

Abstract
BACKGROUND AND PURPOSE
20-Hydroxyeicosatetraenoic acid (20-HETE), formed from arachidonate by cytochrome P450, regulates vascular smooth muscle cell (VSMC) function. Because 20-HETE may activate peroxisome proliferator activator receptors (PPARs) and may participate in inflammatory responses, we asked whether 20-HETE may inhibit cyclooxygenase 2 (COX-2) expression by activating PPARs in VSMC.
EXPERIMENTAL APPROACH
Quiescent neonatal VSMC (R22D cell line), were incubated with 20-HETE, synthetic ligands of PPARs, or inhibitors of the extracellular signal regulated kinase (ERK1/2), c-jun N-terminal kinase and the transcription factor activated protein-1 before adding ATPγS. mRNA and protein expression of COX-2 and the promoter luciferase activity of COX-2 and PPAR response element were determined.
KEY RESULTS
Pretreatment with 20-HETE (5–10 µM) significantly inhibited ATPγS-induced COX-2 mRNA and protein expression in VSMC. The inhibitory effect of 20-HETE on COX-2 expression was mimicked by WY14643, a PPARα ligand and inhibited by MK886, a PPARα inhibitor or by transfection of shRNA for PPARα. Both 20-HETE and WY14643 significantly increased the PPAR-response element luciferase activity. Furthermore, ATPγS-induced activation of the COX-2 promoter containing the activated protein-1 site was also inhibited by pretreatment with 20-HETE, which was reversed by MK886 or by transfection with shRNA for PPARα.
CONCLUSIONS AND IMPLICATIONS
The PPARα may mediate the inhibitory effects of 20-HETE on COX-2 expression through a negative cross-talk between PPARα and the COX-2 promoter.
Keywords: 20-hydroxyeicosatetraenoic acid, peroxisome proliferator activator receptor, vascular smooth muscle cell, cytochrome P450, cyclooxygenase
Introduction
20-Hydroxyeicosatetraenoic acid (20-HETE), a major metabolite of arachidonic acid formed by cytochrome P450 in vascular smooth muscle cell (VSMC), may be an important player in modulating cardiovascular function under physiological and pathological conditions (Roman, 2002). 20-HETE may exert pro-hypertensive effects (Hoagland et al., 2003; Benter et al., 2005), regulate proliferation of VSMC (Uddin et al., 1998; Liang et al., 2008), activate mitogen-induced activation of extracellular signal regulated kinase (ERK1/2) in VSMC (Muthalif et al., 1998; Uddin et al., 1998) and endothelial cells (Ishizuka et al., 2008), and induce pro-inflammatory effects in endothelial cells (Ishizuka et al., 2008). In vasculature, 20-HETE may be converted by cyclooxygenase to prostaglandins (Carroll et al., 1992; Birks et al., 1997; Oyekan, 2005) or by alcohol dehydrogenase to 20-carboxy-arachidonic acid (Collins et al., 2005). Both 20-HETE and 20-carboxy-arachidonic acid may activate peroxisome proliferator-activated receptors (PPARs) (Fang et al., 2007; Ng et al., 2007). However, the subsequent signalling events or biological significance of PPAR activation induced by 20-HETE remains unclear.
The PPARs belong to the nuclear receptor superfamily, which are key players in lipid and glucose metabolism (Bishop-Bailey, 2000). Three PPAR isoforms, α, β, γ, have been identified and all three isoforms are expressed in VSMC (Bishop-Bailey, 2000). Once activated by their ligands, PPARs form heterodimers with retinoid X receptor and bind to PPAR response elements (PPREs) in the promoter region of target genes and modulate gene expression. Alternatively, PPARs may repress transcription factors such as NF-κB (Delerive et al., 1999; Daynes and Jones, 2002) or the activator protein-1 (AP-1) (Delerive et al., 1999; Grau et al., 2006; Konstantinopoulos et al., 2007) leading to inhibition of target genes. Previous studies have demonstrated that PPAR ligands may thus exert an anti-inflammatory effect (Staels et al., 1998; Bishop-Bailey, 2000; Grau et al., 2006). PPARα ligands may inhibit interleukin 1-induced cyclooxygenase 2 (COX-2) expression in human VSMC (Staels et al., 1998), and enhance degradation of inducible nitric oxide synthase in lipopolysaccharide-treated macrophages (Paukkeri et al., 2007). Because COX-2 expression is up-regulated in inflammatory cells and VSMC of human atherosclerotic lesions (Baker et al., 1999; Schonbeck et al., 1999; Belton et al., 2000; Stemme et al., 2000), these results suggest a potential role of PPARα in modulating the process of atherosclerosis and re-stenosis. Although 20-HETE may activate PPAR (Fang et al., 2007; Ng et al., 2007) and PPARα ligands may inhibit COX-2 expression (Staels et al., 1998; Grau et al., 2006), the action of 20-HETE on COX-2 expression remains to be established.
In addition, COX-2-derived prostaglandins may also participate in pathophysiological regulation of vascular tone (Wong and Vanhoutte, 2010). In rat endotoxaemia, endotoxin-induced hypotension and attenuation of systemic/renal 20-HETE levels were restored by COX-2 inhibition (Tunctan et al., 2010), suggesting COX-2 activity may cause attenuation of 20-HETE formation. It appears that interactions between COX-2 and 20-HETE may regulate cardiovascular function.
In addition to its participation in metabolism as the energy source, ATP has been postulated as a critical mediator in the development of cardiovascular diseases (Burnstock, 2002; Di and Solini, 2002). Extracellular ATP may function as an important signalling molecule in pathophysiological processes. In the cardiovascular system, such ATP may be supplied by platelets, inflammatory cells, endothelial cells and VSMC (Gordon, 1986), in concentrations up to several hundred µM (Traut, 1994). By activating purinergic receptors, extracellular ATP may stimulate cell proliferation (Wang et al., 1992)/migration (Chaulet et al., 2001) and secretion of matrix metalloproteinase (Robinson, III et al., 2006), modulate transformation of VSMC phenotype, and regulate inflammatory responses (Di and Solini, 2002). ATPγS can activate, the P2Y receptor (nomenclature follows Alexander et al., 2009) and subsequently induce a transient COX-2 expression in VSMC (Lin et al., 2009). In the present study, we tested the hypothesis that 20-HETE may inhibit COX-2 expression induced by ATPγS via PPARs in VSMC. Our results suggested a potential role of 20-HETE in modulating the pathophysiological function of COX-2 via attenuation of COX-2 expression.
Methods
Cell culture
The R22D cells, a primary culture of VSMC isolated from neonatal rats, was established and selected for abundant production of elastin by Jones et al. (1979). These cells were maintained under 5% CO2 in minimum essential medium with penicillin-streptomycin (1%), tryptose phosphate broth (2%) and fetal bovine serum (10%). In most of the experiments, cultured VSMC were made quiescent by incubation with serum-deprived medium containing transferrin (5 µg·mL−1) and bovine serum albumin (0.05%) for 24 h.
Prostaglandin E2 (PGE2) measurement
Quiescent cells were incubated with ATPγS for different time periods. The conditioned medium was collected for measurement of PGE2 with elisa (Cayman, Ann Arbor, MI, USA) according to manufacture's protocol.
