysiologic processes that include cell growth, cell death, autophagy, angiogenesis, and stress and inflammatory responses. Sphingomyelinases and ceramidases are key enzymes of sphingolipid metabolism that regulate the formation and degradation of ceramide, one of the most intensely studied classes of sphingolipids. Improved understanding of these enzymes that control not only the levels of ceramide but also the complex interconversion of sphingolipid metabolites has provided the foundation for the functional analysis of the roles of sphingolipids. Our current understanding of the roles of various sphingolipids in the regulation of different cellular processes has come from loss-of-function/gain-of-function studies utilizing genetic deletion/downregulation/overexpression of enzymes of sphingolipid metabolism (e.g. knockout animals, RNA interference) and from the use of pharmacologic inhibitors of these same enzymes. While genetic approaches to evaluate the functional roles of sphingolipid enzymes have been instrumental in advancing the field, the use of pharmacologic inhibitors has been equally important in identifying new roles for sphingolipids in important cellular processes.The latter also promises the development of novel therapeutic targets with implications for cancer therapy, inflammation, diabetes, and neurodegeneration. In this review, we focus on the status and use of pharmacologic compounds that inhibit sphingomyelinases and ceramidases, and we will review the history, current uses and future directions for various small molecule inhibitors, and will highlight studies in which inhibitors of sphingolipid metabolizing enzymes have been used to effectively treat models of human disease.
Keywords: sphingolipid, ceramide, sphingomyelinase, ceramidase, sphingomyelinase inhibitor, ceramidase inhibitor, therapeutic development
Introduction – background on sphingolipids
Sphingolipids (SLs) were once considered only structural components of the cellular membranes; however, in the mid 1980s, it was found that sphingosine (Sph) exerted biological and biochemical activities through the inhibition of protein kinase C (Hannun et al., 1986). This was followed by the demonstration of regulated formation of Sph with effects on cell signalling (Merrill et al., 1986; Kolesnick, 1987). Subsequently, it was shown that ceramide functioned as a second messenger (Okazaki et al., 1989). Later, other SLs such as sphingosine-1-phosphate (S1P) and ceramide-1-phosphate (C1P), the phosphorylated products of Sph and ceramide, respectively, were shown also to be bioactive lipids, being involved in the regulation of apoptosis, proliferation, angiogenesis, cell adhesion, differentiation, migration, senescence, and intracellular trafficking (Aguilar et al., 2010). Moreover, ceramide and S1P often exert opposite roles in the cell. Thus, ceramide mediates cell arrest and cell death in response to cell stress, whereas S1P mediates cell survival and proliferation. Therefore, the interconversion of these metabolites through the action of a number of critical enzymes provides for a rich network of cell regulation. As such, these enzymes provide novel approaches to therapeutic strategies that aim to influence SL-mediated biologic and pathobiologic responses.
SLs comprise a family of lipidic compounds derived from (E)-2-amino-4-octadecen-1,3-diol or Sph, the saturated (sphinganine), or the 4-hydroxy forms (phytosphingosine). N-acylation of the sphingoid base with distinct fatty acids of variable length and/or desaturation or other modifications generate diversity of ceramide species. In turn, ceramide is considered the central hub of SL synthesis (Figure 1), and it can serve as substrate for a number of complex SLs. Ceramide is synthesized de novo in the endoplasmic reticulum (ER), and is transported to the Golgi apparatus, where it can be transformed to sphingomyelin (SM) by the transferof a phosphocholine group from phosphatidylcholine totheC1 hydroxyl in ceramide by SM synthases. However, ceramide, in the Golgi apparatus or ER, can also be glycosylated by glucosyl or galactosyl transferases forming the hexosylceramides (HexCer), glucosylceramide (GlcCer) and galactosylceramide respectively. GlcCer serves as the precursor of complex glycosphingolipids, and these glycolipids along with SM are then transported to the plasma membrane (PM), probably primarily through vesicular trafficking. It is not clear how much ceramide or the other ‘simple’ SLs exist at the PM. In fact, a recent study in our group (Canals et al., 2010) suggested that not much ceramide is present at the PM under unstimulated conditions, and PM ceramide would be mainly generated from hydrolysis from SM and probably from HexCer. As to the complex SLs, once at the PM, these molecules may be transported/recycled to lysosomes by endocytic vesicles, where they can be degraded to ceramide. Ceramide, in turn, can be hydrolysed to Sph by ceramidases, and the resulting Sph is then recycled through the salvage pathway by reconverting to ceramide, or via phosphorylation to S1P. Detailed reviews on the biosynthetic and metabolic pathway of SLs can be found elsewhere (Delgado et al., 2007; Hannun and Obeid, 2008; Gangoiti et al., 2010).
Figure 1.
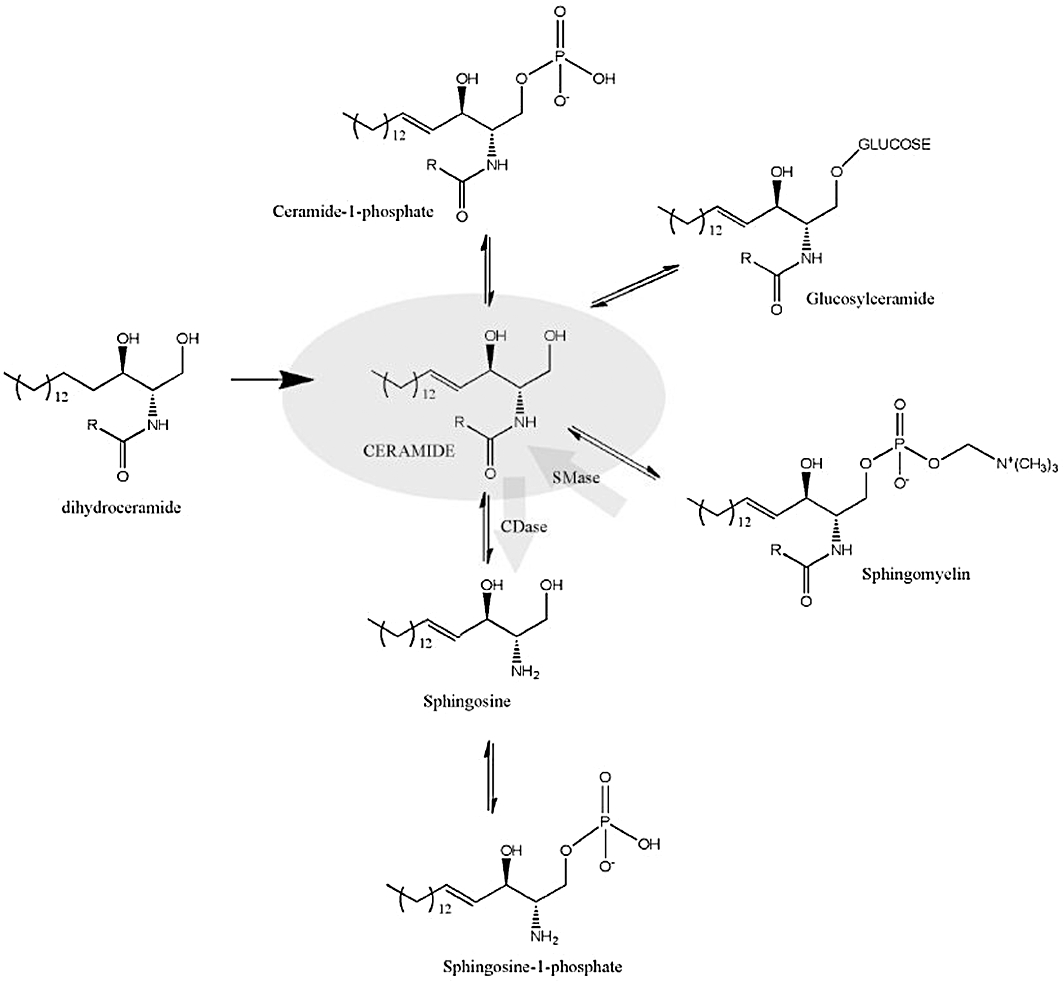
Ceramide is a biosynthetic hub in the sphingolipid (SL) pathway. De novo lineal biosynthesis of SLs leads to the irreversible point of reduction of dihydroceramide to ceramide. Ceramide is a central biosynthetic point from where different SLs are synthesized and might be interconverted. Ceramide can be phosphorylated to the bioactive SL ceramide 1–phosphate or glucosylated (or galactosylated, not shown) to glucosyl-ceramide, which is the first step to complex SLs. Ceramide can also be converted to sphingomyelin, an important structural compound of biological membranes. Finally, ceramide can be hydrolysed by ceramidases to sphingosine, which can be phosphorylated to sphingosine-1-phosphate, a pro-survival cellular signal. Sphingomyelinases (SMase) and ceramidases (CDase) enzyme reactions are indicated in the figure. R- different possible acyl chain length.
Functionally, ceramide serves not only as a structural hub but also as an important bioactive molecule and as a key precursor to produce additional bioactive SLs. Ceramide can be generated from the de novo synthesis, which implies many enzymatic steps, or from hydrolysis of SM or HexCer. The latter can result in the accumulation of ceramide directly, or indirectly via the salvage pathway which involves further hydrolysis of lysosomal ceramide to Sph followed by re-acylation to ceramide. Ceramide deriving from SM has been widely studied. Some stress-signalling molecules, such as tumour necrosis factor (TNF)-α or interleukin-1β (IL-1β) induce an activation of sphingomyelinases (SMases), which can also be activated by other stress stimulus such as exposure to ultraviolet (UV) light or radioactive radiation. These stimuli have been shown to produce an increase of ceramide and subsequent ceramide-dependent responses, such as cell death or cell arrest.
Hydrolysis of ceramide by ceramidases produces another bioactive lipid, Sph, which in turn can be rapidly phosphorylated by sphingosine kinase (SphK) producing S1P. Therefore, the pathways controlling generation of ceramide, Sph and S1P have emerged as key pathways in regulating the formation and interconversion of these bioactive SLs. Importantly, it should be noted that the cellular levels of ceramide are significantly higher than those of Sph, which in turn are significantly higher than those of S1P. Indeed, S1P is bioactive at concentrations two to three orders of magnitude lower than those of ceramide, consistent with their relative cellular concentrations. Thus, even fractional conversion of ceramide to Sph or S1P can have profound cellular effects.
Aberrations in ceramide and bioactive SLs and their metabolism have been linked to various human conditions, including cancer pathogenesis, response to cancer therapeutics, diabetic complications, neurodegeneration, inflammatory responses and ischaemia-reperfusion (heart, liver and brain). Thus, understanding these pathways has significant implications not only to their biochemistry and cell biology, but also for possible therapeutic development.
The present review is focused on the drug targeting of ceramide metabolizing enzymes, notably SMases and ceramidases (Figure 2). Table 1 summarizes the most commonly used inhibitors for those enzymes.
Figure 2.
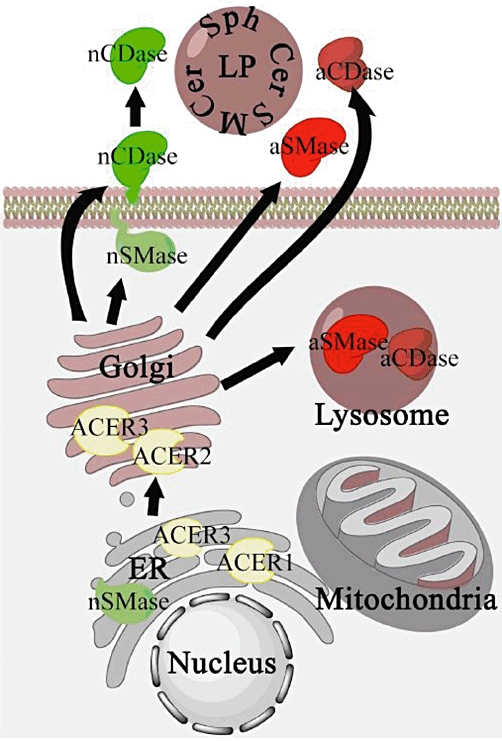
Cellular topology of sphingomyelinases and ceramidases. nSMase have been localized in the endoplasmic reticulum (ER) and attached to the plasma membrane, aSMase, is localized mainly in the lysosomes, and a minor part is also found to be secreted. nCDase is found attached to the plasma membrane, and could have activities in both the inner leaflet and in the intestinal epithelia is attached in the outer leaflet, where can be cleavaged and released to the intestinal lumen. Intestinal secreted nCDase is involved in digestion of dietary sphingolipids (SLs). aCDase follow a similar pattern than aSMase, localized mainly in the lysosome, and secreted in to the extracellular matrix. Althoug less studied, alkCDases have been localized in the ER-Golgi network. Also, mitochondrial sphingomyelinase activities have been described, albeit the frontier between ER and Golgi is diffused, and a specific mitochondria sphingomyelinase have been recently described. The secreted forms of aSMase and aCDase work on the outer leaflet of the plasma membrane, and on lipoproteins (LP) SLs. aCDase, acid ceramidase; aSMase, acid sphingomyelinase; nCDase, neutral ceramidase; nSMase, neutral sphingomyelinase; Sph, sphingosine.
Table 1.
Most used sphingomyelinase and ceramidase inhibitors
| Enzyme targeted | Inhibitor | Comments | |
|---|---|---|---|
| nSMase2 | Scyphostatin | Secondary metabolite from fungi can affect aSMase at high concentrations. | 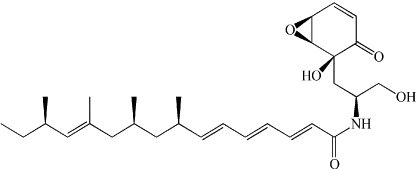 |
| Manumycin A | Also described as Ras farnesyltransferase, and interleukin-1 converting enzyme inhibitor. Irreversible inhibitors of nSMase. | 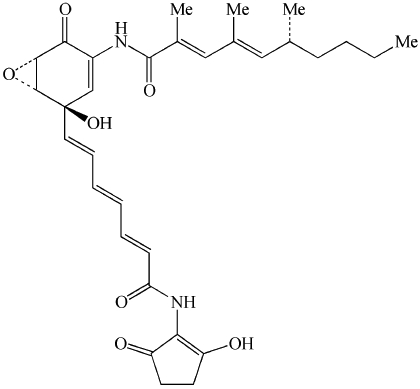 |
|
| GW4869 | Non-competitive. Requires Mg2+ for complete inhibition. | 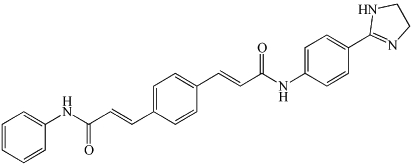 |
|
| C11AG | C11AG was shown to inhibit neutral sphingomyelinase without affecting phospholipase A2, phosphatidylcholine-specific phospholipase C and phospholipase D. | 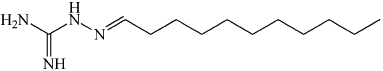 |
|
| aSMase/aCDase | Desipramine | Also affects other lysosomal enzymes. Effect blocked by Cathepsin B/L inhibitors. | 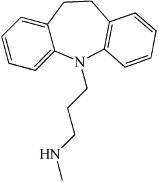 |
| aCDase | NOE | Low potency | 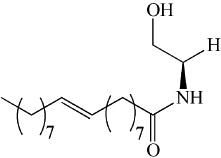 |
| B13 | Potent for aCDase. Low lysosomal uptake. | 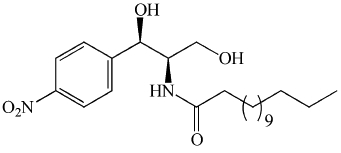 |
|
| LCL385 | Designed for lysosomal uptake. More biological studies needed to confirm effectively | 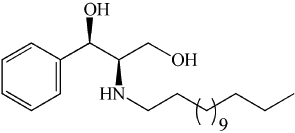 |
|
| nDCDase /alkCDase | D-e-MAPP | Most used inhibitor. Not selective for neutral or alkaline activities. | 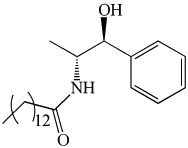 |
aCDase, acid ceramidase; alkCDase, alkaline ceramidase; aSMase, acid sphingomyelinase; C11AG, undecylidene-aminoguanidine; nCDase, neutral ceramidase; nSMase, neutral sphingomyelinase; Sph, sphingosine.