Transfection with short hairpin RNA (shRNA)
The designed sequence of rat PPARα was cloned into pGSH1-GFP shRNA expression vector (Genlantis, San Diego, CA, USA) as previously described (Huang et al., 2004). The plasmid DNA (14 µg) was transfected into VSMC (R22D cells) by electroporation (voltage: 160 V, duration: 7 ms). After transfection for 48 h, the cells were selected for stable expression of the shRNA for PPARα by G418 (600 µg·mL−1). The cells with neomycin-resistance will be collected and cultured for experiments. Equal amounts of total protein (50 µg) were loaded to analyse the efficiency of PPARα knock-down by Western blot, using a PPARα polyclonal antibody (1:1000; Cayman, Ann Arbor, MI, USA).
RNA extraction and reverse transcriptase-PCR
Quiescent R22D cells, PPARα deficient cells (R22D cells with shRNA for PPARα) and the vector control cells were incubated with vehicle or 20-HETE (5 or 10 µM) for 22 h before incubation with ATPγS (100 µM) for additional 2 h. In some experiments, quiescent R22D cells were pretreated with PD98059, SP600125, or tanshinone IIA for 30 min before addition of ATPγS. RNA was extracted using Trizol reagent (Invitrogen; Carlsbad, CA, USA) according to the manufacturer's protocol. Reverse-transcriptase reaction was carried out by using Moloney murine leukaemia virus reverse transcriptase (Invitrogen; Carlsbad, CA, USA). Total RNA (1 µg) was added to a reaction mixture containing oligo-deoxythymidine (oligo-dT; 0.5 µg·µL−1), dNTP (20 mM), dithiothreitol (0.1 M), Tris-HCl (250 mM, pH 8.3), KCl (375 mM) and MgCl2 (15 mM). Reaction mixtures were incubated at 37°C for 90 min. Real-time PCR was carried out with Smart Quant Green Master Mix (Protech Technology, Enterprise, Placentia, CA, USA), the cDNA (50 µg), and following primers: forward 5′-TGGTGCCGGGTCTGATGATG-3′ and reverse 5′-GCAATGCGGTTCTGATACTG-3′ for COX-2; forward 5′-GTAACCCGTTGAACCCCATT-3′ and reverse 5′-CCATCCAATCGGTAGTAGCG-3′ for 18S ribosomal RNA. The reactions were performed with preliminary denaturation at 95°C for 10 min, 40 cycles of denaturation at 95°C for 15 min and annealing/extension at 60°C for 1 min by using ABI Prism 7000 (Applied Biosystems, Carlsbad, CA, USA). The threshold of cycle was used to calculate the relative COX-2 mRNA expression normalized against the 18S RNA.
Western blot
Confluent cells were cultured in serum-free medium for 24 h prior to incubation with ATPγS (30 or 100 µM) for the indicated time. Quiescent R22D, PPARα deficient, or the vector control cells were incubated with vehicle, 20-HETE (5 or 10 µM), or WY14643 (PPARα ligand; 60–250 µM) for 20 h before incubation with ATPγS (100 µM) for additional 4 h. In some experiments, quiescent R22D cells were incubated with 20-HETE, in the presence or absence of MK886 (2, 5, 10 µM), bisphenol A diglycidyl ether (BADGE) (0.5, 1, 2 µM) or GW9662 (0.5, 1, 2 µM) for 20 h prior to addition ATPγS (100 µM). Total protein of each sample was loaded onto 10% sodium dodecyl sulphate polyacrylamide gel and separated by electrophoresis for 2 h. The protein samples were transferred to nitrocellulose membrane in a transfer buffer. The membranes were washed three times with TTBS solution (1% Tween 20, Tris base 50 mM and NaCl 150 mM), incubated overnight with an antibody against COX-2 (1:2000) or GAPDH (1:5000) at 4°C, and then incubated with a peroxidase-conjugated secondary antibody (1:5 000) for 1 h. An enhanced chemiluminescence detection system (Perkin Elmer, Boston, MA, USA) was employed for detection.
Luciferase activity assay
The luciferase reporter constructs of COX-2 (−459 to +9 bp), PPRE (PPRE3-TK-LUC) were kindly provided by L. H. Wang (University of Texas, Houston, TX, USA) and Ronald M. Evans (Salk Institute, San Diego, CA, USA), respectively. For the AP-1 mutation construct, the COX-2 promoter was constructed where the upstream region (+34 to −483) of the COX-2 promoter was cloned into the pGL3-basic vector containing the luciferase reporter system. Introduction of a point mutation into the AP-1-binding site (AP-1 domain; ACAGTCA to ACAACCA) to generate pGL-COX-2-ΔAP1 was performed, using the following (forward) primer: 5′-AAAGAAACAACCATTTCGTC-3 (corresponding to a region from −73 to −54).
Transfection of luciferase reporter plasmid was carried out by using a METAFECTENE reagent (Biontex Laboratories, Martinsreid/Planegg, Germany) according to the manufacturer's instructions. The transfection efficiency was determined as 13% by transfection of green fluorescence protein, followed by flow cytometry. For promoter activity assay, the cells were lysed in Glo lysis buffer (Promega, Madison, WI, USA) and equal amount of cell lysates were subjected to analysis by a luciferase assay system (Promega, Madison, WI, USA). The luciferase activity was then normalized to amount of cellular protein. The cell viability 24 h after transient transfection was 92%.
Electrophoresis mobility shift assay
The nuclear protein was extracted by NE-PER reagent (Pierce, Rockford, IL, USA) according to the manufacturer's protocol. Equal amount of nuclear protein was subjected to analysis AP-1 binding activity by using LightShift Chemiluminescent EMSA Kit (Pierce, Rockford, IL, USA).
Data analysis
All values are presented as mean ± SEM and were analysed with Student's t-test or anova, followed by Duncan's post hoc test. Statistical significance was determined as P < 0.05.
Materials
20-Hydroxyeicosatetraenoic acid (in ethanol), WY14643, troglitazone and antibody against COX-2 and PPARα were purchased from Cayman Chemicals. (Ann Arbor, MI, USA). Rabbit polyclonal IgG against GAPDH was purchased from Santa Cruz (Santa Cruz, CA, USA). Adenosine-5′-o-(3-thiotriphosphate).4Li (ATPγS), BADGE, GW9662, PD98058, SP600125 and MK-886 were purchased from Biomol (Plymouth Meeting, PA, USA). Tryptose phosphate broth was purchased from Sigma-Aldrich (St. Louis, MO, USA). Nitrocellulose membrane was purchased from Pall Life Science (Pensacola, FL, USA). 20-HETE was dried under N2 gas and then resuspended in dimethyl sulphoxide (DMSO) before use. All inhibitors and synthetic PPAR ligands were dissolved in DMSO and the final concentration of DMSO was 0.1%.
Results
20-HETE inhibited ATP-induced COX-2 expression
To determine the effect of ATP on COX-2 expression, the cells were incubated with ATPγS (30 or 100 µM) for the indicated time. As illustrated in Figure 1, ATPγS-induced COX-2 expression was time and concentration dependent. At 100 µM of ATPγS, the COX-2 expression increased significantly and peaked within 4 h (n = 5, P < 0.05). PGE2 release induced by this concentration of ATP was significantly increased (18.1 ± 1 pg·mL−1) after 12 h, compared with that in the vehicle group (3.8 ± 0.3 pg·mL−1; n = 3).
Figure 1.