Sphingomyelinases
Neutral SMases (nSMases)
Background
nSMases are a class of phosphodiesterases, which preferentially hydrolyse SM, producing phosphorylcholine and the bioactive lipid ceramide. These enzymes possess a neutral pH optimum distinguishing them from acid and alkaline SMases. Several genes having nSMase enzymatic activity exist in nature including mammalian, fungal and bacterial versions (Clarke et al., 2006). Pharmacological targeting of mammalian nSMase activity will be discussed with an emphasis on nSMase2.
Diverse stress stimuli such as TNF-α and oxidative stress have been shown to cause an increase in nSMase activity resulting in ceramide production (Adam et al., 1996; Liu et al., 1998). Exactly how this activation occurs is not well understood. Several possibilities exist for the activation of nSMase2, including phosphorylation (Filosto et al., 2011), anionic lipid binding (Marchesini et al., 2003), translocation to the PM, direct redox sensitivity (Liu and Hannun, 1997), protein interaction with factor associated with nSMase activity (Adam-Klages et al., 1996) or embryonic ectoderm development (Philipp et al., 2010), and increased protein levels (Marchesini et al., 2004). nSMase2 contains two hydrophobic domains and multiple palmitoylation sites that anchor it in the membrane (Tani and Hannun, 2007), and it localizes to the inner leaflet of the PM and the Golgi, where a low amount of SM is thought to exist (Marchesini et al., 2007).
The family of nSMases has been implicated in several biological processes including inflammation, cell cycle arrest and apoptosis (further reviewed in Clarke et al., 2006 and Wu et al., 2010a). Furthermore, a role for nSMase2 has been demonstrated in heart failure (Adamy et al., 2007), reactive oxygen species-induced lung damage and Alzheimer disease-related neuronal toxicity with implications for neurodegenerative disorders (Haughey et al., 2004; Jana and Pahan, 2004; Lee et al., 2004). Consistent with its role in cell cycle arrest and apoptosis, mutations or deletions have been found in human leukaemias and in a mouse model of osteosarcoma, suggesting a possible protective role in the context of cancer (Kim et al., 2008). Additionally, from knockout mice studies, it is clear that nSMase2 plays a crucial role in maturation or development of bone during development as these mice display embryonic or juvenile-onset dwarfism (Stoffel et al., 2007).
Small molecule inhibitors of nSMase
Because of the emerging roles of nSMase2 in mediating signals related to inflammation and cell death, targeting of nSMase2 as a novel therapeutic intervention point has been sought after. Inhibitors of nSMase have been identified from natural fungal compounds, high-throughput screening and endogenous cellular substances. Specificity within nSMase isoforms and also between other SMases is a point of emphasis when attempting to implicate a particular nSMase in a biological process especially with the recent identification of mitochondrial-associated nSMase (Yabu et al., 2009; Wu et al., 2010b). It is conceivable that these inhibitors may have different activities and modes of action on each enzyme having nSMase activity.
The earliest nSMase inhibitor to be discovered was Scyphostatin isolated from the fungus Trichopeziza mollissima, a discomycete by Tanaka and Nara et al. (Nara et al., 1999a,b). Scyphostatin displayed mixed inhibition, affecting both Km and Vmax (Nara et al., 1999b), with a 50-fold higher IC50 for acid SMase (aSMase), thus demonstrating specificity for nSMase. Another related natural compound with nSMase inhibitory activity is Manumycin A (Arenz et al., 2001b). Manumycin A and Scyphostatin both have an amide bond and a functionalized cyclohexenone ring structure. Several analogues of Scyphostatin and Manumycin A have been synthesized with modifications of the cyclohexenone ring, the amide side chain and the presence of the primary hydroxy (Arenz and Giannis, 2000; Arenz et al., 2001a; Pitsinos et al., 2003). The main element for inhibition appears to be the cyclohexenone ring, regardless of the functional groups such as hydroxyl or epoxy groups. An unsaturated side chain enhances inhibition (Arenz et al., 2001a) and in one case was required for inhibition (Pitsinos et al., 2003). Interestingly, in Arenz et al., the analogue with a saturated side chain displayed no inhibition without preincubation with the enzyme suggestive of irreversible inhibition, whereas the analogue with an unsaturated side chain had almost complete inhibition regardless of preincubation, implicating the side chain as paramount in determining the type of inhibition. The primary hydroxy appears to enhance inhibition, but was not required in analogues lacking an epoxy group on the cyclohexenone ring (Arenz and Giannis, 2000; Arenz et al., 2001a; Pitsinos et al., 2003).
GW4869 was discovered through high throughput screening for small molecule inhibitors of nSMase2 (Luberto et al., 2002). It displayed noncompetitive inhibition with an IC50 of 1 µM. This structure does not resemble SM, in agreement with it not competing for the substrate. It was shown to have no inhibition on aSMase. Interestingly, even at high doses, GW4869 was not able to completely inhibit nSMase activity at high levels of phosphatidylserine (PS), but at low PS levels it was able to abolish activity.
In an effort to characterize the structural determinants of SM for recognition by nSMase, Lister et al., showed that the C3-OH group is an important site for substrate recognition due to the finding that C3-H (deoxy-SM) was neither a substrate nor an inhibitor. Conversely, C3-O-methylSM (methyl-SM) was not a substrate, yet could inhibit with an IC50 of 50 uM, suggesting that the addition of the methyl group was permissible for binding, but altered the binding in such a way to prevent hydrolysis (Lister et al., 1995).
Amtmann et al. synthesized a series of guanidinium lipophilic compounds showing undecylidene-aminoguanidine (C11AG) to be active (Amtmann et al., 2000). Increased lipophilicity correlated with increased inhibition.
Other inhibitory compounds that could be considered natural and also endogenous include glutathione (GSH) and ubiquinol. From a partially purified nSMase fraction, GSH was shown to inhibit nSMase activity in a reversible fashion as dilution of GSH after preincubation with the enzyme resulted in loss of inhibition, even though preincubation significantly increased inhibition. Inhibition was also shown to be noncompetitive. Interestingly, anologues of GSH such as S-methyl GSH also inhibited demonstrating that a free sulfhydryl group was not necessary for inhibition (Liu and Hannun, 1997). Moreover, it was shown that GSH was able to partially inhibit TNFα-induced ceramide generation and cell death in MCF-7 cells (Liu et al., 1998). GSH is known to be reduced or effluxed by oxidative agents and stresses (Slater et al., 1995), possibly relieving the inhibition of nSMase and resulting in activation. However, as the oxidized form of GSH, gluthatione disulfide (GSSG), also inhibited more potently, it is likely that a decrease in the total levels of GSH/GSSG possibly by efflux would be more likely to result in nSMase activation. Similarly, ubiquinol has been shown to modestly inhibit nSMase isolated from liver PM noncompetitively, which is thought to have roles in PM electron transport and redox regulation (Martin et al., 2001; 2003). Taken together, these antioxidants likely represent an interesting physiological means of regulating nSMase, but due to these being endogenous antioxidants with pleiotropic effects, they should not be employed as specific pharmacological inhibitors. Readers are referred to Delgado et al. (2006) for further review.
Functional effects of inhibitors of nSMases
Scyphostatin has been used in studies as an anti-inflammatory in response to lipopolysaccharide (LPS) and IL-1β, particularly for phosphorylation of Jun Kinase (Nara et al., 1999a; Rutkute et al., 2007). In studies performed in cultured hippocampal neurons, scyphostatin was shown to inhibit neurite outgrowth or cell death depending on the time of culture (Brann et al., 1999; 2002). Manumycin A has been used as an antibiotic, antitumour and anti-inflammatory agent (Zeeck et al., 1987; Hara et al., 1993; Sonoda et al., 1998; Bernier et al., 2006); however, due to its additional inhibitory activity on farnesyltransferase, which is important for activation of Ras and related small G proteins, and other targets (Hara et al., 1993), caution should be exercised with its use.
Many studies have used GW4869 in cell culture and in vivo studies. Treatment of cells with GW4869 was able to block induction of apoptosis in breast carcinoma and glioma cells (Luberto et al., 2002; Peng et al., 2006). nSMase activity and ceramide levels have been shown to be increased in aged endothelial tissue having decreased vessel relaxation, which was reversible by nSMase inhibition with GW4869 (Smith et al., 2006). It has also been used to demonstrate involvement of nSMase2 in hypoxic pulmonary vasoconstriction and chemotaxis of neutrophils (Cogolludo et al., 2009; Sitrin et al., 2010). A role for nSMase2 in various neurological processes, including synaptic plasticity (Wheeler et al., 2009) and spatial memory (Tabatadze et al., 2010) have been suggested based on results with GW4869.
In regard to methyl-SM, it has been shown to inhibit LPS-induced formation of ceramide, Ras activation, NF-kB activation and production of nitric oxide (NO) in rat glioma cells (Won et al., 2004). Interestingly, GW4869 was shown not to inhibit NF-κB activation in response to TNFα (Luberto et al., 2002), contrasting with the above study with methyl-SM. C11AG has been used to show involvement of nSMases in a variety of biologic responses including LPS-induced NO production (Amtmann et al., 2000; 2003; Amtmann and Zoller, 2005).
Potential uses/considerations
From the literature, there appears to be significant potential for the use of nSMase inhibitors in the setting of tissue damage related to inflammation in cardiovascular, pulmonary or neurological systems. However, the full spectrum of use of nSMase inhibitors is only being defined by ongoing studies both in elucidating its physiologic functions and the pathophysiologic implications of this enzyme. In order for nSMase2 to be developed into a therapeutic target, more effort is needed in elaborating its roles in specific disease processes in vivo and in cellular studies with the concomitant development and characterization of lead compounds.
aSMase
Background
aSMase (EC3.1.4.12) is a soluble hydrolase that generates ceramide and phosphorylcholine from the cleavage of the phosphodiester bond of SM at an acid pH optimum (Jenkins et al., 2009). Deficiency of aSMase in humans results in Niemann-Pick disease (NPD), a lysosomal storage disorder (LSD) characterized by pathologic accumulation of SM in cells and tissues (Schuchman, 2007). While absence of aSMase in humans leads to an established pathologic state (i.e. NPD), activation of aSMase has been suggested to play an important role in the development and progression of several pathophysiolgic conditions, including atherosclerosis, cancer, diabetes, Alzheimers's disease, cystic fibrosis and Wilson disease (Smith and Schuchman, 2008; Jenkins et al., 2009; Schuchman, 2010).
Activation of the aSMase/ceramide pathway has been reported in response to a variety of cellular stresses, many of which overlap activators of nSMase2 and including inflammatory cytokines, UV radiation, pathogen exposure and chemotherapeutic agents, leading to production of the bioactive lipid ceramide (Gulbins and Li, 2006; Stancevic and Kolesnick, 2010). In spite of an expanding body of research, the precise role of the aSMase/ceramide pathway in the cellular stress response remains unclear, primarily due to paucity of mechanistic studies and the absence of specific potent inhibitors.
The use of cell lines and animals lacking aSMase activity has been integral in the advancement of the field of ceramide signalling. However, cells lacking aSMase develop a subsequent SM storage phenotype that can have indirect effects on cell biology (Lozano et al., 2001). To better understand the precise role of aSMase in specific pathophysiologic processes, small molecule inhibitors are needed to inhibit aSMase acutely, in the absence of SM storage and other indirect effects of chronic aSMase deficiency. Discussed below are several endogenous and exogenous inhibitors of aSMase, their reported effects and mechanism of action, as well as future directions and the need for more specific agents. For a more comprehensive discussion of aSMase inhibitors, readers are referred to reviews by Arenz (2010) and Kornhuber et al. (2010).
Small molecule inhibitors of aSMase
Endogenous inhibitors
A variety of endogenous molecules have been shown to inhibit aSMase activity. Phosphate-containing molecules, including inorganic phosphate, nucleotides (e.g. cAMP), dolichol phosphate and phosphoinositides, are known to potently and non-competitively inhibit aSMase activity (Callahan et al., 1983; Watanabe et al., 1983). Later, specific phosphoinositides, PtdIns-3,5-P2 (Kolzer et al., 2003) and PtdIns-3,4,5-P3 (Testai et al., 2004), were demonstrated to inhibit aSMase activity in the low µM range. Importantly, and in contrast, another phosphate-containing lipid, bismonoacylgycerophophate (BMP) is a potent activator of aSMase (Linke et al., 2001a). Of note, BMP is enriched in lysosomes and the activating effect of BMP requires an acidic pH.
Studies on NPD type C (NPD-C) revealed that another endogenous lipid – cholesterol – potently inhibits aSMase activity (Reagan et al., 2000). While mutations in the SMPD1 gene that encodes the aSMase protein gives rise to NPD type A and type B, NPD-C results from mutations in NPC-1 and NPC-2 genes that encode proteins involved in cholesterol trafficking within the endo-lysosomal compartment (Sturley et al., 2004). Patients with NPD-C have a functional aSMase deficiency via post-translational inhibition of aSMase activity by cholesterol. Removal of lipoproteins from culture media of NPD-C dermal fibroblasts is sufficient to remove the inhibition of aSMase (Thomas et al., 1989), indicating that exogenous sterols play an important role in the modulation of in situ aSMase activity. Moreover, various sterols (e.g. 7-ketocholesterol) have been reported to inhibit aSMase activity in vitro (Maor et al., 1995), indicating that sterol derivatives may represent another class of molecules that can be utilized to develop inhibitors of aSMase.
The overall effect of these lipids on aSMase activity in situ is likely a function of relative concentrations of each of these molecules in specific subcellular compartments, with additional influence of local pH. In addition, aSMase itself is subject to post-translational modification, which may alter subcellular localization and/or specific activity. Moreover, the relevance of these in vitro findings must be considered in the context of intracellular, extracellular and local concentrations. For example, as lysosomes mature, cholesterol concentrations decrease, pH increases, the concentration of BMP increases, thereby promoting a favourable environment for the action of aSMase.
Exogeous inhibitors
In vitro inhibitors
Building on these studies mentioned above, several groups have synthesized compounds based on the structure of SM or of endogenous aSMase regulators. The first class are difluoromethylene analogoues of SM, referred to as SMAs (Yokomatsu et al., 2001). Administration of SMA-7 protected mice against dextran sulfate sodium-induced colitis (Sakata et al., 2007a), a model of inflammatory bowel disease. Cell culture studies demonstrated that inhibition of aSMase by SMA-7 disrupted LPS-induced formation of ceramide and disrupted activation of NF-κB and subsequent release of pro-inflammatory cytokines from macrophages (Sakata et al., 2007a), as well as decreased interleukin-8 production by epithelial cells (Sakata et al., 2007b). The latter effect was related to impaired neutral SMase activation, so the specificity of these SMAs remains unclear. Phosphoinositide-based inhibitors (Roth et al., 2009b), bisphosphonate-based inhibitors (Roth et al., 2009a) and carbohydrate-based inhibitors (Roth et al., 2010) have all been synthesized and characterized, although these inhibitors have not been widely utilized and therefore data on their efficacy is limited.
In vivo inhibitors
Several pharmacologic agents have been shown to induce functional loss of aSMase activity, most notably the tricyclic antidepressants (e.g. desipramine, imipramine and amitryptiline) (Albouz et al., 1981; Kornhuber et al., 2010). These agents inhibit aSMase activity in vivo but do not act as in vitro enzyme inhibitors. The calmodulin antagonist W-7 (Masson et al., 1989), cocaine (Nassogne et al., 2004), SR33557 (Jaffrezou et al., 1991), and most recently, the S1P receptor antagonist FTY720 (Dawson and Qin, 2011), induce a functional loss of aSMase, along with other cationic amphiphilic drugs (Yoshida et al., 1985). With the tricyclics, the mechanism proposed is that of preventing the interaction of aSMase with anionic lipids in the lysosome, especially BMP, resulting in the enzyme becoming a substrate for lysosomal proteases. For a more comprehensive discussion of functional aSMase inhibitors, readers are referred to the review by Kornhuber et al. (2010).
Importantly, these drugs are not specific for aSMase, and have been shown to inhibit acid ceramidase (aCDase) (Zeidan et al., 2006) and lysosomal phospholipases (Pappu and Hostetler, 1984). Importantly, several other lysosomal enzymes, including acid lipase, arylsulfatases A and B, and hexosaminidases, are unaffected by tricyclic compounds (Albouz et al., 1981), suggesting a certain degree of selectivity. The mechanism of tricyclic antidepressant action on aSMase appears to be by promoting proteolysis of mature, lysosomal aSMase (Hurwitz et al., 1994), which distinguishes these agents from other lysosomotropic agents, such as chloroquine, which induce global lysosomal dysfunction and would presumably affect all lysosomal hydrolases.
Current uses, and future directions, for aSMase inhibitors
For several years, anti-inflammatory properties of tricyclic compounds have been reported (Martelli et al., 1967; Roumestan et al., 2007). In recent years, functional aSMase inhibitors have been shown to reduce the severity of several different mouse models of human disease, including cystic fibrosis (Teichgraber et al., 2008; Becker et al., 2010), Wilson disease (Lang et al., 2007), acute lung injury (Yang et al., 2010) and hepatic ischaemia-reperfusion injury (Llacuna et al., 2006). Recently, Roth et al. demonstrated that several bisphosphonate compounds potently inhibited aSMase (Roth et al., 2009a).The most potent of these bisphosphate derivates exhibited an IC50 of 20 nM towards aSMase. Bisphosphonates, such as zoledronic acid, are already in clinical use for the treatment of osteoporosis, and may also prove to be functional aSMase inhibitors in vivo.
However, it is important to emphasize that aSMase represents multiple enzymatic forms that exist in different cellular and extracellular compartments. A common protein precursor (pro-aSMase) is differentially trafficked to form lysosomal aSMase (L-SMase) or secretory aSMase (S-SMase) (Schissel et al., 1998; Tabas, 1999). While L-SMase resides in the endo-lysosomal compartment, S-SMase is an extracellular enzyme. An additional form of aSMase has been reported in close association with the outer leaflet of the PM where it can form ceramide-rich platforms (Stancevic and Kolesnick, 2010). As future research uncovers the specific roles of these different forms of aSMase, it will likely become increasingly important to target aSMase in a compartment-specific fashion to maximize the therapeutic potential of aSMase inhibitors.
Summary
A variety of endogenous and exogenous molecules regulate the activity of aSMase. While endogenous regulators – including nucleotides, phospholipids, sterols – may serve as a foundation for the development of more selective inhibitors of aSMase in the future, exogenous compounds, namely the functional aSMase inhibitors (e.g. tricyclic antidepressants), are emerging as attractive therapeutic options in the treatment of several pathophysiologic conditions. Clinically approved agents such as tricyclic antidepressants and bisphosphonates represent attractive agents to evaluate the role of aSMase in several pathophysiologic conditions. Re-evaluation of these compounds as functional inhibitors of aSMase signalling may provide an inroad for the treatment and/or improved clinical management of several disease states. However, as noted, these are not totally specific for aSMase, and therefore, an urgent need still exists for the development of potent and selective small molecule inhibitors of this enzyme.
Ceramidases
Background
Thus far, we have described how ceramide is generated from SM by SMases, and how an excess or defect in ceramide or sphingomyelinase activity may result in pathobiologic effects. However, ceramide and S1P have been shown to often exert opposite effects (Cuvillier O et al., 1998; Huwiler and Pfeilschifter, 2006; Taha et al., 2006b; Canals et al., 2010). There are only two enzymatic steps separating ceramide and S1P: ceramidases provide the first node of this regulatory connection, and SphK the second one. It should not be surprising that both of these enzymes have also been involved in cancer progression and inflammation (Horton, 1999; Nemoto et al., 2009; Pyne and Pyne, 2010; Snider et al., 2010). This section will discuss the ceramidases; the reader is referred to other reviews for discussion of SphKs (Taha et al., 2006a; Pitman and Pitson, 2010).
Five different human genes have been identified that encode proteins that hydrolyse ceramide to Sph and free fatty acid (N-acylsphingosine amidohydrolase). They have been grouped as acid, neutral or alkaline ceramidases (EC 3.5.1.23), depending on the optimum pH of their ceramidase activity being pH 4.5 for aCDase (gene name: ASAH1, NAAA), pH 7–9 for neutral ceramidase (nCDase, ASAH2) and pH 8.5–9.5 for alkaline ceramidases (alkCDase, three genes: ASAH3/ACER1, ACER2 and ACER3). A reverse activity has also been described for most of these ceramidases with that for aCDase showing a pH optimum slightly less acidic (5.5) than that of the forward reaction (Okino et al., 2003). Interestingly, and as predicted from their mechanism, the reverse activity of ceramidases uses a free fatty acid as a substrate, unlike ceramide synthases (EC 2.3.1.24) that use acyl-CoAs as substrates. These different ceramidases are found in diverse cellular compartments. Thus, aCDase resides in the lysosomes (Koch et al., 1996), nCDase at the PM, intracellular compartments and secreted in the intestinal lumen (El Bawab et al., 2000) and ACER family in the ER-Golgi network (Mao et al., 2001; Xu et al., 2006).
Acid ceramidase
Acid ceramidase activity, as well as the reverse reaction, was first described in 1963 by Shimon Gatt using rat tissues (Gatt, 1963). In the early 1990s, human aCDase was purified to homogeneity from human urine (Bernardo et al., 1995), and a few years later, the cDNA for the gene was cloned (Li et al., 1998). aCDase has subsequently been purified and characterized from other tissues (Linke et al., 2001b). aCDase consists of a single precursor polypeptide of 53–55 kDa (Ferlinz et al., 2001) that is proteolytically processed into α- and β-subunits that migrate at 13 kDa and 40 kDa respectively. The α-subunit can be reduced to 29 KDa by N-glycanase F treatment (Koch et al., 1996). Of six individual potential N-glycosylation sites, five of them are used (Schulze et al., 2007), and some of them are required for correct lysosomal processing or enzymatic activity, and for the formation of the heterodimeric enzyme form (Ferlinz et al., 2001). Purification of human aCDase revealed that at least two β-subunits could be generated for aCDase differing by 2–4 kDa at the C-terminus (He et al., 2003). Although aCDase has been localized mainly in lysosomes, a portion of aCDase is secreted to the medium as a 47 kDa monomer (Bernardo et al., 1995).
Little is known about how the secretory form is regulated and processed. The generation of the lysosomal mature form involves cleavage of the precursor in endosonal/lysosomal compartments, as well as trafficking through the mannose-6-phosphate receptor. Interestingly, mutation of putative N-glycosylation sites did not alter the ratio of secreted : lysosomal aCDase (Ferlinz et al., 2001). The use of insect Sf21 cells overexpressing human aCDase showed that the secreted precursor can be processed to a mature form upon acidification of the cell culture supernatant to pH 4.2–4.3m causing the processing of the precursor and resulting in a homogeneous sample of mature aCDase.
aCDase and aSMase are metabolically consecutive enzymes in SM hydrolysis, and their expression and activity are regulated by each other. For example, aCDase activity enhances aSMase secretion (He et al., 2003). More recent work has shown that secreted aCDase can be found forming a multi-enzymatic complex with aSMase and β-galactosidase (He et al., 2003). The results showed strong enough interaction among these enzymes to allow co-precipitation, and there was significant specificity that other lysosomal enzymes were not detected in those complexes.
Interestingly, as seen for nSMase and aSMase, stress stimuli such as TNF-α also activate aCDase. Other stress stimuli such as UV and ionizing radiation have also been reported to induce aCDase activity. Moreover, aCDase requires anionic lysosomal lipids and SL activator proteins (saposin) as cofactors for efficient hydrolysis of ceramide in vivo (Azuma et al., 1994; Linke et al., 2001a).
Abnormal overexpression of aCDase has been related to tumour progression and protection from cell death; on the other hand, deficiency of aCDase results in Farber disease (first described in 1957 by Sidney Farber), an autosomal-recessively inherited LSD resulting in accumulation of ceramide. Farber disease is associated with distinct clinical phenotypes, involving painful swelling of the joints and tendons, pulmonary insufficiency, neurological and general deficient development and a shortened lifespan (Levade et al., 1995; Koch et al., 1996). Several point mutations and exon skipping in the aCDase gene have been identified in Farber disease. Moreover, although certain mutations in the aCDase gene mimic Farber disease, the generation of aCDase knockout mice (Li et al., 2002) resulted in embryonic lethality for the homozygous mice. Furthermore, aCDase has been implicated in other metabolic complications; for example, aCDase was shown to have a role in preventing type 2 diabetes and modulating insulin signalling (Chavez et al., 2005).
Neutral ceramidase
nCDase activity, and the reverse activity, were described as early as 1969 as a ceramide-cleaving activity found in humanduodenal contents, with an optimal pH of 7.6 (Nilsson, 1969), and in 1980, in microsomes from rat liver (Stoffel and Melzner, 1980). nCDases have been cloned from bacteria (Okino et al., 1999; 2010), human (El Bawab et al., 2000), mouse (Tani et al., 2000), rat (Mitsutake et al., 2001), Drosophila (Yoshimura et al., 2002), amoeba (Monjusho et al., 2003), Zebra fish (Yoshimura et al., 2004), plants (Pata et al., 2008) and fungus (Tada et al., 2009). Note that the Aspergillus and the amoeba nCDases showed an optimum acidic pH. nCDase does not share significant sequence identity with either acid or alkCDases, as well as other amidases, including proteases (Galadari et al., 2006). Northern blotting analysis also revealed that nCDase is expressed ubiquitously (El Bawab et al., 2000), but highly expressed in kidney, liver, heart (Tani et al., 2000) and intestine (Choi et al., 2003). The activity of purified intestinal rat (Olsson et al., 2004) and human (Ohlsson et al., 2008) nCDase, a glycosylated protein of molecular weight of 116 kDa, is not affected by Ca2+, Mg2+or Mn2+, but inhibited by Zn2+, Fe2+ and Cu2+ (Galadari et al., 2006). No cations are required to activate nCDase. nCDase was found to be secreted into the intestine lumen for SL digestion, being resistant to pancreatic proteases (Olsson et al., 2004; Ohlsson et al., 2008) and, as with aSMase, was found extracellularly attached to the PM, exposing the catalytic site to the intestinal lumen (Duan et al., 2007). In other tissues and in cell culture, nCDase can be secreted or localized at the PM (Hwang et al., 2005) as a type II integral membrane protein (Tani et al., 2003). In addition, it has also been localized in endosome-like structures (Mitsutake et al., 2001), and in mitochondria in MCF-7 and HEK293 cells (El Bawab et al., 2000). Thus, intestinal and intracellular nCDase are identical enzymes (Duan and Nilsson, 2009). Moreover, nCDase is also highly glycosylated. Even though deglycosylation did not affect its activity, mutants lacking the mucine box or O-glycosylation sites in the mucine box were secreted and not localized at the cell surface in HEK cells (Tani et al., 2003). This is consistent with bacterial and invertebrate nCDase, which lack the mucin box, being secreted proteins (Inoue et al., 2009). Overexpression of nCDase did not result in Sph accumulation in unstimulated human platelets, unless the PM SM was previously hydrolysed (Tani et al., 2005), suggesting that not much ceramide is found at PM in unstimulated conditions. The same conclusion was shown using combination of recombinant bacterial sphingomyelinase and recombinant bacterial ceramidase (Canals et al., 2010).
Galadari et al. identified a nCDase motif comprised of six amino acids core (GDVSPN) from a comparison of diverse nCDase sequences, and identified the serine residue (Ser 354) in that hexapeptide as the nucleophile attacking the amide bond of ceramide in the catalytic site. The mutation of that serine, as well as the aspartate or the cysteine of the hexapeptide led to complete loss of nCDase activity (Galadari et al., 2006). However, Inoue et al., based on Pseusomona aeruginosa nCDase crystal structure, suggested that Ser-354 is involved in Zn2+ binding (Inoue et al., 2009). The crystal structure of bacterial P. aeruginosa nCDase also allowed the identification of the Zn2+ and the Mg2+/Ca2+ binding sites. Modelling the rat sequence on the P. aeruginosa crystal structure, other amino acids were identified in rat nCDase to be indispensable for Zn2+ binding and formation of the active site (His 175 and Tyr 160), for catalysis (Arg 238) and for the ceramide binding site (Tyr 572). Single mutations of these residues led to the loss of rat nCDase forward and reverse activity (Inoue et al., 2009).
The substrate specificity of nCDase was studied by El Bawab et al. (El Bawab et al., 2000), finding that only the natural d-erythro-ceramide isomer was used as substrate of the four stereoisomers of ceramide. Dihydroceramide or phytoceramide forms, shortening the length of the alkyl backbone, methylation of the primary or secondary hydroxyl groups resulted in reduction or loss of the nCDase activity (el Bawab et al., 2002).
Physiologically, IL-1β increased the activity of acid, but also neutral ceramidase in rat hepatocytes (Nikolova-Karakashian et al., 1997) and in renal mesangial cells, describing not only an increase in nCDase activity but also in mRNA levels and protein synthesis. This protein induction by IL-1β was blocked by the p38 mitogen-activated protein kinase inhibitor SB 202190 (Franzen et al., 2001). Moreover, other cytokines such as TNF and interferon-gamma also increased activity, mRNA and nCDase protein synthesis, where this elevation was reported to protect cells from cytokine-induced cell death (Zhu et al., 2008).
Alkaline ceramidases
In 1975, it was reported that the hydrolysis of ceramide was observed not only at pH 4.0 but also at pH 9.0 in normal cerebellum (Sugita et al., 1975). Acid and alkCDase activity were also found in fibroblasts and leukocytes (Dulaney et al., 1976), and in different rat tissues (Spence et al., 1986).
In a study in yeast, aimed at molecular identification of enzymes of ceramide metabolism, Mao et al. identified two novel alkaline ceramidases (YPC1 and YDC1). These then became the founding members of a novel family of alkCDases. Indeed, three different genes comprise the known human alkCDases, ACER1, 2 and 3, which contain 264 (Sun et al., 2008), 275 (Xu et al., 2006) and 267 (Mao et al., 2001) amino acid, respectively, with a similar molecular weight around 31 kDa. All of them present multiple transmembrane domains. ACER1 is localized in the ER and is mainly expressed in the skin (Mao et al., 2003). ACER2 is localized in the Golgi apparatus and is highly expressed in the placenta and modestly expressed in many other tissues. ACER3 is localized in both ER and Golgi apparatus, and is highly expressed in most tissues, especially in the placenta (Mao and Obeid, 2008). All three ACER activities are enhanced by Ca2+ (Mao et al., 2001).