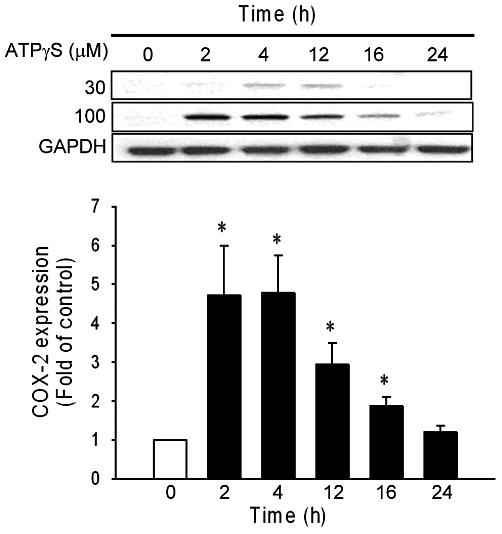
ATPγS-induced COX-2 expression in vascular smooth muscle cell. Confluent vascular smooth muscle cell were made quiescent for 24 h before incubation with ATPγS (30 and 100 µM) for indicated times. COX-2 expression in response to ATPγS (100 µM) is shown in the lower panel (n = 5). Values are mean ± SE. *P < 0.05 compared with the control group.
To determine whether 20-HETE inhibited this ATPγS-induced COX-2 expression, the cells were pre-incubated with 20-HETE (5 or 10 µM) for 20 h and subsequently incubated with ATPγS for 4 h. Although lower concentrations of 20-HETE (0.1–3 µM) did not alter ATP-induced COX-2 expression in VSMC (n = 3; data not shown), 20-HETE at 5 and 10 µM significantly inhibited ATPγS-induced COX-2 expression (Figures 2A, n = 6–11, P < 0.05). In contrast, COX-1 was constitutively expressed in VSMC and its level was not altered by ATPγS (Figure S1A). Quantitative PCR revealed that 20-HETE exerted a similar inhibitory effect on COX-2 mRNA expression (Figure 2B); pre-incubation with 5 and 10 µM of 20-HETE significantly inhibited ATPγS-induced COX-2 mRNA expression (n = 3, P < 0.05). These results suggested that inhibition by 20-HETE of the ATPγS-induced COX-2 expression may be via transcriptional regulation.
Figure 2.
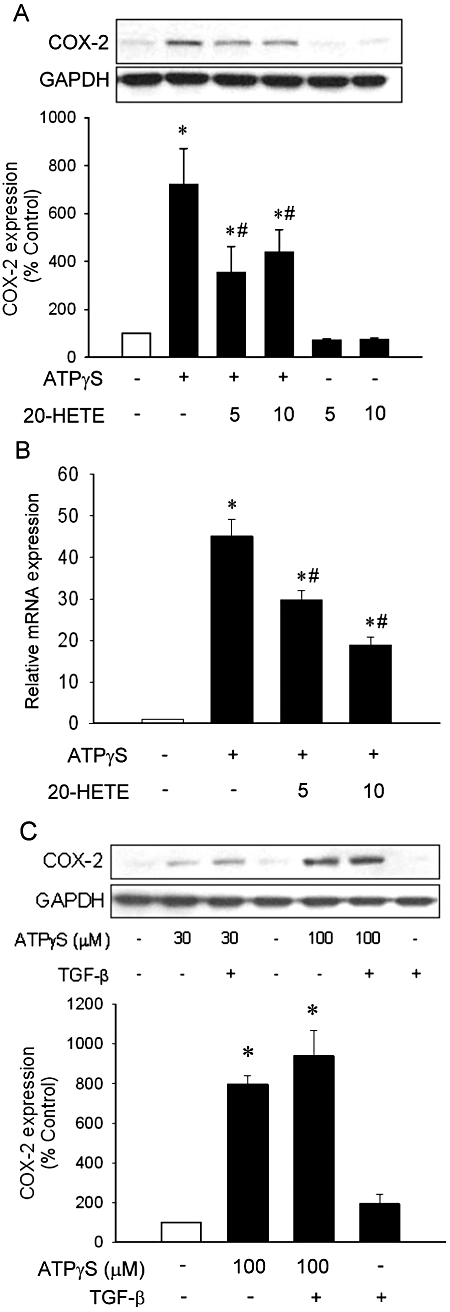
20-Hydroxyeicosatetraenoic acid (20-HETE) attenuated ATPγS-induced COX-2 expression in vascular smooth muscle cell. Quiescent vascular smooth muscle cell were pre-incubated with 20-HETE (5 or 10 µM) or TGF-β (200 pg·mL−1) for 20 h (A,C) or 22 h (B) before incubation with ATPγS (100 µM) for 2 h (B) or 4 h (A,C). Total protein and RNA was subjected to analysis of COX-2 expression by Western blot (A, n = 6–11; C, n = 3) and quantitative-PCR (B, n = 3), respectively. Values are mean ± SE. *P < 0.05 compared with corresponding control groups. #P < 0.05 compared with corresponding group without 20-HETE.
As we had previously demonstrated that transforming growth factor-β (TGF-β) may mediate the growth inhibitory effect of 20-HETE (Liang et al., 2008), we asked whether the effect of 20-HETE on ATPγS-induced COX-2 expression was mediated by TGF-β. However, exogenous addition of TGF-β (200 pg·mL−1) to mimic additional TGF-β released in response to 20-HETE exerted no effect on ATPγS-induced COX-2 expression (Figure 2C).
PPARα mediated the inhibitory effect of 20-HETE on COX-2 expression
As 20-HETE may activate PPARα and γ (Fang et al., 2007; Ng et al., 2007), we next examined the effects of reagents affecting PPAR activity on ATPγS-induced COX-2 expression. WY14643, a PPARα activator, inhibited ATPγS-induced COX-2 expression in a concentration-dependent manner (Figure 3A, P < 0.05). Both 20-HETE (10 µM) and WY14643 (100 µM) induced a minor, but significant increase in PPRE luciferase activity (Figure 3B, n = 6), probably due to low transfection efficiency in these cells. In addition, MK886, a PPARα inhibitor, effectively reversed the inhibitory effects of 20-HETE on ATPγS-induced COX-2 expression (n = 4, P < 0.05; Figure 3C). Cells transfected with shRNA against PPARα reduced the levels of PPARα protein, whereas transfection with control vector had no effect (Figure 3D). Transfection with shRNA of PPARα effectively reversed the inhibitory effect of 20-HETE on ATPγS-induced COX-2 mRNA (Figure 3E) and protein (Figure 3F) expression; whereas transfection with control vector had no effect on ATPγS-induced COX-2 expression. In contrast, incubation with PPARγ inhibitors GW9662 and BADGE did not affect the inhibitory effect of 20-HETE on ATPγS-induced COX-2 expression (Figure 3G). Furthermore, incubation with MK886, GW9662 and BADGE did not affect ATPγS-induced COX-2 expression (Figure S1B). These results suggested that the inhibitory effect of 20-HETE on ATPγS-induced COX-2 expression was mediated by PPARα.
Figure 3.