The substrate specificity among aCDases, nCDases and alkCDases was first studied in 1982, finding short acyl-chain ceramides (i.e. C16 acyl chain) being poor substrates for acid, but not for neutral and alkCDases. Both, alkaline and nCDase have been reported to be inhibited by phosphatidylcholine and SM (Yada et al., 1995).
Recently, plasma S1P has been reported to derive from Sph generated from alkCDase activity in erythrocytes. Moreover, this activity is the only ceramidase activity found in erythrocytes. Furthermore, alkCDase has also been shown to be important for erythroid differentiation inK562 erythroleukaemic cells (Xu et al., 2010).
ACER1 is upregulated in epidermal keratinocytes in response to increasing the concentration of Ca2+ in tissue culture medium. Using RNAi technology, ACER1 has been implicated in mediating extracellular Ca2+-induced differentiation of human keratinocytes. The mechanism by which ACER1 mediates keratinocyte differentiation remains unclear although an increase in the generation of Sph, S1P or both may be involved. ACER2 has been shown to be upregulated in tumours, and its upregulation promotes tumour cell proliferation and survival likely through increasing the generation of S1P and activating the S1P receptor S1P1. Interestingly, over-expression of ACER2 may also result in cell proliferation inhibition or cell death due to an accumulation of Sph, which is highly cytotoxic. ACER2 has also been implicated in the regulation ofβ1 integrin maturation and cell adhesion (Sun et al., 2009).
The cytotoxic compound retinoid N- (4-hydroxyphenyl)retinamide (4-HPR), an inhibitor of dihydroceramide desaturaseactivity (Kraveka et al., 2007), has recently been shown to increase ACER2 activity and mRNA, but not ACER 1 or 3. Over-expression of ACER2 (but neither ACER3, nor aCDase nor nCDase) enhanced 4-HPR-induced (same with GT11 another dihydorceramide desaturase inhibitor) dihydroSph formation, and cell death (Mao et al., 2010), suggesting that ACER2 also regulates the levels of dihdyrosphingosine (DHS) as well as DHS-mediated cell death by controling the hydrolysis of certain dihydroceramides with unsaturated acyl chains. ACER3 prefers unsaturated long-chain ceramides, minor ceramide species in mammalian cells and tissues, so ACER3 knockdown results in an increase in the levels of unsaturated long-chain ceramides (d-e-C18:1-ceramide and d-e-C20:1-ceramide) in tumour cells. Interestingly, ACER3 down-regulation decreases the levels of other ceramide species while increasing the levels of both SPH and S1P by increasing the expression of ACER2, which hydrolyses most mammalian ceramide species. ACER3 knockdown inhibited cell proliferation by up-regulation of the cyclin-dependent kinase inhibitor p21 (CIP1/WAF1), but also inhibited serum-deprivation induced apoptosis (Hu et al., 2010).
Endogenous inhibitors
The physiologic regulation of ceramidase activity depends on the specific ceramidase (aCDase, nCDase or alkaline) and their localization in the cell. Thus, aCDase has been the most studied ceramidase, shown to be inhibited by anionic lipids such as phosphatidic acid (PA) and phosphatidylserine (PS) for the forward reaction, but these lipids appear to promote the reverse reaction (El Bawab et al., 1999). Conversely, SM activates aCDase in the forward reaction, inhibiting the reverse activity. Zinc cations strongly inhibit the reverse activity. In addition, the product of the forward activity, Sph and oleic acid, as well as other complex SLs such as HexCers have been found to inhibit forward ceramidase activity (Sugita et al., 1975). Furthermore, pH also plays an important regulatory role in ceramidase activity as aCDase can reach both endosome-lysosomal and extracellular matrix compartments. Not only the optimum pH of the enzyme is important for its activity, but also the processing requires low pH to produce the mature form, as seen before.
In vitro studies have shown that nCDase activity was inhibited by reducing agents such as dithiothreitol ans β-mercaptoethanol (Galadari et al., 2006). Natural Sph acted as a competitive inhibitor, and the reverse activity of nCDase was inhibited by L-erythro-Sph and myristaldehyde in a competitive mechanism (El Bawab et al., 2001). Phosphatidic acid and cardiolipin showed moderate inhibition as well. Interestingly, cardiolipin enhanced ceramidase activity, and, importantly, the reverse activity was not inhibited by fumonisin B1, an inhibitor of the CoA-dependent ceramide synthase enzymes (El Bawab et al., 2001). As seen for aSMase, low concentrations of cholesterol also inhibited intestinal nCDase (Ohlsson et al., 2007).
Small molecule inhibitors of ceramidases
As seen for aSMase, some amphiphilic tricyclic agents such as desipramine, chlorpromazine and chloroquine, also exert an indirect inhibitory effect on lysosomal ceramidase activity by down-regulating the aCDase protein, but not modifying RNA message level. The inhibition by desipramine is blocked by the cathepsin protease inhibitors leupeptin and CA074ME, but not by pepstatin A. This inhibition by the tricylics has been observed in vivo (Elojeimy et al., 2006) but not in vitro (Zeidan et al., 2006). Thus, desipramine, induces down-regulatioin of aCDase by activation of proteolysis of aCDase by cathepsin B/L (Elojeimy et al., 2006). However, not all lysosomotropic agents have been found to have this effect; ammonium chloride and bafilomycin A1 have no effect on the amount of aCDase protein (Elojeimy et al., 2006). The diacylglycerol analogue, phorbol myristate acetate (PMA) also induced down-regulation of ACER2 (Sun et al., 2009).
Synthetic ceramidases inhibitors
The design and screening of structural analogues of the sphingoid bases and ceramides has led to a few specific inhibitors for ceramidases.
The first aCDase inhibitor described was N-oleoylethanolamine (NOE) (Sugita et al., 1975), now better known as an endocannabinoid-related molecule, and has been one of the most used in vitro inhibitors for aCDase. In vitrostudies showed that NOE increased ceramide levels and enhanced apoptosis in L929 cells (Strelow et al., 2000), glioma cells (Hara et al., 2004), primary placenta trophoblast (Payne et al., 1999), and dendritic cells (Kanto et al., 2001). Although those effects were attributed to an increase in ceramide levels, they cannot be specificallyattributed to inhibition of aCDase, as NOE, afterwards, was found to inhibit ceramide glucosylation in neuroepithelioma cells as well (Spinedi et al., 1999). Furthermore, structural analogs of NOE were explored as aCDase inhibitors by Fabrias G (Grijalvo et al., 2006), defining NOE as a weak aCDase inhibitorin vitro and in vivo. Thus, NOE structure was improved for the development of aCDase inhibitors using shorter oxoacyl groups, adding the missing sphingoid alkyl tail and adding also a C3 hydroxyl group. Although the (E)-4,5 double bound was not required for inhibition, a (Z)-4,5 geometry was found to revert the inhibition. Due to its lack of selectivity (NOE can also inhibit neutral and alkaline ceramidase activities), and low potency, the canonical structure of NOE has been avoided for therapeutic use, although structural analogues of it are current candidates for aCDase inhibitors.
Not long after, another ceramidase inhibitor was developed; d-e-MAPP [(1S, 2R)-D-erythro-2- (N-Myristoylamino)-1-phenyl-1-propanol], was synthesized by Bielawska et al. as a ceramide analogue, causing cell cycle arrest in HL-60 cells, elevating intracellular ceramide levels up to 3-fold. In vitro studies showed that d-e-MAPP inhibited, at low micromolar concentrations, alkaline and neutral ceramidase, but had no effect on aCDase activity. Moreover, its enantiomer L-e-MAPP, although undergoing similar cellular uptake, was metabolized and had no effect on ceramidase activity (Bielawska et al., 1996). A recent study demonstrated that d-e-MAPP inhibits cellular activities of ACER1-3. Other authors have also reported increases in cellular ceramide levels after d-e-MAPP treatment (Raisova et al., 2002; Rodriguez-Lafrasse et al., 2002; Alphonse et al., 2004), and this increase in ceramide levels by d-e-MAPP has been involved in apoptosis induction (Rodriguez-Lafrasse et al., 2002; Choi et al., 2003; Lepine et al., 2004) and in radiation treatment, overcoming the resistance to radiation of cancer cells over-expressing ceramidases. Furthermore, d-e-MAPP was seen to block the mitogenic effect of oxidized low density lipoprotein (oxLDL), suggesting S1P as the responsible molecule of the mitogenic effect, coming from LDL-ceramide (Auge et al., 1999). Other pathways requiring S1P were also blocked by d-e-MAPP, such as DNA synthesis in smooth muscle cells (Maupas-Schwalm et al., 2004). In other cases, an acid ceramidase inhibitor was required to induce apoptosis, where d-e-MAPP had no effect on induction of apoptosis, although ceramide levels increased in both treatments (Payne et al., 1999). In fact, d-e-MAPP has been shown to protect cells from apoptosis in response to serum deprivation, the synthetic glucocorticoid dexamethasone, and the herbicide paraquat by preventing the production of Sph. d-e-MAPP resulted in blockage of progestin and adiponectinreceptor (PAQR), involving SLs, and ceramidases downstream of PAQR signalling, an important hormone receptor related to pathological conditions, including obesity, diabetes and coronary artery disease (Kupchak et al., 2009). Other studies have shown d-e-MAPP, as well as exogenous short length acyl-ceramides, to enhance the A23187 (Ca2+ionophore)-induced release of arachidonic acid, associated with an increase of endogenous ceramide accumulation (Shimizu et al., 2009). Modifications of the structure of d-e-MAPP led to B13 ((1R,2R)-2- (N-tetradecanoylamino)-1- (4-nitrophenyl)-1,3- propanediol), a more water soluble form. Interestingly, B13,as shown for d-e-MAPP, also increased intracellular levels of ceramide, but unlike d-e-MAPP, B13 was shown to be an inhibitor of acid ceramidase (Bielawska et al., 2008).
Although B13 showed a high potency in aCDase inhibition in vitro, the effect on the enzyme in vivo may not be direct; its neutral nature makes it difficult to accumulate in the lysosome. Thus, B13 was taken as a scaffold to design new ceramidase inhibitor structures, and the uptake by the lysosome was enhanced with a series of lysomotrophic molecules such as LCL204 (also known as Ad 2646). Thus, LCL204 significantly decreased cell migration caused by over-expressing aCDase, and it also sensitized head and neck cancer cells to FAS induced apoptosis.LCL204 overcame the resistance to apoptin mediated by the over-expression of aCDase in DU145, PC-3 and LNCaP cells. However, LCL204, although targeted to the lysosomes, was found to eventually destroy the lysosome and to cause degradation of aCDase by cathepsin B/L, and it also inhibited aSMase (Holman et al., 2008), as previously was observed with desipramine. A new family of LCL204 analogues, which also target the lysosome without destabilizing them and without inducing degradation of aCDase were developed, and these include LCL 433, 449, 463, 464, 488 and LCL506 (Bai et al., 2009). Another B13 analogue, LCL385 (Mahdy et al., 2009) showed aCDase inhibition in vivo, with similar effects as seen with knocking down aCDase, sensitizing prostatic cancer cells to radiation, and reducing xenografts tumour growth.
A series of modifications in ceramide and Sph were tested on nCDase activity, all stereoisomers of d-erythro-ceramide and Sph (L-threo, d-threo, and L-erythro isomers), N-methyl-D-erythro-Sph and d-erythro-urea-C16-ceramide showed significant inhibitory effects, whereas other ceramide and Sph analogues such as N-methyl ceramide, 1-O-methyl ceramide, cis-D-erythro ceramide or d N,N-dimethyl-D-erythro-sphingosine had no effect. Of note, C1P and S1P stimulated the enzyme (Usta et al., 2001). Diverse detergents such as taurocholate (TC), taurodeoxycholate (TDC), glycodeoxycholate (GDC), and (3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulphonate) (CHAPS)inhibited in vitro nCDase activity (Ohlsson et al., 2007).
Functional effects of inhibitors of ceramidases
Together with aSMase, aCDase has also been related to the pathogenesis of cystic fibrosis, suggesting ceramide accumulation mediating inflammation and cell death in lungs, and enhancing bacterial infection which is the major cause of mortality in CF patients. Thus, CF patients treated with amitriptyline, another tricyclic antidepressant, showed improvement in their lung function (Riethmuller et al., 2009).
ACDase is involved in many cancers such as breast cancer (Ruckhaberle et al., 2008; 2009), prostate cancer (Saad et al., 2007; Holman et al., 2008; Mahdy et al., 2009), leukaemia (Furlong et al., 2008; Shah et al., 2008), colon cancer (Selzner et al., 2001), head and neck squamous cell tumours (Elojeimy et al., 2007), and individuals developing asbestos-induced malignant pleural mesothelioma (Archimandriti et al., 2009). Therefore, one of the important goals to control cancer progression has become the design of drugs targeting aCDase. Endogenous over-expression of aCDase has been shown to reduce ceramide levels and increase S1P levels, and both effects have been related to stimulate cancer progression (Huwiler and Pfeilschifter, 2006). Moreover, resistance to radiation and/or chemotherapy used in cancer treatment has been showed to be caused by induction of aCDase synthesis in some cancer cell culture models (Mahdy et al., 2009), thus pharmacological inhibition of aCDase sensitizes cancer cells to radiation and chemotherapy. The aCDase inhibitor B13 induced up to 90% apoptosis in colon cancer cells (Liu et al., 2008) and reduction of colon tumor growth in nude mice (Selzner et al., 2001).Furthermore, theB13 analogue, LCL 385, sensitized prostate cancer cells to radiation, and also decreased growth of xenograft tumour (Mahdy et al., 2009),and the analogue LCL204 sensitized head and neck squamous cell tumours to Fas-induced cell death in both in vitro an in vivo systems (Elojeimy et al., 2007).
On the other hand, the role of nCDase in cancer has not been studied. However, nCDase has been implicated in protective role in inflammation (Franzen et al., 2001) and pro-proliferation, as down-regulation of nCDase led to cell cycle arrest. ACER2 up-regulation has been implicated in tumour cell proliferation and survival whereas the role of ACER1 and ACER3 in cancer has not been studied.
Drug targeting of human ceramidases summary
The protein structure, the pH optimum, and the sub-cellular localization and topology of the ceramidases create, for one single reaction, different possible physiologies. Ceramidases have been implicated in many diseases, including various types of cancer, thus rendering ceramidases as attractive targets for drug developing. Thus, inhibitors of aCDase and nCDase have been shown to be promising drugs to overcome cell death resistance after prolonged anti-cancer treatments. Nevertheless, at the moment there are no inhibitors that combine specificity and the ability to reach the right cell compartment. For example, desipramine is used to inhibit aCDase, but it also inhibits aSMase, and other lysosomal enzymes. The inhibitor B13 is not efficient enough to reach the lysosome; encouragingly, other molecules such as LCL385 have been developed to solve this issue, and more studies are needed to show their in vivo specificity. There are no highly specific inhibitors for neutral or alkCDases, although d-e-MAPP seems to inhibit the latter enzymes, having no effect in the acidic form. More research is needed for selective inhibitors that would help to understand not only the function of different ceramidases, but also their development into more efficacious and promising therapeutics to control ceramide-related pathologies.
Discussion
Dysregulation of SL metabolism has been reported for several human disease states; therefore, drugs that target enzymes of SL metabolism are attractive candidates for therapeutic development. Because ceramide has emerged as a key bioactive SL, enzymes of ceramide metabolism have emerged as key cellular regulators in stress responses. Enhanced accumulation of ceramide has been implicated in neuro-degeneration, diabetic complications, and increased responses to ischaemia/reperfusion injury. Therefore, targeting enzymes of ceramide production is an attractive target to ameliorate these conditions. Reciprocally, insufficient ceramide levels may allow cancer cells to escape cell death and to acquire resistance to chemotherapeutics; therefore, enhancing ceramide formation, primarily by inhibiting ceramidases (and other enzymes of ceramide metabolism, such as glucosylceramide synthase) are emerging as targets in cancer therapy.