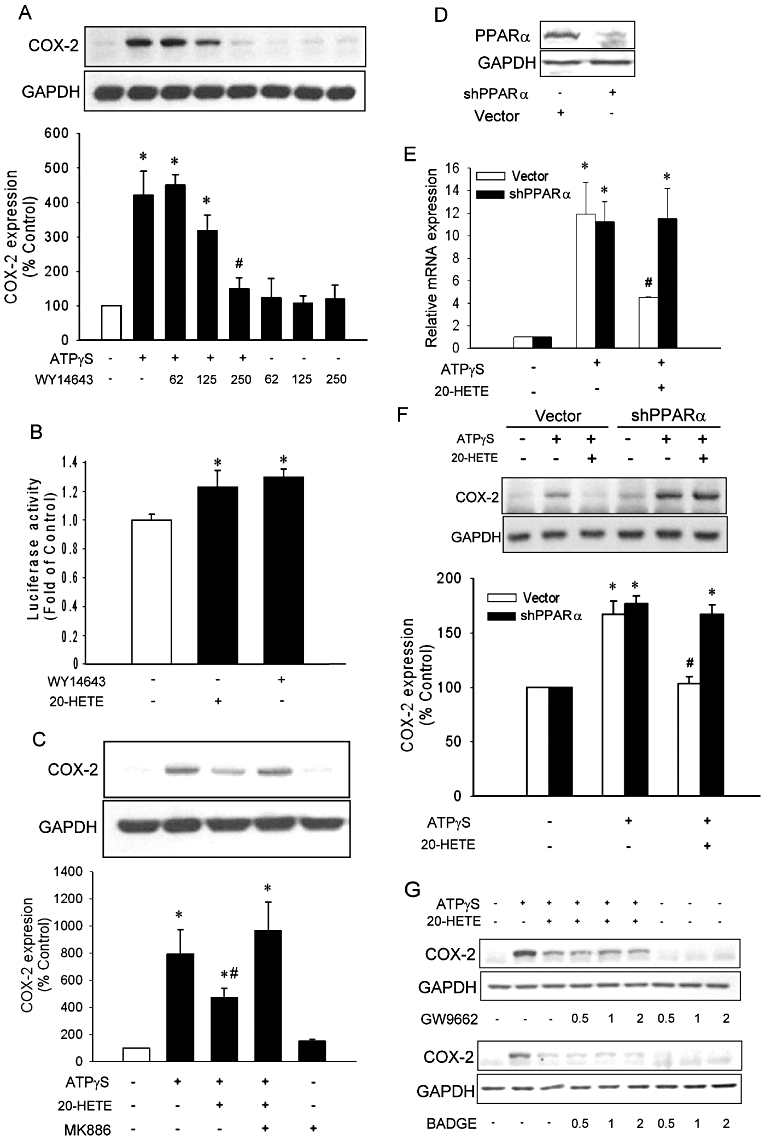
Peroxisome proliferator activator receptor-α (PPARα) mediated the inhibitory effects of 20-hydroxyeicosatetraenoic acid (20-HETE) on COX-2 expression. (A) Quiescent vascular smooth muscle cell (VSMC) were pre-incubated with WY14643 for 20 h followed by incubation with or without ATPγS (100 µM) for additional 4 h (n = 3). (B) VSMC was transfected with PPAR response element promoter luciferase for 24 h before incubation with or without 20-HETE (10 µM) or WY14643 (100 µM) for 24 h (n = 6). Quiescent VSMC were pre-incubated with or without MK886 (2 µM; C), GW9662 or bisphenol A diglycidyl ether (in µM; G) in the presence or absence of 20-HETE for 20 h before incubation with ATPγS (n = 4). (D) The VSMC were transfected with shRNA for PPARα or control vector and the PPARα expression level was determined by Western blot. The PPARα deficient or control cells were incubated with or without 20-HETE before incubation with ATPγS (E,F). COX-2 expression was analysed with quantitative PCR (E) or Western blot (F). Values of luciferase activity or COX-2 protein expression are presented as mean ± SEM. *P < 0.05 compared with corresponding control groups. #P < 0.05 compared with the corresponding group without WY-14643 (B) or 20-HETE (E).
ATP-induced COX-2 expression via activation of MAPK/AP-1 pathway
To determine the potential target which was negatively regulated by 20-HETE, we examined whether the transcription factor AP-1 was involved in ATPγS-induced COX-2 expression. As illustrated in Figure 4, pre-incubation with a ERK1/2 kinase (MEK) inhibitor PD98059 (Figure 4A) or the c-jun N-terminal kinase (JNK) inhibitor SP600125 (Figure 4B) significantly inhibited ATPγS-induced COX-2 expression in a concentration-dependent manner (n = 3, P < 0.05), suggesting that the mitogen-activated protein kinase (MAPK) cascade may mediate COX-2 induction by ATPγS. In addition, pre-incubation with the AP-1 inhibitor, tanshinone IIA (Park et al., 1999), inhibited ATPγS-induced COX-2 expression in a concentration-dependent manner (Figures 4C, n = 3, P < 0.05). Furthermore, ATPγS induced a time-dependent phosphorylation of c-jun but not Elk-1. The phosphorylation of c-jun increased within 10 min and reached a maximal response within 60 min during the observation periods after addition of ATPγS (Figure 4D, n = 3). The mRNA expression induced by ATPγS was also inhibited by pre-incubation with inhibitors of ERK1/2, JNK and AP-1 (Figures 4E, n = 3, P < 0.05). In addition, ATPγS increased COX-2 promoter activity in cells transfected with wild type COX-2 promoter. However, transfection with AP-1 mutation promoter significantly attenuated the promoter activity induced by ATPγS (Figures 4F, n = 8, P < 0.05). These results suggested that ATPγS-induced COX-2 expression in our cell system is likely to be mediated through the MAPK/AP-1 pathway.
Figure 4.
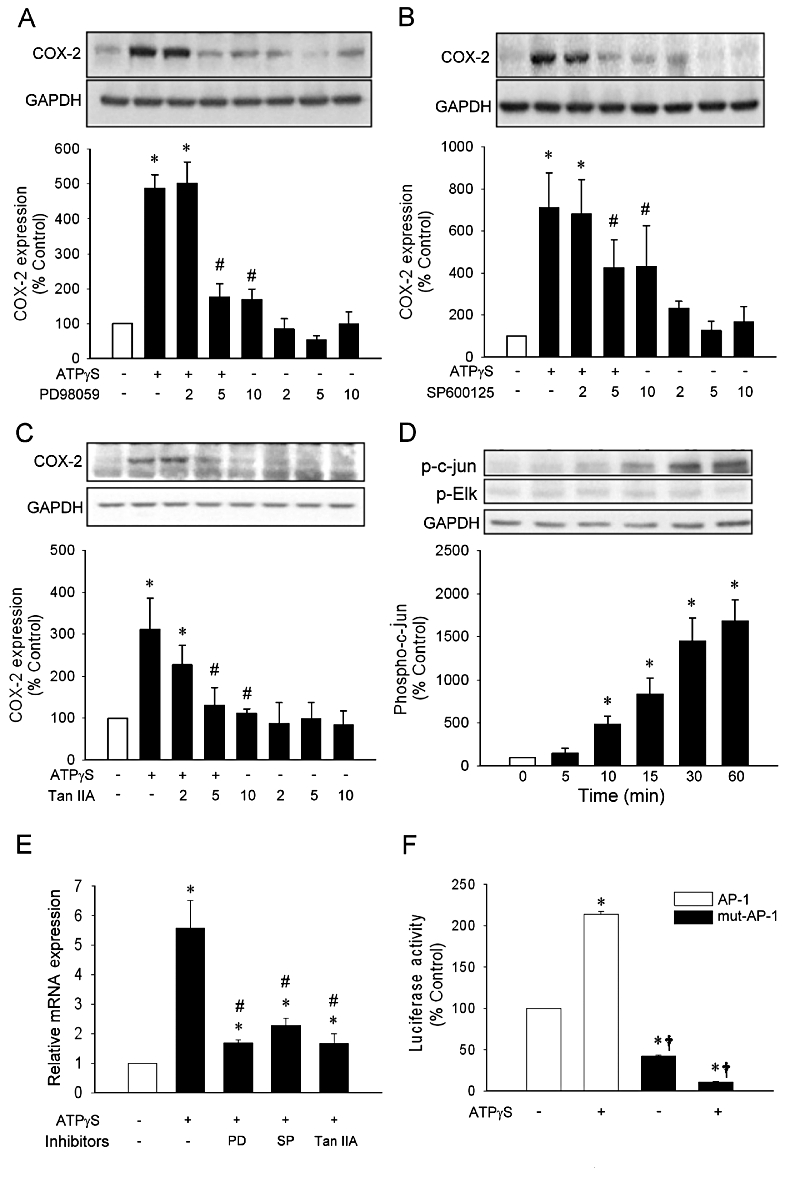
Effects of inhibitors on ATPγS-induced COX-2 expression. Quiescent vascular smooth muscle cell (VSMC) was incubated with concentrations (µM) of inhibitors (PD98059, A; SP600125, B; tanshinone IIA, C) prior to addition of ATPγS (100 µM). The expression of COX-2 was determined by Western blot. Quiescent VSMC were incubated with ATPγS at indicated times prior to cell harvest for determination of phosphorylation levels of c-jun and Elk 1 by Western blot (D). Quiescent VSMC were incubated with PD98059, SP600125 or tanshinone IIA for 30 min before incubation with ATPγS for 2 h (E). The cells were transiently transfected with the COX-2 promoter luciferase plasmid containing wild type or mutated activated protein-1 (AP-1) site followed by addition of ATPγS for 4 h (F). Values are presented as mean ± SEM. *P < 0.05 compared with corresponding control groups. #P < 0.05 compared with corresponding group without inhibitor. †P < 0.05 compared with ATPγS group in wild type.
PPARα mediated the inhibitory effect of 20-HETE on COX-2 transcription
We next asked whether 20-HETE inhibited AP-1-mediated COX-2 expression via PPARα. VSMC were transfected with the COX-2 luciferase plasmid containing AP-1 site. Pre-incubation with 20-HETE (10 µM) significantly attenuated ATPγS-induced COX-2 promoter activity (Figures 5A, n = 3, P < 0.05). In addition, MK886 effectively reversed the inhibitory effect of 20-HETE on ATPγS-induced COX-2 promoter activity (Figure 5B, n = 3, P < 0.05). Furthermore, transfection with shRNA for PPARα effectively reversed the inhibitory effects of 20-HETE on ATPγS-induced promoter activity (Figure 5C, n = 3); whereas transfection with control vector did not. Similar results were also obtained from the cells incubated with WY14643 (100 µM, n = 3; Figure 5E). In addition, pre-incubation with WY14643 significantly attenuated ATPγS-induced COX-2 promoter activity, which was reversed by MK886 (Figures 5D, n = 3, P < 0.05). In addition, 20-HETE or WY14643 did not alter AP-1/DNA binding induced by ATPγS (Figure S1C). These results suggested that PPARα may mediate the inhibitory effect of 20-HETE on ATPγS-induced COX-2 expression.
Figure 5.
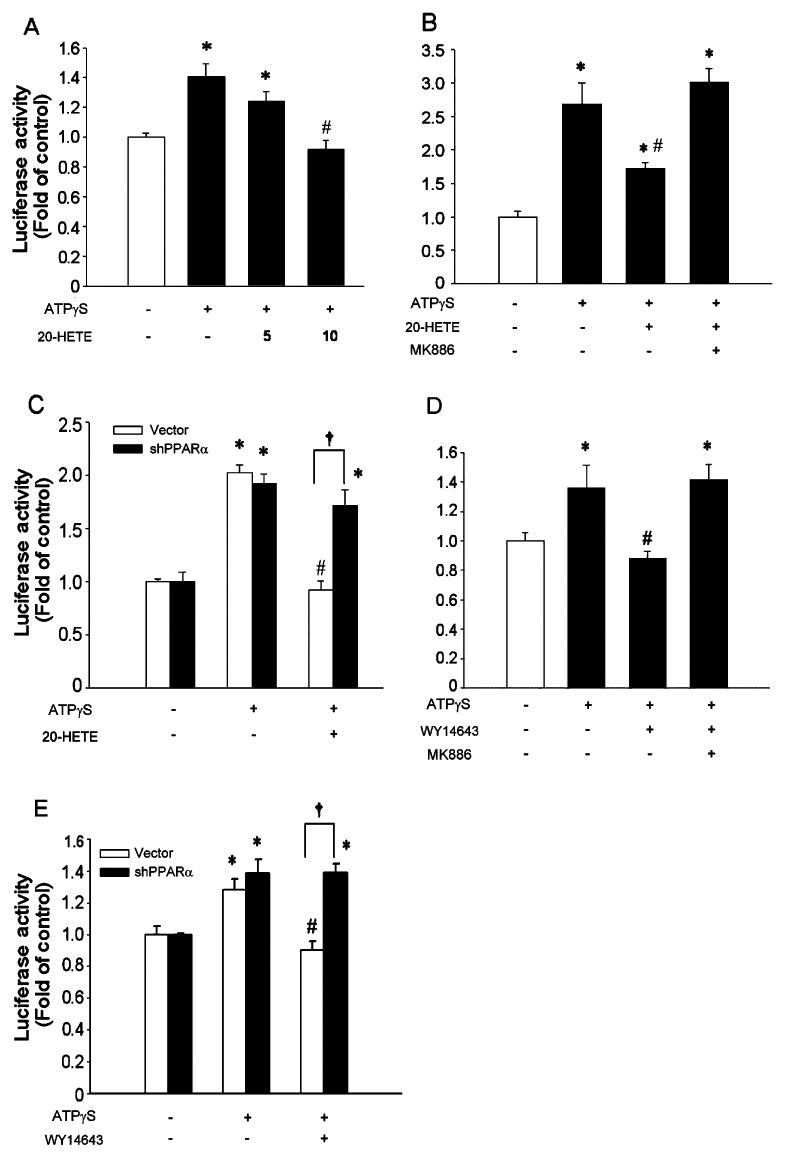
20-Hydroxyeicosatetraenoic acid (20-HETE) inhibited activated protein-1 (AP-1) mediated transcription of COX-2 via peroxisome proliferator activator receptor-α (PPARα). The cells were transiently transfected with the COX-2 promoter luciferase plasmid containing AP-1 binding site for 24 h, followed by pre-incubation with 20-HETE for 20 h before incubation with ATPγS (100 µM; A). After transfection, quiescent vascular smooth muscle cell were pre-incubated with MK886 (2 µM) in the presence or absence of 20-HETE (10 µM; B) or WY14643 (100 µM; D) for 20 h before incubation with ATPγS (100 µM) and incubated for additional 4 h. The PPARα deficient or control cells were transfected with the COX-2 promoter luciferase plasmid containing AP-1 binding site for 24 h before incubation with ATPγS in the presence or absence of 20-HETE (C) or WY14643 (E). Values are presented as mean ± SEM. *P < 0.05 compared with corresponding control groups. #P < 0.05 compared with corresponding group without 20-HETE. †P < 0.05 compared with the indicated group.