Classically, ceramide has been studied as a single effector molecule; however emerging evidence clearly demonstrates that the generation or hydrolysis of ceramide (and bioactive lipids in general) might have different effects depending on where this ceramide is produced or further metabolized. For that reason, developing specific drugs against individual and distinct enzymes and targeted to the right compartment has emerged as a key and important goal that will help to understand ceramide function depending onits cellular localization and topology. This, in turn, should lead to more specific and rational therapeutics.
Acknowledgments
We gratefully acknowledge Drs Cungui Mao and Christopher Clarke for the careful reading of this manuscript. This work was supported by NIH grants CA87584 and CA97132.
Glossary
Abbreviations
- aCDase
acid ceramidase
- alkCDase
alkaline ceramidase
- aSMase
acid sphingomyelinase
- C1P
ceramide-1-phosphate
- nCDase
neutral ceramidase
- NPD
Niemann-Pick disease
- nSMase
neutral sphingomyelinase
- PM
plasma membrane
- S1P
sphingosine-1-phosphate
- SL
sphingolipid
- SM
sphingomyelin
- Sph
sphingosine
- SphK
sphingosine kinase
Conflict of interest statement
None.
References
- Adam D, Wiegmann K, Adam-Klages S, Ruff A, Kronke M. A novel cytoplasmic domain of the p55 tumor necrosis factor receptor initiates the neutral sphingomyelinase pathway. J Biol Chem. 1996;271:14617–14622. doi: 10.1074/jbc.271.24.14617. [DOI] [PubMed] [Google Scholar]
- Adam-Klages S, Adam D, Wiegmann K, Struve S, Kolanus W, Schneider-Mergener J, et al. FAN, a novel WD-repeat protein, couples the p55 TNF-receptor to neutral sphingomyelinase. Cell. 1996;86:937–947. doi: 10.1016/s0092-8674(00)80169-5. [DOI] [PubMed] [Google Scholar]
- Adamy C, Mulder P, Khouzami L, Andrieu-abadie N, Defer N, Candiani G, et al. Neutral sphingomyelinase inhibition participates to the benefits of N-acetylcysteine treatment in post-myocardial infarction failing heart rats. J Mol Cell Cardiol. 2007;43:344–353. doi: 10.1016/j.yjmcc.2007.06.010. [DOI] [PubMed] [Google Scholar]
- Aguilar PS, Frohlich F, Rehman M, Shales M, Ulitsky I, Olivera-Couto A, et al. A plasma-membrane E-MAP reveals links of the eisosome with sphingolipid metabolism and endosomal trafficking. Nat Struct Mol Biol. 2010;17:901–908. doi: 10.1038/nsmb.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albouz S, Hauw JJ, Berwald-Netter Y, Boutry JM, Bourdon R, Baumann N. Tricyclic antidepressants induce sphingomyelinase deficiency in fibroblast and neuroblastoma cell cultures. Biomedicine. 1981;35:218–220. [PubMed] [Google Scholar]
- Alphonse G, Bionda C, Aloy MT, Ardail D, Rousson R, Rodriguez-Lafrasse C. Overcoming resistance to gamma-rays in squamous carcinoma cells by poly-drug elevation of ceramide levels. Oncogene. 2004;23:2703–2715. doi: 10.1038/sj.onc.1207357. [DOI] [PubMed] [Google Scholar]
- Amtmann E, Zoller M. Stimulation of CD95-induced apoptosis in T-cells by a subtype specific neutral sphingomyelinase inhibitor. Biochem Pharmacol. 2005;69:1141–1148. doi: 10.1016/j.bcp.2004.12.014. [DOI] [PubMed] [Google Scholar]
- Amtmann E, Zoller M, Schilling G. Neutral sphingomyelinase-inhibiting guanidines prevent herpes simplex virus-1 replication. Drugs Exp Clin Res. 2000;26:57–65. [PubMed] [Google Scholar]
- Amtmann E, Baader W, Zoller M. Neutral sphingomyelinase inhibitor C11AG prevents lipopolysaccharide-induced macrophage activation. Drugs Exp Clin Res. 2003;29:5–13. [PubMed] [Google Scholar]
- Archimandriti DT, Dalavanga YA, Cianti R, Bianchi L, Manda-Stachouli C, Armini A, et al. Proteome analysis of bronchoalveolar lavage in individuals from Metsovo, nonoccupationally exposed to asbestos. J Proteome Res. 2009;8:860–869. doi: 10.1021/pr800370n. [DOI] [PubMed] [Google Scholar]
- Arenz C. Small molecule inhibitors of acid sphingomyelinase. Cell Physiol Biochem. 2010;26:1–8. doi: 10.1159/000315100. [DOI] [PubMed] [Google Scholar]
- Arenz C, Giannis A. Synthesis of the first selective irreversible inhibitor of neutral sphingomyelinase. Angew Chem Int Ed Engl. 2000;39:1440–1442. doi: 10.1002/(sici)1521-3773(20000417)39:8<1440::aid-anie1440>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Arenz C, Gartner M, Wascholowski V, Giannis A. Synthesis and biochemical investigation of scyphostatin analogues as inhibitors of neutral sphingomyelinase. Bioorg Med Chem. 2001a;9:2901–2904. doi: 10.1016/s0968-0896(01)00165-1. [DOI] [PubMed] [Google Scholar]
- Arenz C, Thutewohl M, Block O, Waldmann H, Altenbach HJ, Giannis A. Manumycin A and its analogues are irreversible inhibitors of neutral sphingomyelinase. Chembiochem. 2001b;2:141–143. doi: 10.1002/1439-7633(20010202)2:2<141::AID-CBIC141>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Auge N, Nikolova-Karakashian M, Carpentier S, Parthasarathy S, Negre-Salvayre A, Salvayre R, et al. Role of sphingosine 1-phosphate in the mitogenesis induced by oxidized low density lipoprotein in smooth muscle cells via activation of sphingomyelinase, ceramidase, and sphingosine kinase. J Biol Chem. 1999;274:21533–21538. doi: 10.1074/jbc.274.31.21533. [DOI] [PubMed] [Google Scholar]
- Azuma N, O'Brien JS, Moser HW, Kishimoto Y. Stimulation of acid ceramidase activity by saposin D. Arch Biochem Biophys. 1994;311:354–357. doi: 10.1006/abbi.1994.1248. [DOI] [PubMed] [Google Scholar]
- Bai A, Szulc ZM, Bielawski J, Mayroo N, Liu X, Norris J, et al. Synthesis and bioevaluation of omega-N-amino analogs of B13. Bioorg Med Chem. 2009;17:1840–1848. doi: 10.1016/j.bmc.2009.01.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el Bawab S, Mao C, Obeid LM, Hannun YA. Ceramidases in the regulation of ceramide levels and function. Subcell Biochem. 2002;36:187–205. doi: 10.1007/0-306-47931-1_10. [DOI] [PubMed] [Google Scholar]
- Becker KA, Riethmuller J, Luth A, Doring G, Kleuser B, Gulbins E. Acid sphingomyelinase inhibitors normalize pulmonary ceramide and inflammation in cystic fibrosis. Am J Respir Cell Mol Biol. 2010;42:716–724. doi: 10.1165/rcmb.2009-0174OC. [DOI] [PubMed] [Google Scholar]
- Bernardo K, Hurwitz R, Zenk T, Desnick RJ, Ferlinz K, Schuchman EH, et al. Purification, characterization, and biosynthesis of human acid ceramidase. J Biol Chem. 1995;270:11098–11102. doi: 10.1074/jbc.270.19.11098. [DOI] [PubMed] [Google Scholar]
- Bernier M, Kwon YK, Pandey SK, Zhu TN, Zhao RJ, Maciuk A, et al. Binding of manumycin A inhibits IkappaB kinase beta activity. J Biol Chem. 2006;281:2551–2561. doi: 10.1074/jbc.M511878200. [DOI] [PubMed] [Google Scholar]
- Bielawska A, Greenberg MS, Perry D, Jayadev S, Shayman JA, McKay C, et al. 1S,2R)-D-erythro-2-(N-myristoylamino)-1-phenyl-1-propanol as an inhibitor of ceramidase. J Biol Chem. 1996;271:12646–12654. doi: 10.1074/jbc.271.21.12646. [DOI] [PubMed] [Google Scholar]
- Bielawska A, Bielawski J, Szulc ZM, Mayroo N, Liu X, Bai A, et al. Novel analogs of d-e-MAPP and B13. Part 2: signature effects on bioactive sphingolipids. Bioorg Med Chem. 2008;16:1032–1045. doi: 10.1016/j.bmc.2007.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brann AB, Scott R, Neuberger Y, Abulafia D, Boldin S, Fainzilber M, et al. Ceramide signaling downstream of the p75 neurotrophin receptor mediates the effects of nerve growth factor on outgrowth of cultured hippocampal neurons. J Neurosci. 1999;19:8199–8206. doi: 10.1523/JNEUROSCI.19-19-08199.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brann AB, Tcherpakov M, Williams IM, Futerman AH, Fainzilber M. Nerve growth factor-induced p75-mediated death of cultured hippocampal neurons is age-dependent and transduced through ceramide generated by neutral sphingomyelinase. J Biol Chem. 2002;277:9812–9818. doi: 10.1074/jbc.M109862200. [DOI] [PubMed] [Google Scholar]
- Callahan JW, Jones CS, Davidson DJ, Shankaran P. The active site of lysosomal sphingomyelinase: evidence for the involvement of hydrophobic and ionic groups. J Neurosci Res. 1983;10:151–163. doi: 10.1002/jnr.490100205. [DOI] [PubMed] [Google Scholar]
- Canals D, Jenkins RW, Roddy P, Hernandez-Corbacho MJ, Obeid LM, Hannun YA. Differential effects of ceramide and sphingosine-1-phosphate on ERM phosphorylation: probing sphingolipid signaling at the outer plasma membrane. J Biol Chem. 2010;285:32476–32485. doi: 10.1074/jbc.M110.141028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez JA, Holland WL, Bar J, Sandhoff K, Summers SA. Acid ceramidase overexpression prevents the inhibitory effects of saturated fatty acids on insulin signaling. J Biol Chem. 2005;280:20148–20153. doi: 10.1074/jbc.M412769200. [DOI] [PubMed] [Google Scholar]
- Choi MS, Anderson MA, Zhang Z, Zimonjic DB, Popescu N, Mukherjee AB. Neutral ceramidase gene: role in regulating ceramide-induced apoptosis. Gene. 2003;315:113–122. doi: 10.1016/s0378-1119(03)00721-2. [DOI] [PubMed] [Google Scholar]
- Clarke CJ, Snook CF, Tani M, Matmati N, Marchesini N, Hannun YA. The extended family of neutral sphingomyelinases. Biochemistry. 2006;45:11247–11256. doi: 10.1021/bi061307z. [DOI] [PubMed] [Google Scholar]
- Cogolludo A, Moreno L, Frazziano G, Moral-Sanz J, Menendez C, Castaneda J, et al. Activation of neutral sphingomyelinase is involved in acute hypoxic pulmonary vasoconstriction. Cardiovasc Res. 2009;82:296–302. doi: 10.1093/cvr/cvn349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuvillier O, Rosenthal DS, Smulson ME, Spiegel S. Sphingosine 1-phosphate inhibits activation of caspases that cleave poly(ADP-ribose) polymerase and lamins during fas- and ceramide-mediated apoptosis in jurkat T lymphocytes*. J Biol Chem. 1998;273:2910–2916. doi: 10.1074/jbc.273.5.2910. [DOI] [PubMed] [Google Scholar]
- Dawson G, Qin J. Gilenya (FTY720) inhibits acid sphingomyelinase by a mechanism similar to tricyclic antidepressants. Biochem Biophys Res Commun. 2011;404:321–323. doi: 10.1016/j.bbrc.2010.11.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado A, Casas J, Llebaria A, Abad JL, Fabrias G. Inhibitors of sphingolipid metabolism enzymes. Biochim Biophys Acta. 2006;1758:1957–1977. doi: 10.1016/j.bbamem.2006.08.017. [DOI] [PubMed] [Google Scholar]
- Delgado A, Casas J, Llebaria A, Abad JL, Fabrias G. Chemical tools to investigate sphingolipid metabolism and functions. ChemMedChem. 2007;2:580–606. doi: 10.1002/cmdc.200600195. [DOI] [PubMed] [Google Scholar]
- Duan RD, Nilsson A. Metabolism of sphingolipids in the gut and its relation to inflammation and cancer development. Prog Lipid Res. 2009;48:62–72. doi: 10.1016/j.plipres.2008.04.003. [DOI] [PubMed] [Google Scholar]
- Duan RD, Verkade HJ, Cheng Y, Havinga R, Nilsson A. Effects of bile diversion in rats on intestinal sphingomyelinases and ceramidase. Biochim Biophys Acta. 2007;1771:196–201. doi: 10.1016/j.bbalip.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Dulaney JT, Milunsky A, Sidbury JB, Hobolth N, Moser HW. Diagnosis of lipogranulomatosis (Farber disease) by use of cultured fibroblasts. J Pediatr. 1976;89:59–61. doi: 10.1016/s0022-3476(76)80927-4. [DOI] [PubMed] [Google Scholar]
- El Bawab S, Bielawska A, Hannun YA. Purification and characterization of a membrane-bound nonlysosomal ceramidase from rat brain. J Biol Chem. 1999;274:27948–27955. doi: 10.1074/jbc.274.39.27948. [DOI] [PubMed] [Google Scholar]
- El Bawab S, Roddy P, Qian T, Bielawska A, Lemasters JJ, Hannun YA. Molecular cloning and characterization of a human mitochondrial ceramidase. J Biol Chem. 2000;275:21508–21513. doi: 10.1074/jbc.M002522200. [DOI] [PubMed] [Google Scholar]
- El Bawab S, Birbes H, Roddy P, Szulc ZM, Bielawska A, Hannun YA. Biochemical characterization of the reverse activity of rat brain ceramidase. A CoA-independent and fumonisin B1-insensitive ceramide synthase. J Biol Chem. 2001;276:16758–16766. doi: 10.1074/jbc.M009331200. [DOI] [PubMed] [Google Scholar]
- Elojeimy S, Holman DH, Liu X, El-Zawahry A, Villani M, Cheng JC, et al. New insights on the use of desipramine as an inhibitor for acid ceramidase. FEBS Lett. 2006;580:4751–4756. doi: 10.1016/j.febslet.2006.07.071. [DOI] [PubMed] [Google Scholar]
- Elojeimy S, Liu X, McKillop JC, El-Zawahry AM, Holman DH, Cheng JY, et al. Role of acid ceramidase in resistance to FasL: therapeutic approaches based on acid ceramidase inhibitors and FasL gene therapy. Mol Ther. 2007;15:1259–1263. doi: 10.1038/sj.mt.6300167. [DOI] [PubMed] [Google Scholar]
- Ferlinz K, Kopal G, Bernardo K, Linke T, Bar J, Breiden B, et al. Human acid ceramidase: processing, glycosylation, and lysosomal targeting. J Biol Chem. 2001;276:35352–35360. doi: 10.1074/jbc.M103066200. [DOI] [PubMed] [Google Scholar]
- Filosto S, Castillo S, Danielson A, Franzi L, Khan E, Kenyon N, et al. Neutral Sphingomyelinase 2: a novel target in cigarette smoke-induced apoptosis and lung injury. Am J Respir Cell Mol Biol. 2011;44:350–360. doi: 10.1165/rcmb.2009-0422OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzen R, Pautz A, Brautigam L, Geisslinger G, Pfeilschifter J, Huwiler A. Interleukin-1beta induces chronic activation and de novo synthesis of neutral ceramidase in renal mesangial cells. J Biol Chem. 2001;276:35382–35389. doi: 10.1074/jbc.M102153200. [DOI] [PubMed] [Google Scholar]
- Furlong SJ, Ridgway ND, Hoskin DW. Modulation of ceramide metabolism in T-leukemia cell lines potentiates apoptosis induced by the cationic antimicrobial peptide bovine lactoferricin. Int J Oncol. 2008;32:537–544. [PubMed] [Google Scholar]
- Galadari S, Wu BX, Mao C, Roddy P, El Bawab S, Hannun YA. Identification of a novel amidase motif in neutral ceramidase. Biochem J. 2006;393:687–695. doi: 10.1042/BJ20050682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangoiti P, Camacho L, Arana L, Ouro A, Granado MH, Brizuela L, et al. Control of metabolism and signaling of simple bioactive sphingolipids: implications in disease. Prog Lipid Res. 2010;49:316–334. doi: 10.1016/j.plipres.2010.02.004. [DOI] [PubMed] [Google Scholar]
- Gatt S. Enzymic hydrolysis and synthesis of ceramides. J Biol Chem. 1963;238:3131–3133. [PubMed] [Google Scholar]
- Grijalvo S, Bedia C, Triola G, Casas J, Llebaria A, Teixido J, et al. Design, synthesis and activity as acid ceramidase inhibitors of 2-oxooctanoyl and N-oleoylethanolamine analogues. Chem Phys Lipids. 2006;144:69–84. doi: 10.1016/j.chemphyslip.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Gulbins E, Li PL. Physiological and pathophysiological aspects of ceramide. Am J Physiol Regul Integr Comp Physiol. 2006;290:R11–R26. doi: 10.1152/ajpregu.00416.2005. [DOI] [PubMed] [Google Scholar]
- Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008;9:139–150. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- Hannun YA, Loomis CR, Merrill AH, Jr, Bell RM. Sphingosine inhibition of protein kinase C activity and of phorbol dibutyrate binding in vitro and in human platelets. J Biol Chem. 1986;261:12604–12609. [PubMed] [Google Scholar]
- Hara M, Akasaka K, Akinaga S, Okabe M, Nakano H, Gomez R, et al. Identification of Ras farnesyltransferase inhibitors by microbial screening. Proc Natl Acad Sci U S A. 1993;90:2281–2285. doi: 10.1073/pnas.90.6.2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara S, Nakashima S, Kiyono T, Sawada M, Yoshimura S, Iwama T, et al. Ceramide triggers caspase activation during gamma-radiation-induced apoptosis of human glioma cells lacking functional p53. Oncol Rep. 2004;12:119–123. [PubMed] [Google Scholar]
- Haughey NJ, Cutler RG, Tamara A, McArthur JC, Vargas DL, Pardo CA, et al. Perturbation of sphingolipid metabolism and ceramide production in HIV-dementia. Ann Neurol. 2004;55:257–267. doi: 10.1002/ana.10828. [DOI] [PubMed] [Google Scholar]
- He X, Okino N, Dhami R, Dagan A, Gatt S, Schulze H, et al. Purification and characterization of recombinant, human acid ceramidase. Catalytic reactions and interactions with acid sphingomyelinase. J Biol Chem. 2003;278:32978–32986. doi: 10.1074/jbc.M301936200. [DOI] [PubMed] [Google Scholar]
- Holman DH, Turner LS, El-Zawahry A, Elojeimy S, Liu X, Bielawski J, et al. Lysosomotropic acid ceramidase inhibitor induces apoptosis in prostate cancer cells. Cancer Chemother Pharmacol. 2008;61:231–242. doi: 10.1007/s00280-007-0465-0. [DOI] [PubMed] [Google Scholar]
- Horton JW. Tumor necrosis factor-alpha, sphingosine, ceramide: which is the appropriate marker of inflammation? Crit Care Med. 1999;27:2580–2581. doi: 10.1097/00003246-199911000-00048. [DOI] [PubMed] [Google Scholar]
- Hu W, Xu R, Sun W, Szulc ZM, Bielawski J, Obeid LM, et al. Alkaline ceramidase 3 (ACER3) hydrolyzes unsaturated long-chain ceramides, and its down-regulation inhibits both cell proliferation and apoptosis. J Biol Chem. 2010;285:7964–7976. doi: 10.1074/jbc.M109.063586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurwitz R, Ferlinz K, Sandhoff K. The tricyclic antidepressant desipramine causes proteolytic degradation of lysosomal sphingomyelinase in human fibroblasts. Biol Chem Hoppe Seyler. 1994;375:447–450. doi: 10.1515/bchm3.1994.375.7.447. [DOI] [PubMed] [Google Scholar]
- Huwiler A, Pfeilschifter J. Altering the sphingosine-1-phosphate/ceramide balance: a promising approach for tumor therapy. Curr Pharm Des. 2006;12:4625–4635. doi: 10.2174/138161206779010422. [DOI] [PubMed] [Google Scholar]
- Hwang YH, Tani M, Nakagawa T, Okino N, Ito M. Subcellular localization of human neutral ceramidase expressed in HEK293 cells. Biochem Biophys Res Commun. 2005;331:37–42. doi: 10.1016/j.bbrc.2005.03.134. [DOI] [PubMed] [Google Scholar]
- Inoue T, Okino N, Kakuta Y, Hijikata A, Okano H, Goda HM, et al. Mechanistic insights into the hydrolysis and synthesis of ceramide by neutral ceramidase. J Biol Chem. 2009;284:9566–9577. doi: 10.1074/jbc.M808232200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffrezou JP, Herbert JM, Levade T, Gau MN, Chatelain P, Laurent G. Reversal of multidrug resistance by calcium channel blocker SR33557 without photoaffinity labeling of P-glycoprotein. J Biol Chem. 1991;266:19858–19864. [PubMed] [Google Scholar]
- Jana A, Pahan K. Human immunodeficiency virus type 1 gp120 induces apoptosis in human primary neurons through redox-regulated activation of neutral sphingomyelinase. J Neurosci. 2004;24:9531–9540. doi: 10.1523/JNEUROSCI.3085-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins RW, Canals D, Hannun YA. Roles and regulation of secretory and lysosomal acid sphingomyelinase. Cell Signal. 2009;21:836–846. doi: 10.1016/j.cellsig.2009.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanto T, Kalinski P, Hunter OC, Lotze MT, Amoscato AA. Ceramide mediates tumor-induced dendritic cell apoptosis. J Immunol. 2001;167:3773–3784. doi: 10.4049/jimmunol.167.7.3773. [DOI] [PubMed] [Google Scholar]
- Kim WJ, Okimoto RA, Purton LE, Goodwin M, Haserlat SM, Dayyani F, et al. Mutations in the neutral sphingomyelinase gene SMPD3 implicate the ceramide pathway in human leukemias. Blood. 2008;111:4716–4722. doi: 10.1182/blood-2007-10-113068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch J, Gartner S, Li CM, Quintern LE, Bernardo K, Levran O, et al. Molecular cloning and characterization of a full-length complementary DNA encoding human acid ceramidase. Identification of the first molecular lesion causing Farber disease. J Biol Chem. 1996;271:33110–33115. doi: 10.1074/jbc.271.51.33110. [DOI] [PubMed] [Google Scholar]
- Kolesnick RN. 1,2-Diacylglycerols but not phorbol esters stimulate sphingomyelin hydrolysis in GH3 pituitary cells. J Biol Chem. 1987;262:16759–16762. [PubMed] [Google Scholar]
- Kolzer M, Arenz C, Ferlinz K, Werth N, Schulze H, Klingenstein R, et al. Phosphatidylinositol-3,5-Bisphosphate is a potent and selective inhibitor of acid sphingomyelinase. Biol Chem. 2003;384:1293–1298. doi: 10.1515/BC.2003.144. [DOI] [PubMed] [Google Scholar]
- Kornhuber J, Tripal P, Reichel M, Muhle C, Rhein C, Muehlbacher M, et al. Functional Inhibitors of Acid Sphingomyelinase (FIASMAs): a novel pharmacological group of drugs with broad clinical applications. Cell Physiol Biochem. 2010;26:9–20. doi: 10.1159/000315101. [DOI] [PubMed] [Google Scholar]
- Kraveka JM, Li L, Szulc ZM, Bielawski J, Ogretmen B, Hannun YA, et al. Involvement of dihydroceramide desaturase in cell cycle progression in human neuroblastoma cells. J Biol Chem. 2007;282:16718–16728. doi: 10.1074/jbc.M700647200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupchak BR, Garitaonandia I, Villa NY, Smith JL, Lyons TJ. Antagonism of human adiponectin receptors and their membrane progesterone receptor paralogs by TNFalpha and a ceramidase inhibitor. Biochemistry. 2009;48:5504–5506. doi: 10.1021/bi9006258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang PA, Schenck M, Nicolay JP, Becker JU, Kempe DS, Lupescu A, et al. Liver cell death and anemia in Wilson disease involve acid sphingomyelinase and ceramide. Nat Med. 2007;13:164–170. doi: 10.1038/nm1539. [DOI] [PubMed] [Google Scholar]
- Lee JT, Xu J, Lee JM, Ku G, Han X, Yang DI, et al. Amyloid-beta peptide induces oligodendrocyte death by activating the neutral sphingomyelinase-ceramide pathway. J Cell Biol. 2004;164:123–131. doi: 10.1083/jcb.200307017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepine S, Lakatos B, Courageot MP, Le Stunff H, Sulpice JC, Giraud F. Sphingosine contributes to glucocorticoid-induced apoptosis of thymocytes independently of the mitochondrial pathway. J Immunol. 2004;173:3783–3790. doi: 10.4049/jimmunol.173.6.3783. [DOI] [PubMed] [Google Scholar]
- Levade T, Vidal F, Vermeersch S, Andrieu N, Gatt S, Salvayre R. Degradation of fluorescent and radiolabelled sphingomyelins in intact cells by a non-lysosomal pathway. Biochim Biophys Acta. 1995;1258:277–287. doi: 10.1016/0005-2760(95)00132-v. [DOI] [PubMed] [Google Scholar]
- Li CM, Hong SB, Kopal G, He X, Linke T, Hou WS, et al. Cloning and characterization of the full-length cDNA and genomic sequences encoding murine acid ceramidase. Genomics. 1998;50:267–274. doi: 10.1006/geno.1998.5334. [DOI] [PubMed] [Google Scholar]
- Li CM, Park JH, Simonaro CM, He X, Gordon RE, Friedman AH, et al. Insertional mutagenesis of the mouse acid ceramidase gene leads to early embryonic lethality in homozygotes and progressive lipid storage disease in heterozygotes. Genomics. 2002;79:218–224. doi: 10.1006/geno.2002.6686. [DOI] [PubMed] [Google Scholar]
- Linke T, Wilkening G, Lansmann S, Moczall H, Bartelsen O, Weisgerber J, et al. Stimulation of acid sphingomyelinase activity by lysosomal lipids and sphingolipid activator proteins. Biol Chem. 2001a;382:283–290. doi: 10.1515/BC.2001.035. [DOI] [PubMed] [Google Scholar]
- Linke T, Wilkening G, Sadeghlar F, Mozcall H, Bernardo K, Schuchman E, et al. Interfacial regulation of acid ceramidase activity. Stimulation of ceramide degradation by lysosomal lipids and sphingolipid activator proteins. J Biol Chem. 2001b;276:5760–5768. doi: 10.1074/jbc.M006846200. [DOI] [PubMed] [Google Scholar]
- Lister MD, Ruan ZS, Bittman R. Interaction of sphingomyelinase with sphingomyelin analogs modified at the C-1 and C-3 positions of the sphingosine backbone. Biochim Biophys Acta. 1995;1256:25–30. doi: 10.1016/0005-2760(94)00249-x. [DOI] [PubMed] [Google Scholar]
- Liu B, Hannun YA. Inhibition of the neutral magnesium-dependent sphingomyelinase by glutathione. J Biol Chem. 1997;272:16281–16287. doi: 10.1074/jbc.272.26.16281. [DOI] [PubMed] [Google Scholar]
- Liu B, Hassler DF, Smith GK, Weaver K, Hannun YA. Purification and characterization of a membrane bound neutral pH optimum magnesium-dependent and phosphatidylserine-stimulated sphingomyelinase from rat brain. J Biol Chem. 1998;273:34472–34479. doi: 10.1074/jbc.273.51.34472. [DOI] [PubMed] [Google Scholar]
- Liu G, Wang W, Sun G, Ma X, Liu Z, Yang J. Nystatin interferes with the effects of N-methyl-N'-nitro-N-nitrosoguanidine on sphingolipid metabolism in human FL cells. Lipids. 2008;43:867–875. doi: 10.1007/s11745-008-3209-y. [DOI] [PubMed] [Google Scholar]
- Llacuna L, Mari M, Garcia-Ruiz C, Fernandez-Checa JC, Morales A. Critical role of acidic sphingomyelinase in murine hepatic ischemia-reperfusion injury. Hepatology. 2006;44:561–572. doi: 10.1002/hep.21285. [DOI] [PubMed] [Google Scholar]
- Lozano J, Morales A, Cremesti A, Fuks Z, Tilly JL, Schuchman E, et al. Niemann-Pick Disease versus acid sphingomyelinase deficiency. Cell Death Differ. 2001;8:100–103. doi: 10.1038/sj.cdd.4400775. [DOI] [PubMed] [Google Scholar]
- Luberto C, Hassler DF, Signorelli P, Okamoto Y, Sawai H, Boros E, et al. Inhibition of tumor necrosis factor-induced cell death in MCF7 by a novel inhibitor of neutral sphingomyelinase. J Biol Chem. 2002;277:41128–41139. doi: 10.1074/jbc.M206747200. [DOI] [PubMed] [Google Scholar]
- Mahdy AE, Cheng JC, Li J, Elojeimy S, Meacham WD, Turner LS, et al. Acid ceramidase upregulation in prostate cancer cells confers resistance to radiation: AC inhibition, a potential radiosensitizer. Mol Ther. 2009;17:430–438. doi: 10.1038/mt.2008.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao C, Obeid LM. Ceramidases: regulators of cellular responses mediated by ceramide, sphingosine, and sphingosine-1-phosphate. Biochim Biophys Acta. 2008;1781:424–434. doi: 10.1016/j.bbalip.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao C, Xu R, Szulc ZM, Bielawska A, Galadari SH, Obeid LM. Cloning and characterization of a novel human alkaline ceramidase. A mammalian enzyme that hydrolyzes phytoceramide. J Biol Chem. 2001;276:26577–26588. doi: 10.1074/jbc.M102818200. [DOI] [PubMed] [Google Scholar]
- Mao C, Xu R, Szulc ZM, Bielawski J, Becker KP, Bielawska A, et al. Cloning and characterization of a mouse endoplasmic reticulum alkaline ceramidase: an enzyme that preferentially regulates metabolism of very long chain ceramides. J Biol Chem. 2003;278:31184–31191. doi: 10.1074/jbc.M303875200. [DOI] [PubMed] [Google Scholar]
- Mao Z, Sun W, Xu R, Novgorodov S, Szulc ZM, Bielawski J, et al. Alkaline ceramidase 2 (ACER2) and its product dihydrosphingosine mediate the cytotoxicity of N-(4-hydroxyphenyl)retinamide in tumor cells. J Biol Chem. 2010;285:29078–29090. doi: 10.1074/jbc.M110.105296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maor I, Mandel H, Aviram M. Macrophage uptake of oxidized LDL inhibits lysosomal sphingomyelinase, thus causing the accumulation of unesterified cholesterol-sphingomyelin-rich particles in the lysosomes. A possible role for 7-Ketocholesterol. Arterioscler Thromb Vasc Biol. 1995;15:1378–1387. doi: 10.1161/01.atv.15.9.1378. [DOI] [PubMed] [Google Scholar]