Discussion and conclusions
In the present study, we have demonstrated that 20-HETE inhibited ATPγS-induced COX-2 expression via PPARα activation in a VSMC cell line (R22D cells). To our knowledge, this is the first report demonstrating that 20-HETE may regulate COX-2 expression. 20-HETE inhibited ATPγS-induced COX-2 mRNA as well as protein expression. The inhibitory effect of 20-HETE appeared to be mediated by activation of PPARα, because an inhibitor of PPARα and shRNA for PPARα reversed the effect of 20-HETE on COX-2 expression and promoter activity.
Our results indicated that ATPγS induced expression of COX-2 in VSMC, in a concentration- and time-dependent manner. Although a previous study had indicated that the COX-2 expression may be sustained over 18 h in response to stimulation (Callejas et al., 2002), our results are consistent with previous findings that ATPγS induced a transient expression of COX-2 protein in VSMC (Lin et al., 2009). Mutation of the AP-1 site effectively attenuated ATPγS-induced COX-2 promoter activity, suggesting that the transcription factor AP-1 was required for COX-2 transcription in response to ATPγS (Figure 6). Because 20-HETE did not inhibit ERK/JNK phosphorylation in R22D cells (Liang et al., 2008), 20-HETE may act on AP-1, the downstream molecule of ERK/JNK, to exert its inhibitory effect on COX-2 expression (Figure 6). The luciferase activity assay with an AP-1 containing COX-2 promoter further demonstrated that 20-HETE inhibited ATPγS-induced transcription via PPARα (Figure 5). In addition, our previous study in VSMC demonstrated that 20-HETE may increase TGF-β levels (Liang et al., 2008), which has been shown to regulate stability of COX-2 mRNA (Harding et al., 2006). However, our current results indicated that exogenous addition of TGF-β did not affect ATP-induced COX-2 expression, suggesting that effects of 20-HETE on COX-2 expression is unlikely to be mediated via regulation of COX-2 mRNA stability. Nevertheless, we cannot rule out the possibility that 20-HETE may enhance degradation of COX-2 protein or mRNA via activation of PPARs, because PPAR ligands may enhance degradation of other inflammation-related molecules, such as iNOS (Paukkeri et al., 2007).
Figure 6.
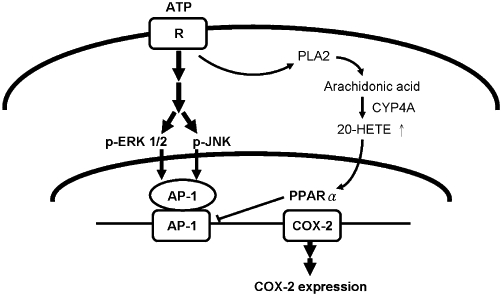
Diagram of mechanism(s) underlying the inhibitory effects of 20-hydroxyeicosatetraenoic acid (20-HETE) on COX-2 expression in vascular smooth muscle cell. ATP binds to purinergic receptors and activates mitogen-activated protein kinase/activated protein-1 (AP-1) pathways to induce COX-2 expression. 20-HETE activates peroxisome proliferator activator receptor-α (PPARα) to suppress AP-1-mediated COX-2 transcription.
Although 20-HETE may activate both PPARα and γ (Fang et al., 2007; Ng et al., 2007), little is known about the biological consequences of PPAR activation by 20-HETE. Previous studies indicated that activation of PPARα or γ may exert an anti-inflammatory effect via suppression of target gene expression (Bishop-Bailey, 2000). In the current study, 20-HETE inhibited ATPγS-induced COX-2 expression, and the effects of 20-HETE were reversed by shRNA for PPARα or a PPARα inhibitor but not by a PPARγ inhibitor. These results suggested that 20-HETE inhibited ATPγS-induced COX-2 expression that was mediated by PPARα but not PPARγ, although both forms were present in VSMC (Staels et al., 1998). This is consistent with previous findings that activation of PPARα by synthetic ligands inhibited COX-2 expression in VSMC (Staels et al., 1998; Hu et al., 2002). In addition, 20-HETE may be further converted to 20-carboxy-arachidonic acid (Collins et al., 2005), which may act as a more potent activator of PPARs with a higher binding affinity to PPARα than that to PPARγ (Fang et al., 2007). In vascular tissue, cyclooxygenase prostanoid metabolites of 20-HETE may activate PPAR. However, the effect of 20-HETE on PPAR activation is not likely to be mediated by its potential PGE or PGF metabolites (Carroll et al., 1992; Birks et al., 1997; Oyekan, 2005), because neither activates PPAR (Bishop-Bailey, 2000). Although it is not known whether 20-HETE exerts different binding affinity for PPARs, the expression level of PPARα is much higher than that of PPARγ in VSMC (Staels et al., 1998).
Several molecular mechanisms have been proposed for the repressive effects of PPAR on target gene expression. PPAR may mediate repression of NF-κB (Delerive et al., 1999; Daynes and Jones, 2002) or AP-1 (Delerive et al., 1999; Grau et al., 2006; Konstantinopoulos et al., 2007). Our results demonstrated that PPARα mediated the inhibitory effect of 20-HETE on COX-2 expression, at least in part, by repressing transcription mediated by AP-1 (Figure 5). Cross-talk between PPARα and AP-1 is known. Thus, PPARα may directly interact with c-jun (Delerive et al., 1999), a component of AP-1, and subsequently attenuate AP-1 binding to the promoter of target genes. Nevertheless, our results (Figure S1C) indicated that AP-1/DNA binding induced by ATP was not attenuated by 20-HETE or WY14643. Alternatively, 20-HETE-activated PPARα may compete for the limited numbers of co-activators that are essential for the AP-1-mediated transcription (Delerive et al., 1999; Daynes and Jones, 2002; Mochizuki et al., 2002). In addition, 20-HETE may be converted by cyclooxygenase to prostaglandins in the vasculature (Carroll et al., 1992; Birks et al., 1997; Oyekan, 2005) and, in this way, 20-HETE may down-regulate the enzyme that metabolizes itself. Recent studies demonstrated that endotoxin reduced systemic and renal 20-HETE levels, which were restored by COX-2 inhibition (Tunctan et al., 2010), suggesting that COX-2 may directly or indirectly attenuate 20-HETE production in certain vascular beds. However, the pathophysiological role of this negative feedback of 20-HETE on COX-2 expression in VSMC remains to be further investigated. In contrast to our findings, 20-HETE at lower concentrations may induce pro-inflammatory effects via activation of NF-κB in endothelial cells (Ishizuka et al., 2008), whereas activation of PPARα by endogenous polyunsaturated fatty acids requires concentrations in the µM range (Robinson and Grieve, 2009). The different effects of 20-HETE on inflammation is likely to be due to cell-specific effects.
In conclusion, results of the present study demonstrated that activation of PPARα by 20-HETE inhibited ATPγS-induced COX-2 expression by suppressing its transcription, perhaps via a negative interaction with the COX-2 promoter. Our results also revealed a potential role of 20-HETE in modulating inflammatory responses of VSMC.
Acknowledgments
This work was supported by grants from the National Science Council of Taiwan (NSC93-2320-B-182-036), Chang Gung University (CMRPD32046), and Ministry of Education of Taiwan (EMRPD170061). The authors thank the RNAi Core Laboratory, Chang Gung University for providing the shRNA for PPARα and control plasmids.
Glossary
Abbreviations
- 20-HETE
20-hydroxyeicosatetraenoic acid
- AP-1
activated protein-1
- BADGE
bisphenol A diglycidyl ether
- COX-2
cyclooxygenase 2
- PPAR
peroxisome proliferator activator receptor
- VSMC
vascular smooth muscle cell
Conflict of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 (A) 20-HETE did not alter COX-1 expression in VSMC. Quiescent VSMC was pre-incubated with 20-HETE (10 µM) for 20 h prior to addition of ATPγS (100 µM) for 4 h (B) Inhibitors did not alter ATPγS-induced COX-2 expression. Quiescent VSMC was pre-incubated with MK (MK886, 2 µM), GW (GW9662, 2 µM) and BA (BADGE, 2 µM) for 20 h prior to addition of ATPγS for 4 h. (C) ATPγS-induced AP-1 activation was not inhibited by pre-incubation of 20-HETE (5 and 10 µM) or WY14643 (250 µM) for 1 h. The nuclear protein was extracted and subjected to analysis of AP-1 activation by EMSA.
{kind=link}
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 4th edn. Br J Pharmacol. 2009;158:S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker CSR, Hall RJC, Evans TJ, Pomerance A, Maclouf J, Creminon C, et al. Cyclooxygenase-2 is widely expressed in atherosclerotic lesions affecting native and transplanted human coronary arteries and colocalizes with inducible nitric oxide synthase and nitrotyrosine particularly in macrophages. Arterioscler Thromb Vasc Biol. 1999;19:646–655. doi: 10.1161/01.atv.19.3.646. [DOI] [PubMed] [Google Scholar]
- Belton O, Byrne D, Kearney D, Leahy A, Fitzgerald DJ. Cyclooxygenase-1 and -2-dependent prostacyclin formation in patients with atherosclerosis. Circulation. 2000;102:840–845. doi: 10.1161/01.cir.102.8.840. [DOI] [PubMed] [Google Scholar]
- Benter IF, Francis I, Cojocel C, Juggi JS, Yousif MH, Canatan H. Contribution of cytochrome P450 metabolites of arachidonic acid to hypertension and end-organ damage in spontaneously hypertensive rats treated with L-NAME. Auton Autacoid Pharmacol. 2005;25:143–154. doi: 10.1111/j.1474-8673.2005.00343.x. [DOI] [PubMed] [Google Scholar]
- Birks EK, Bousamra M, Presberg K, Marsh JA, Effros RM, Jacobs ER. Human pulmonary arteries dilate to 20-HETE, an endogenous eicosanoid of lung tissue. Am J Physiol. 1997;272:L823–L829. doi: 10.1152/ajplung.1997.272.5.L823. [DOI] [PubMed] [Google Scholar]
- Bishop-Bailey D. Peroxisome proliferator-activated receptors in the cardiovascular system. Br J Pharmacol. 2000;129:823–834. doi: 10.1038/sj.bjp.0703149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnstock G. Purinergic signaling and vascular cell proliferation and death. Arterioscler Thromb Vasc Biol. 2002;22:364–373. doi: 10.1161/hq0302.105360. [DOI] [PubMed] [Google Scholar]
- Callejas NA, Casado M, Bosca L, Martin-Sanz P. Absence of nuclear factor kappaB inhibition by NSAIDs in hepatocytes. Hepatology. 2002;35:341–348. doi: 10.1053/jhep.2002.31163. [DOI] [PubMed] [Google Scholar]
- Carroll MA, Garcia MP, Falck JR, McGiff JC. Cyclooxygenase dependency of the renovascular actions of cytochrome P450-derived arachidonate metabolites. J Pharmacol Exp Ther. 1992;260:104–109. [PubMed] [Google Scholar]
- Chaulet H, Desgranges C, Renault MA, Dupuch F, Ezan G, Peiretti F, et al. Extracellular nucleotides induce arterial smooth muscle cell migration via osteopontin. Circ Res. 2001;89:772–778. doi: 10.1161/hh2101.098617. [DOI] [PubMed] [Google Scholar]
- Collins XH, Harmon SD, Kaduce TL, Berst KB, Fang X, Moore SA, et al. ω-Oxidation of 20-hydroxyeicosatetraenoic acid (20-HETE) in cerebral microvascular smooth muscle and endothelium by alcohol dehydrogenase 4. J Biol Chem. 2005;280:33157–33164. doi: 10.1074/jbc.M504055200. [DOI] [PubMed] [Google Scholar]
- Daynes RA, Jones DC. Emerging roles of PPARs in inflammation and immunity. Nat Rev Immunol. 2002;2:748–759. doi: 10.1038/nri912. [DOI] [PubMed] [Google Scholar]
- Delerive P, De Bosscher K, Besnard S, Vanden Berghe W, Peters JM, Gonzalez FJ, et al. Peroxisome proliferator-activated receptor α negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-κB and AP-1. J Biol Chem. 1999;274:32048–32054. doi: 10.1074/jbc.274.45.32048. [DOI] [PubMed] [Google Scholar]
- Di VF, Solini A. P2 receptors: new potential players in atherosclerosis. Br J Pharmacol. 2002;135:831–842. doi: 10.1038/sj.bjp.0704524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X, Dillon JS, Hu S, Harmon SD, Yao J, Anjaiah S, et al. 20-Carboxy-arachidonic acid is a dual activator of peroxisome proliferator-activated receptors α and γ. Prostaglandins Other Lipid Mediat. 2007;82:175–184. doi: 10.1016/j.prostaglandins.2006.05.002. [DOI] [PubMed] [Google Scholar]
- Gordon JL. Extracellular ATP: effects, sources and fate. Biochem J. 1986;233:309–319. doi: 10.1042/bj2330309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grau R, Punzon C, Fresno M, Iniguez MA. Peroxisome-proliferator-activated receptor α agonists inhibit cyclo-oxygenase 2 and vascular endothelial growth factor transcriptional activation in human colorectal carcinoma cells via inhibition of activator protein-1. Biochem J. 2006;395:81–88. doi: 10.1042/BJ20050964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding P, Balasubramanian L, Swegan J, Stevens A, Glass WF. Transforming growth factor beta regulates cyclooxygenase-2 in glomerular mesangial cells. Kidney Int. 2006;69:1578–1585. doi: 10.1038/sj.ki.5000323. [DOI] [PubMed] [Google Scholar]
- Hoagland KM, Maier KG, Roman RJ. Contributions of 20-HETE to the antihypertensive effects of Tempol in Dahl salt-sensitive rats. Hypertension. 2003;41:697–702. doi: 10.1161/01.HYP.0000047881.15426.DC. [DOI] [PubMed] [Google Scholar]