- Marchesini N, Luberto C, Hannun YA. Biochemical properties of mammalian neutral sphingomyelinase 2 and its role in sphingolipid metabolism. J Biol Chem. 2003;278:13775–13783. doi: 10.1074/jbc.M212262200. [DOI] [PubMed] [Google Scholar]
- Marchesini N, Osta W, Bielawski J, Luberto C, Obeid LM, Hannun YA. Role for mammalian neutral sphingomyelinase 2 in confluence-induced growth arrest of MCF7 cells. J Biol Chem. 2004;279:25101–25111. doi: 10.1074/jbc.M313662200. [DOI] [PubMed] [Google Scholar]
- Marchesini N, Jones JA, Hannun YA. Confluence induced threonine41/serine45 phospho-beta-catenin dephosphorylation via ceramide-mediated activation of PP1cgamma. Biochim Biophys Acta. 2007;1771:1418–1428. doi: 10.1016/j.bbalip.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martelli EA, Toth E, Segre AD, Corsico N. Mechanism of inhibition of experimental inflammation by antidepressant drugs. Eur J Pharmacol. 1967;2:229–233. doi: 10.1016/0014-2999(67)90092-1. [DOI] [PubMed] [Google Scholar]
- Martin SF, Navarro F, Forthoffer N, Navas P, Villalba JM. Neutral magnesium-dependent sphingomyelinase from liver plasma membrane: purification and inhibition by ubiquinol. J Bioenerg Biomembr. 2001;33:143–153. doi: 10.1023/a:1010704715979. [DOI] [PubMed] [Google Scholar]
- Martin SF, Gomez-Diaz C, Bello RI, Navas P, Villalba JM. Inhibition of neutral Mg2+-dependent sphingomyelinase by ubiquinol-mediated plasma membrane electron transport. Protoplasma. 2003;221:109–116. doi: 10.1007/s00709-002-0070-3. [DOI] [PubMed] [Google Scholar]
- Masson M, Albouz S, Boutry JM, Spezzatti B, Castagna M, Baumann N. Calmodulin antagonist W-7 inhibits lysosomal sphingomyelinase activity in C6 glioma cells. J Neurochem. 1989;52:1645–1647. doi: 10.1111/j.1471-4159.1989.tb09221.x. [DOI] [PubMed] [Google Scholar]
- Maupas-Schwalm F, Auge N, Robinet C, Cambus JP, Parsons SJ, Salvayre R, et al. The sphingomyelin/ceramide pathway is involved in ERK1/2 phosphorylation, cell proliferation, and uPAR overexpression induced by tissue-type plasminogen activator. FASEB J. 2004;18:1398–1400. doi: 10.1096/fj.03-1123fje. [DOI] [PubMed] [Google Scholar]
- Merrill AH, Jr, Sereni AM, Stevens VL, Hannun YA, Bell RM, Kinkade JM., Jr Inhibition of phorbol ester-dependent differentiation of human promyelocytic leukemic (HL-60) cells by sphinganine and other long-chain bases. J Biol Chem. 1986;261:12610–12615. [PubMed] [Google Scholar]
- Mitsutake S, Tani M, Okino N, Mori K, Ichinose S, Omori A, et al. Purification, characterization, molecular cloning, and subcellular distribution of neutral ceramidase of rat kidney. J Biol Chem. 2001;276:26249–26259. doi: 10.1074/jbc.M102233200. [DOI] [PubMed] [Google Scholar]
- Monjusho H, Okino N, Tani M, Maeda M, Yoshida M, Ito M. A neutral ceramidase homologue from Dictyostelium discoideum exhibits an acidic pH optimum. Biochem J. 2003;376:473–479. doi: 10.1042/BJ20030652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nara F, Tanaka M, Hosoya T, Suzuki-Konagai K, Ogita T. Scyphostatin, a neutral sphingomyelinase inhibitor from a discomycete, Trichopeziza mollissima: taxonomy of the producing organism, fermentation, isolation, and physico-chemical properties. J Antibiot. 1999a;52:525–530. doi: 10.7164/antibiotics.52.525. [DOI] [PubMed] [Google Scholar]
- Nara F, Tanaka M, Masuda-Inoue S, Yamasato Y, Doi-Yoshioka H, Suzuki-Konagai K, et al. Biological activities of scyphostatin, a neutral sphingomyelinase inhibitor from a discomycete, Trichopeziza mollissima. J Antibiot. 1999b;52:531–535. doi: 10.7164/antibiotics.52.531. [DOI] [PubMed] [Google Scholar]
- Nassogne MC, Lizarraga C, N'Kuli F, Van Bambeke F, Van Binst R, Wallemacq P, et al. Cocaine induces a mixed lysosomal lipidosis in cultured fibroblasts, by inactivation of acid sphingomyelinase and inhibition of phospholipase A1. Toxicol Appl Pharmacol. 2004;194:101–110. doi: 10.1016/j.taap.2003.09.026. [DOI] [PubMed] [Google Scholar]
- Nemoto S, Nakamura M, Osawa Y, Kono S, Itoh Y, Okano Y, et al. Sphingosine kinase isoforms regulate oxaliplatin sensitivity of human colon cancer cells through ceramide accumulation and Akt activation. J Biol Chem. 2009;284:10422–10432. doi: 10.1074/jbc.M900735200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolova-Karakashian MN, Russell RW, Booth RA, Jenden DJ, Merrill AH., Jr Sphingomyelin metabolism in rat liver after chronic dietary replacement of choline by N-aminodeanol. J Lipid Res. 1997;38:1764–1770. [PubMed] [Google Scholar]
- Nilsson A. The presence of spingomyelin- and ceramide-cleaving enzymes in the small intestinal tract. Biochim Biophys Acta. 1969;176:339–347. doi: 10.1016/0005-2760(69)90192-1. [DOI] [PubMed] [Google Scholar]
- Ohlsson L, Palmberg C, Duan RD, Olsson M, Bergman T, Nilsson A. Purification and characterization of human intestinal neutral ceramidase. Biochimie. 2007;89:950–960. doi: 10.1016/j.biochi.2007.03.009. [DOI] [PubMed] [Google Scholar]
- Ohlsson L, Hjelte L, Huhn M, Scholte BJ, Wilke M, Flodstrom-Tullberg M, et al. Expression of intestinal and lung alkaline sphingomyelinase and neutral ceramidase in cystic fibrosis f508del transgenic mice. J Pediatr Gastroenterol Nutr. 2008;47:547–554. doi: 10.1097/MPG.0b013e3181826daf. [DOI] [PubMed] [Google Scholar]
- Okazaki T, Bell RM, Hannun YA. Sphingomyelin turnover induced by vitamin D3 in HL-60 cells. Role in cell differentiation. J Biol Chem. 1989;264:19076–19080. [PubMed] [Google Scholar]
- Okino N, Ichinose S, Omori A, Imayama S, Nakamura T, Ito M. Molecular cloning, sequencing, and expression of the gene encoding alkaline ceramidase from Pseudomonas aeruginosa. Cloning of a ceramidase homologue from Mycobacterium tuberculosis. J Biol Chem. 1999;274:36616–36622. doi: 10.1074/jbc.274.51.36616. [DOI] [PubMed] [Google Scholar]
- Okino N, He X, Gatt S, Sandhoff K, Ito M, Schuchman EH. The reverse activity of human acid ceramidase. J Biol Chem. 2003;278:29948–29953. doi: 10.1074/jbc.M303310200. [DOI] [PubMed] [Google Scholar]
- Okino N, Ikeda R, Ito M. Expression, purification, and characterization of a recombinant neutral ceramidase from Mycobacterium tuberculosis. Biosci Biotechnol Biochem. 2010;74:316–321. doi: 10.1271/bbb.90645. [DOI] [PubMed] [Google Scholar]
- Olsson M, Duan RD, Ohlsson L, Nilsson A. Rat intestinal ceramidase: purification, properties, and physiological relevance. Am J Physiol Gastrointest Liver Physiol. 2004;287:G929–G937. doi: 10.1152/ajpgi.00155.2004. [DOI] [PubMed] [Google Scholar]
- Pappu A, Hostetler KY. Effect of cationic amphiphilic drugs on the hydrolysis of acidic and neutral phospholipids by liver lysosomal phospholipase A. Biochem Pharmacol. 1984;33:1639–1644. doi: 10.1016/0006-2952(84)90286-7. [DOI] [PubMed] [Google Scholar]
- Pata MO, Wu BX, Bielawski J, Xiong TC, Hannun YA, Ng CK. Molecular cloning and characterization of OsCDase, a ceramidase enzyme from rice. Plant J. 2008;55:1000–1009. doi: 10.1111/j.1365-313X.2008.03569.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne SG, Brindley DN, Guilbert LJ. Epidermal growth factor inhibits ceramide-induced apoptosis and lowers ceramide levels in primary placental trophoblasts. J Cell Physiol. 1999;180:263–270. doi: 10.1002/(SICI)1097-4652(199908)180:2<263::AID-JCP14>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Peng CH, Huang CN, Hsu SP, Wang CJ. Penta-acetyl geniposide induce apoptosis in C6 glioma cells by modulating the activation of neutral sphingomyelinase-induced p75 nerve growth factor receptor and protein kinase Cdelta pathway. Mol Pharmacol. 2006;70:997–1004. doi: 10.1124/mol.106.022178. [DOI] [PubMed] [Google Scholar]
- Philipp S, Puchert M, Adam-Klages S, Tchikov V, Winoto-Morbach S, Mathieu S, et al. The Polycomb group protein EED couples TNF receptor 1 to neutral sphingomyelinase. Proc Natl Acad Sci U S A. 2010;107:1112–1117. doi: 10.1073/pnas.0908486107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitman MR, Pitson SM. Inhibitors of the sphingosine kinase pathway as potential therapeutics. Curr Cancer Drug Targets. 2010;10:354–367. doi: 10.2174/156800910791208599. [DOI] [PubMed] [Google Scholar]
- Pitsinos EN, Wascholowski V, Karaliota S, Rigou C, Couladouros EA, Giannis A. Synthesis and evaluation of three novel scyphostatin analogues as neutral sphingomyelinase inhibitors. Chembiochem. 2003;4:1223–1225. doi: 10.1002/cbic.200300667. [DOI] [PubMed] [Google Scholar]
- Pyne NJ, Pyne S. Sphingosine 1-phosphate and cancer. Nat Rev Cancer. 2010;10:489–503. doi: 10.1038/nrc2875. [DOI] [PubMed] [Google Scholar]
- Raisova M, Goltz G, Bektas M, Bielawska A, Riebeling C, Hossini AM, et al. Bcl-2 overexpression prevents apoptosis induced by ceramidase inhibitors in malignant melanoma and HaCaT keratinocytes. FEBS Lett. 2002;516:47–52. doi: 10.1016/s0014-5793(02)02472-9. [DOI] [PubMed] [Google Scholar]
- Reagan JW, Jr, Hubbert ML, Shelness GS. Posttranslational regulation of acid sphingomyelinase in niemann-pick type C1 fibroblasts and free cholesterol-enriched chinese hamster ovary cells. J Biol Chem. 2000;275:38104–38110. doi: 10.1074/jbc.M005296200. [DOI] [PubMed] [Google Scholar]
- Riethmuller J, Anthonysamy J, Serra E, Schwab M, Doring G, Gulbins E. Therapeutic efficacy and safety of amitriptyline in patients with cystic fibrosis. Cell Physiol Biochem. 2009;24:65–72. doi: 10.1159/000227814. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Lafrasse C, Alphonse G, Aloy MT, Ardail D, Gerard JP, Louisot P, et al. Increasing endogenous ceramide using inhibitors of sphingolipid metabolism maximizes ionizing radiation-induced mitochondrial injury and apoptotic cell killing. Int J Cancer. 2002;101:589–598. doi: 10.1002/ijc.10652. [DOI] [PubMed] [Google Scholar]
- Roth AG, Drescher D, Yang Y, Redmer S, Uhlig S, Arenz C. Potent and selective inhibition of acid sphingomyelinase by bisphosphonates. Angew Chem Int Ed Engl. 2009a;48:7560–7563. doi: 10.1002/anie.200903288. [DOI] [PubMed] [Google Scholar]
- Roth AG, Redmer S, Arenz C. Potent inhibition of acid sphingomyelinase by phosphoinositide analogues. Chembiochem. 2009b;10:2367–2374. doi: 10.1002/cbic.200900281. [DOI] [PubMed] [Google Scholar]
- Roth AG, Redmer S, Arenz C. Development of carbohydrate-derived inhibitors of acid sphingomyelinase. Bioorg Med Chem. 2010;18:939–944. doi: 10.1016/j.bmc.2009.11.030. [DOI] [PubMed] [Google Scholar]
- Roumestan C, Michel A, Bichon F, Portet K, Detoc M, Henriquet C, et al. Anti-inflammatory properties of desipramine and fluoxetine. Respir Res. 2007;8:35. doi: 10.1186/1465-9921-8-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruckhaberle E, Rody A, Engels K, Gaetje R, von Minckwitz G, Schiffmann S, et al. Microarray analysis of altered sphingolipid metabolism reveals prognostic significance of sphingosine kinase 1 in breast cancer. Breast Cancer Res Treat. 2008;112:41–52. doi: 10.1007/s10549-007-9836-9. [DOI] [PubMed] [Google Scholar]
- Ruckhaberle E, Holtrich U, Engels K, Hanker L, Gatje R, Metzler D, et al. Acid ceramidase 1 expression correlates with a better prognosis in ER-positive breast cancer. Climacteric. 2009;12:502–513. doi: 10.3109/13697130902939913. [DOI] [PubMed] [Google Scholar]
- Rutkute K, Karakashian AA, Giltiay NV, Dobierzewska A, Nikolova-Karakashian MN. Aging in rat causes hepatic hyperresposiveness to interleukin-1beta which is mediated by neutral sphingomyelinase-2. Hepatology. 2007;46:1166–1176. doi: 10.1002/hep.21777. [DOI] [PubMed] [Google Scholar]
- Saad AF, Meacham WD, Bai A, Anelli V, Elojeimy S, Mahdy AE, et al. The functional effects of acid ceramidase overexpression in prostate cancer progression and resistance to chemotherapy. Cancer Biol Ther. 2007;6:1455–1460. doi: 10.4161/cbt.6.9.4623. [DOI] [PubMed] [Google Scholar]
- Sakata A, Ochiai T, Shimeno H, Hikishima S, Yokomatsu T, Shibuya S, et al. Acid sphingomyelinase inhibition suppresses lipopolysaccharide-mediated release of inflammatory cytokines from macrophages and protects against disease pathology in dextran sulphate sodium-induced colitis in mice. Immunology. 2007a;122:54–64. doi: 10.1111/j.1365-2567.2007.02612.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakata A, Yasuda K, Ochiai T, Shimeno H, Hikishima S, Yokomatsu T, et al. Inhibition of lipopolysaccharide-induced release of interleukin-8 from intestinal epithelial cells by SMA, a novel inhibitor of sphingomyelinase and its therapeutic effect on dextran sulphate sodium-induced colitis in mice. Cell Immunol. 2007b;245:24–31. doi: 10.1016/j.cellimm.2007.03.005. [DOI] [PubMed] [Google Scholar]
- Schissel SL, Keesler GA, Schuchman EH, Williams KJ, Tabas I. The cellular trafficking and zinc dependence of secretory and lysosomal sphingomyelinase, two products of the acid sphingomyelinase gene. J Biol Chem. 1998;273:18250–18259. doi: 10.1074/jbc.273.29.18250. [DOI] [PubMed] [Google Scholar]
- Schuchman EH. The pathogenesis and treatment of acid sphingomyelinase-deficient Niemann-Pick disease. J Inherit Metab Dis. 2007;30:654–663. doi: 10.1007/s10545-007-0632-9. [DOI] [PubMed] [Google Scholar]