- Hu ZW, Kerb R, Shi XY, Wei-Lavery T, Hoffman BB. Angiotensin II increases expression of cyclooxygenase-2: implications for the function of vascular smooth muscle cells. J Pharmacol Exp Ther. 2002;303:563–573. doi: 10.1124/jpet.102.037705. [DOI] [PubMed] [Google Scholar]
- Huang CL, Cheng JC, Liao CH, Stern A, Hsieh JT, Wang CH, et al. Disabled-2 is a negative regulator of integrin {alpha}IIb{beta}3-mediated fibrinogen adhesion and cell signaling. J Biol Chem. 2004;279:42279–42289. doi: 10.1074/jbc.M402540200. [DOI] [PubMed] [Google Scholar]
- Ishizuka T, Cheng J, Singh H, Vitto MD, Manthati VL, Falck JR, et al. 20-Hydroxyeicosatetraenoic acid stimulates nuclear factor-κB activation and the production of inflammatory cytokines in human endothelial cells. J Pharmacol Exp Ther. 2008;324:103–110. doi: 10.1124/jpet.107.130336. [DOI] [PubMed] [Google Scholar]
- Jones PA, Scott-Burden T, Gevers W. Glycoprotein, elastin, and collagen secretion by rat smooth muscle cells. Proc Natl Acad Sci U S A. 1979;76:353–357. doi: 10.1073/pnas.76.1.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinopoulos PA, Vandoros GP, Sotiropoulou-Bonikou G, Kominea A, Papavassiliou AG. NF-κB/PPAR γ and/or AP-1/PPAR γ‘on/off’ switches and induction of CBP in colon adenocarcinomas: correlation with COX-2 expression. Int J Colorectal Dis. 2007;22:57–68. doi: 10.1007/s00384-006-0112-y. [DOI] [PubMed] [Google Scholar]
- Liang CJ, Ives HE, Yang CM, Ma YH. 20-HETE inhibits the proliferation of vascular smooth muscle cells via transforming growth factor-β. J Lipid Res. 2008;49:66–73. doi: 10.1194/jlr.M700155-JLR200. [DOI] [PubMed] [Google Scholar]
- Lin CC, Lin WN, Wang WJ, Sun CC, Tung WH, Wang HH, et al. Functional coupling expression of COX-2 and cPLA2 induced by ATP in rat vascular smooth muscle cells: role of ERK1/2, p38 MAPK, and NF-kappaB. Cardiovasc Res. 2009;82:522–531. doi: 10.1093/cvr/cvp069. [DOI] [PubMed] [Google Scholar]
- Mochizuki K, Suruga K, Sakaguchi N, Takase S, Goda T. Major intestinal coactivator p300 strongly activates peroxisome proliferator-activated receptor in intestinal cell line, Caco-2. Gene. 2002;291:271–277. doi: 10.1016/s0378-1119(02)00625-x. [DOI] [PubMed] [Google Scholar]
- Muthalif MM, Benter IF, Karzoun N, Fatima S, Harper J, Uddin MR, et al. 20-Hydroxyeicosatetraenoic acid mediates calcium/calmodulin-dependent protein kinase II-induced mitogen-activated protein kinase activation in vascular smooth muscle cells. Proc Natl Acad Sci U S A. 1998;95:12701–12706. doi: 10.1073/pnas.95.21.12701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng VY, Huang Y, Reddy LM, Falck JR, Lin ET, Kroetz DL. Cytochrome P450 eicosanoids are activators of peroxisome proliferator-activated receptor α. Drug Metab Dispos. 2007;35:1126–1134. doi: 10.1124/dmd.106.013839. [DOI] [PubMed] [Google Scholar]
- Oyekan AO. Differential effects of 20-hydroxyeicosatetraenoic acid on intrarenal blood flow in the rat. J Pharmacol Exp Ther. 2005;313:1289–1295. doi: 10.1124/jpet.104.080218. [DOI] [PubMed] [Google Scholar]
- Park S, Song JS, Lee DK, Yang CH. Suppression of AP-1 activity by tanshinone. Bull Korean Chem Soc. 1999;20:925–929. [Google Scholar]
- Paukkeri EL, Leppanen T, Sareila O, Vuolteenaho K, Kankaanranta H, Moilanen E. PPAR-α agonists inhibit nitric oxide production by enhancing iNOS degradation in LPS-treated macrophages. Br J Pharmacol. 2007;152:1081–1091. doi: 10.1038/sj.bjp.0707477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson E, Grieve DJ. Significance of peroxisome proliferator-activated receptors in the cardiovascular system in health and disease. Pharmacol Ther. 2009;122:246–263. doi: 10.1016/j.pharmthera.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Robinson WP, III, Douillet CD, Milano PM, Boucher RC, Patterson C, Rich PB. ATP stimulates MMP-2 release from human aortic smooth muscle cells via JNK signaling pathway. Am J Physiol Heart Circ Physiol. 2006;290:H1988–H1996. doi: 10.1152/ajpheart.00344.2005. [DOI] [PubMed] [Google Scholar]
- Roman RJ. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev. 2002;82:131–185. doi: 10.1152/physrev.00021.2001. [DOI] [PubMed] [Google Scholar]
- Schonbeck U, Sukhova GK, Graber P, Coulter S, Libby P. Augmented expression of cyclooxygenase-2 in human atherosclerotic lesions. Am J Pathol. 1999;155:1281–1291. doi: 10.1016/S0002-9440(10)65230-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staels B, Koenig W, Habib A, Merval R, Lebret M, Torra IP, et al. Activation of human aortic smooth-muscle cells is inhibited by PPAR α but not by PPAR γ activators. Nature. 1998;393:790–793. doi: 10.1038/31701. [DOI] [PubMed] [Google Scholar]
- Stemme V, Swedenborg J, Claesson H, Hansson GK. Expression of cyclo-oxygenase-2 in human atherosclerotic carotid arteries. Eur J Vasc Endovasc Surg. 2000;20:146–152. doi: 10.1053/ejvs.2000.1145. [DOI] [PubMed] [Google Scholar]
- Traut TW. Physiological concentrations of purines and pyrimidines. Mol Cell Biochem. 1994;140:1–22. doi: 10.1007/BF00928361. [DOI] [PubMed] [Google Scholar]
- Tunctan B, Korkmaz B, Cuez T, Kemal BC, Sahan-Firat S, Falck J, et al. Contribution of vasoactive eicosanoids and nitric oxide production to the effect of selective cyclooxygenase-2 inhibitor, NS-398, on endotoxin-induced hypotension in rats. Basic Clin Pharmacol Toxicol. 2010;107:877–882. doi: 10.1111/j.1742-7843.2010.00589.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uddin MR, Muthalif MM, Karzoun NA, Benter IF, Malik KU. Cytochrome P-450 metabolites mediate norepinephrine-induced mitogenic signaling. Hypertension. 1998;31:242–247. doi: 10.1161/01.hyp.31.1.242. [DOI] [PubMed] [Google Scholar]
- Wang DJ, Huang NN, Heppel LA. Extracellular ATP and ADP stimulate proliferation of porcine aortic smooth muscle cells. J Cell Physiol. 1992;153:221–233. doi: 10.1002/jcp.1041530202. [DOI] [PubMed] [Google Scholar]
- Wong MS, Vanhoutte PM. COX-mediated endothelium-dependent contractions: from the past to recent discoveries. Acta Pharmacol Sin. 2010;31:1095–1102. doi: 10.1038/aps.2010.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.