- Schuchman EH. Acid sphingomyelinase, cell membranes and human disease: lessons from Niemann-Pick disease. FEBS Lett. 2010;584:1895–1900. doi: 10.1016/j.febslet.2009.11.083. [DOI] [PubMed] [Google Scholar]
- Schulze H, Schepers U, Sandhoff K. Overexpression and mass spectrometry analysis of mature human acid ceramidase. Biol Chem. 2007;388:1333–1343. doi: 10.1515/BC.2007.152. [DOI] [PubMed] [Google Scholar]
- Selzner M, Bielawska A, Morse MA, Rudiger HA, Sindram D, Hannun YA, et al. Induction of apoptotic cell death and prevention of tumor growth by ceramide analogues in metastatic human colon cancer. Cancer Res. 2001;61:1233–1240. [PubMed] [Google Scholar]
- Shah C, Yang G, Lee I, Bielawski J, Hannun YA, Samad F. Protection from high fat diet-induced increase in ceramide in mice lacking plasminogen activator inhibitor 1. J Biol Chem. 2008;283:13538–13548. doi: 10.1074/jbc.M709950200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu M, Tada E, Makiyama T, Yasufuku K, Moriyama Y, Fujino H, et al. Effects of ceramide, ceramidase inhibition and expression of ceramide kinase on cytosolic phospholipase A2alpha; additional role of ceramide-1-phosphate in phosphorylation and Ca2+ signaling. Cell Signal. 2009;21:440–447. doi: 10.1016/j.cellsig.2008.11.014. [DOI] [PubMed] [Google Scholar]
- Sitrin RG, Sassanella TM, Petty HR. An obligate role for membrane-associated neutral sphingomyelinase activity in orienting chemotactic migration of human neutrophils. Am J Respir Cell Mol Biol. 2010;44:205–212. doi: 10.1165/rcmb.2010-0019OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slater AF, Stefan C, Nobel I, van den Dobbelsteen DJ, Orrenius S. Signalling mechanisms and oxidative stress in apoptosis. Toxicol Lett. 1995;82–83:149–153. doi: 10.1016/0378-4274(95)03474-9. [DOI] [PubMed] [Google Scholar]
- Smith EL, Schuchman EH. The unexpected role of acid sphingomyelinase in cell death and the pathophysiology of common diseases. Faseb J. 2008;22:3419–3431. doi: 10.1096/fj.08-108043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AR, Visioli F, Frei B, Hagen TM. Age-related changes in endothelial nitric oxide synthase phosphorylation and nitric oxide dependent vasodilation: evidence for a novel mechanism involving sphingomyelinase and ceramide-activated phosphatase 2A. Aging Cell. 2006;5:391–400. doi: 10.1111/j.1474-9726.2006.00232.x. [DOI] [PubMed] [Google Scholar]
- Snider AJ, Orr Gandy KA, Obeid LM. Sphingosine kinase: role in regulation of bioactive sphingolipid mediators in inflammation. Biochimie. 2010;92:707–715. doi: 10.1016/j.biochi.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda K, Sakamoto T, Yoshikawa H, Ashizuka S, Ohshima Y, Kishihara K, et al. Inhibition of corneal inflammation by the topical use of Ras farnesyltransferase inhibitors: selective inhibition of macrophage localization. Invest Ophthalmol Vis Sci. 1998;39:2245–2251. [PubMed] [Google Scholar]
- Spence MW, Beed S, Cook HW. Acid and alkaline ceramidases of rat tissues. Biochem Cell Biol. 1986;64:400–404. doi: 10.1139/o86-056. [DOI] [PubMed] [Google Scholar]
- Spinedi A, Di Bartolomeo S, Piacentini M. N-Oleoylethanolamine inhibits glucosylation of natural ceramides in CHP-100 neuroepithelioma cells: possible implications for apoptosis. Biochem Biophys Res Commun. 1999;255:456–459. doi: 10.1006/bbrc.1999.0230. [DOI] [PubMed] [Google Scholar]
- Stancevic B, Kolesnick R. Ceramide-rich platforms in transmembrane signaling. FEBS Lett. 2010;584:1728–1740. doi: 10.1016/j.febslet.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoffel W, Melzner I. Studies in vitro on the biosynthesis of ceramide and sphingomyelin. A reevaluation of proposed pathways. Hoppe Seylers Z Physiol Chem. 1980;361:755–771. doi: 10.1515/bchm2.1980.361.1.755. [DOI] [PubMed] [Google Scholar]
- Stoffel W, Jenke B, Holz B, Binczek E, Gunter RH, Knifka J, et al. Neutral sphingomyelinase (SMPD3) deficiency causes a novel form of chondrodysplasia and dwarfism that is rescued by Col2A1-driven smpd3 transgene expression. Am J Pathol. 2007;171:153–161. doi: 10.2353/ajpath.2007.061285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strelow A, Bernardo K, Adam-Klages S, Linke T, Sandhoff K, Kronke M, et al. Overexpression of acid ceramidase protects from tumor necrosis factor-induced cell death. J Exp Med. 2000;192:601–612. doi: 10.1084/jem.192.5.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturley SL, Patterson MC, Balch W, Liscum L. The pathophysiology and mechanisms of NP-C disease. Biochim Biophys Acta. 2004;1685:83–87. doi: 10.1016/j.bbalip.2004.08.014. [DOI] [PubMed] [Google Scholar]
- Sugita M, Willians M, Dulaney JT, Moser HW. Ceramidase and ceramide synthesis in human kidney and cerebellum. Description of a new alkaline ceramidase. Biochim Biophys Acta. 1975;398:125–131. doi: 10.1016/0005-2760(75)90176-9. [DOI] [PubMed] [Google Scholar]
- Sun W, Xu R, Hu W, Jin J, Crellin HA, Bielawski J, et al. Upregulation of the human alkaline ceramidase 1 and acid ceramidase mediates calcium-induced differentiation of epidermal keratinocytes. J Invest Dermatol. 2008;128:389–397. doi: 10.1038/sj.jid.5701025. [DOI] [PubMed] [Google Scholar]
- Sun W, Hu W, Xu R, Jin J, Szulc ZM, Zhang G, et al. Alkaline ceramidase 2 regulates beta1 integrin maturation and cell adhesion. FASEB J. 2009;23:656–666. doi: 10.1096/fj.08-115634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I. Secretory sphingomyelinase. Chem Phys Lipids. 1999;102:123–130. doi: 10.1016/s0009-3084(99)00080-8. [DOI] [PubMed] [Google Scholar]
- Tabatadze N, Savonenko A, Song H, Bandaru VV, Chu M, Haughey NJ. Inhibition of neutral sphingomyelinase-2 perturbs brain sphingolipid balance and spatial memory in mice. J Neurosci Res. 2010;88:2940–2951. doi: 10.1002/jnr.22438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada S, Matsushita-Morita M, Suzuki S, Kusumoto K, Kashiwagi Y. Characterization of a neutral ceramidase orthologue from Aspergillus oryzae. FEMS Microbiol Lett. 2009;298:157–165. doi: 10.1111/j.1574-6968.2009.01713.x. [DOI] [PubMed] [Google Scholar]
- Taha TA, Hannun YA, Obeid LM. Sphingosine kinase: biochemical and cellular regulation and role in disease. J Biochem Mol Biol. 2006a;39:113–131. doi: 10.5483/bmbrep.2006.39.2.113. [DOI] [PubMed] [Google Scholar]
- Taha TA, Mullen TD, Obeid LM. A house divided: ceramide, sphingosine, and sphingosine-1-phosphate in programmed cell death. Biochim Biophys Acta. 2006b;1758:2027–2036. doi: 10.1016/j.bbamem.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tani M, Hannun YA. Neutral sphingomyelinase 2 is palmitoylated on multiple cysteine residues. Role of palmitoylation in subcellular localization. J Biol Chem. 2007;282:10047–10056. doi: 10.1074/jbc.M611249200. [DOI] [PubMed] [Google Scholar]
- Tani M, Okino N, Mori K, Tanigawa T, Izu H, Ito M. Molecular cloning of the full-length cDNA encoding mouse neutral ceramidase. A novel but highly conserved gene family of neutral/alkaline ceramidases. J Biol Chem. 2000;275:11229–11234. doi: 10.1074/jbc.275.15.11229. [DOI] [PubMed] [Google Scholar]
- Tani M, Iida H, Ito M. O-glycosylation of mucin-like domain retains the neutral ceramidase on the plasma membranes as a type II integral membrane protein. J Biol Chem. 2003;278:10523–10530. doi: 10.1074/jbc.M207932200. [DOI] [PubMed] [Google Scholar]
- Tani M, Igarashi Y, Ito M. Involvement of neutral ceramidase in ceramide metabolism at the plasma membrane and in extracellular milieu. J Biol Chem. 2005;280:36592–36600. doi: 10.1074/jbc.M506827200. [DOI] [PubMed] [Google Scholar]
- Teichgraber V, Ulrich M, Endlich N, Riethmuller J, Wilker B, De Oliveira-Munding CC, et al. Ceramide accumulation mediates inflammation, cell death and infection susceptibility in cystic fibrosis. Nat Med. 2008;14:382–391. doi: 10.1038/nm1748. [DOI] [PubMed] [Google Scholar]
- Testai FD, Landek MA, Goswami R, Ahmed M, Dawson G. Acid sphingomyelinase and inhibition by phosphate ion: role of inhibition by phosphatidyl-myo-inositol 3,4,5-triphosphate in oligodendrocyte cell signaling. J Neurochem. 2004;89:636–644. doi: 10.1046/j.1471-4159.2004.02374.x. [DOI] [PubMed] [Google Scholar]
- Thomas GH, Tuck-Muller CM, Miller CS, Reynolds LW. Correction of sphingomyelinase deficiency in Niemann-Pick type C fibroblasts by removal of lipoprotein fraction from culture media. J Inherit Metab Dis. 1989;12:139–151. doi: 10.1007/BF01800716. [DOI] [PubMed] [Google Scholar]
- Usta J, El Bawab S, Roddy P, Szulc ZM, Yusuf HA, et al. Structural requirements of ceramide and sphingosine based inhibitors of mitochondrial ceramidase. Biochemistry. 2001;40:9657–9668. doi: 10.1021/bi010535k. [DOI] [PubMed] [Google Scholar]
- Watanabe K, Sakuragawa N, Arima M, Satoyoshi E. Partial purification and properties of acid sphingomyelinase from rat liver. J Lipid Res. 1983;24:596–603. [PubMed] [Google Scholar]
- Wheeler D, Knapp E, Bandaru VV, Wang Y, Knorr D, Poirier C, et al. Tumor necrosis factor-alpha-induced neutral sphingomyelinase-2 modulates synaptic plasticity by controlling the membrane insertion of NMDA receptors. J Neurochem. 2009;109:1237–1249. doi: 10.1111/j.1471-4159.2009.06038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won JS, Im YB, Khan M, Singh AK, Singh I. The role of neutral sphingomyelinase produced ceramide in lipopolysaccharide-mediated expression of inducible nitric oxide synthase. J Neurochem. 2004;88:583–593. doi: 10.1046/j.1471-4159.2003.02165.x. [DOI] [PubMed] [Google Scholar]
- Wu BX, Clarke CJ, Hannun YA. Mammalian neutral sphingomyelinases: regulation and roles in cell signaling responses. Neuromolecular Med. 2010a;4:320–330. doi: 10.1007/s12017-010-8120-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu BX, Rajagopalan V, Roddy PL, Clarke CJ, Hannun YA. Identification and characterization of murine mitochondria-associated neutral sphingomyelinase (MA-nSMase), the mammalian sphingomyelin phosphodiesterase 5. J Biol Chem. 2010b;285:17993–18002. doi: 10.1074/jbc.M110.102988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu R, Jin J, Hu W, Sun W, Bielawski J, Szulc Z, et al. Golgi alkaline ceramidase regulates cell proliferation and survival by controlling levels of sphingosine and S1P. FASEB J. 2006;20:1813–1825. doi: 10.1096/fj.05-5689com. [DOI] [PubMed] [Google Scholar]
- Xu R, Sun W, Jin J, Obeid LM, Mao C. Role of alkaline ceramidases in the generation of sphingosine and its phosphate in erythrocytes. FASEB J. 2010;24:2507–2515. doi: 10.1096/fj.09-153635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabu T, Shimuzu A, Yamashita M. A novel mitochondrial sphingomyelinase in zebrafish cells. J Biol Chem. 2009;284:20349–20363. doi: 10.1074/jbc.M109.004580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yada Y, Higuchi K, Imokawa G. Purification and biochemical characterization of membrane-bound epidermal ceramidases from guinea pig skin. J Biol Chem. 1995;270:12677–12684. doi: 10.1074/jbc.270.21.12677. [DOI] [PubMed] [Google Scholar]
- Yang J, Qu JM, Summah H, Zhang J, Zhu YG, Jiang HN. Protective effects of imipramine in murine endotoxin-induced acute lung injury. Eur J Pharmacol. 2010;638:128–133. doi: 10.1016/j.ejphar.2010.04.005. [DOI] [PubMed] [Google Scholar]
- Yokomatsu T, Takechi H, Akiyama T, Shibuya S, Kominato T, Soeda S, et al. Synthesis and evaluation of a difluoromethylene analogue of sphingomyelin as an inhibitor of sphingomyelinase. Bioorg Med Chem Lett. 2001;11:1277–1280. doi: 10.1016/s0960-894x(01)00179-2. [DOI] [PubMed] [Google Scholar]
- Yoshida Y, Arimoto K, Sato M, Sakuragawa N, Arima M, Satoyoshi E. Reduction of acid sphingomyelinase activity in human fibroblasts induced by AY-9944 and other cationic amphiphilic drugs. J Biochem. 1985;98:1669–1679. doi: 10.1093/oxfordjournals.jbchem.a135438. [DOI] [PubMed] [Google Scholar]
- Yoshimura Y, Okino N, Tani M, Ito M. Molecular cloning and characterization of a secretory neutral ceramidase of Drosophila melanogaster. J Biochem. 2002;132:229–236. doi: 10.1093/oxfordjournals.jbchem.a003215. [DOI] [PubMed] [Google Scholar]
- Yoshimura Y, Tani M, Okino N, Iida H, Ito M. Molecular cloning and functional analysis of zebrafish neutral ceramidase. J Biol Chem. 2004;279:44012–44022. doi: 10.1074/jbc.M405598200. [DOI] [PubMed] [Google Scholar]
- Zeeck A, Schroder K, Frobel K, Grote R, Thiericke R. The structure of manumycin. I. Characterization, structure elucidation and biological activity. J Antibiot. 1987;40:1530–1540. doi: 10.7164/antibiotics.40.1530. [DOI] [PubMed] [Google Scholar]
- Zeidan YH, Pettus BJ, Elojeimy S, Taha T, Obeid LM, Kawamori T, et al. Acid ceramidase but not acid sphingomyelinase is required for tumor necrosis factor–induced PGE2 production. J Biol Chem. 2006;281:24695–24703. doi: 10.1074/jbc.M604713200. [DOI] [PubMed] [Google Scholar]
- Zhu Q, Jin JF, Shan XH, Liu CP, Mao XD, Xu KF, et al. Chronic activation of neutral ceramidase protects beta-cells against cytokine-induced apoptosis. Acta Pharmacol Sin. 2008;29:593–599. doi: 10.1111/j.1745-7254.2008.00781.x. [DOI] [PubMed] [Google Scholar]