

Abstract
BACKGROUND AND PURPOSE
Exposure to drugs of abuse or stress results in adaptation in the brain involving changes in gene expression and transcription factors. Morphine withdrawal modulates gene expression through various second-messenger signal transduction systems. Here, we investigated changes in activation of the transcription factor, cAMP-response element binding protein (CREB), in the hypothalamic paraventricular nucleus (PVN) and the kinases that may mediate the morphine withdrawal-triggered activation of CREB and the response of the hypothalamic-pituitary-adrenocortical (HPA) axis after naloxone-induced morphine withdrawal.
EXPERIMENTAL APPROACH
The effects of morphine dependence and withdrawal, phosphorylated CREB (pCREB), corticotrophin-releasing factor (CRF) expression in the PVN and HPA axis activity were measured using immunoblotting, immunohistochemistry and radioimmunoassay in controls and in morphine-dependent rats, withdrawn with naloxone and pretreated with vehicle, calphostin C, chelerythrine (inhibitors of protein kinase C (PKC) or SL-327 [inhibitor of extracellular signal regulated kinase (ERK) kinase]. In addition, changes in PKCα and PKCγ immunoreactivity were measured after 60 min of withdrawal.
KEY RESULTS
In morphine-withdrawn rats, pCREB immunoreactivity was increased within CRF immunoreactive neurons in the PVN and plasma corticosterone levels were raised. SL-327, at doses that reduced the augmented pERK levels in the PVN, did not attenuate the rise in pCREB immunoreactivity or plasma corticosterone secretion. In contrast, PKC inhibition reduced the withdrawal-triggered rise in pCREB, pERK1/2 and corticosterone secretion.
CONCLUSIONS AND IMPLICATIONS
PKC mediated, in part, both CREB activation and the HPA response to morphine withdrawal. The ERK kinase/ERK pathway might not be necessary for either activation of CREB or HPA axis hyperactivity.
Keywords: morphine withdrawal, CREB, CRF, PVN, HPA axis, PKC
Introduction
The paraventricular nucleus of the hypothalamus (PVN) is the final common pathway linking excitatory stress response signals to activation of the pituitary-adrenocortical axis. The stress response is a reflex that incorporates both neuronal and endocrine components and that can influence different facets of drug addiction, including the rewarding properties of drugs of abuse and relapse vulnerability after cessation of drug use (Koob, 2008). In the CNS, the role of integrating stress signals producing a neuroendocrine output to the hypothalamic-pituitary-adrenocortical (HPA) axis is played by neurons secreting corticotrophin-releasing factor (CRF) (Swanson et al., 1983). This 41-amino-acid peptide is the most powerful adrenocorticotropic hormone (ACTH) secretagogue and also causes a wide spectrum of neuroendocrine, autonomic, and behavioural changes typical of a stress reaction. These diverse functions are achieved by CRF acting not only as a hormone but also as a neurotransmitter and a neuromodulator in a number of brain structures.
Like stressors, morphine withdrawal activates the HPA axis in rats, which results in neuronal activation of stress-related neurosecretory neurons in the parvocellular cells of the PVN as well as corticosterone secretion (Laorden et al., 2002a; Núñez et al. 2007a; Cleck and Blendy, 2008). Previous studies showed that morphine withdrawal is able to modulate the activity of cells within the different divisions of the PVN and to increase corticosterone release (Laorden et al., 2002a). Complementary to these findings are data demonstrating that hypothalamic levels of CRF mRNA are increased in response to morphine withdrawal, as are plasma ACTH and corticosterone levels. Thus, we have previously shown that activation of HPA during naloxone-precipitated morphine withdrawal involves transcriptional up-regulation of hypophyseotropic CRF and arginine-vasopressin (AVP) expression in the PVN (Núñez et al., 2007a).
In addition to these neuroendocrine responses, morphine dependence exerts long-lasting effects on gene expression (Blendy and Maldonado, 1998; McClung and Nestler, 2008). Phosphorylation and subsequent activation of the transcription factor, cAMP-response element binding protein (CREB), is a site of convergence for several signal transduction cascades, including the cAMP pathway via protein kinase A (PKA), intracellular Ca2+ via Ca2+-calmodulin-dependent kinases (CaMK), the Ras/extracellular signal regulated kinase (ERK) protein kinase pathway and the phosphatidylinositol-3-kinase (PI3K)/Akt (Chao and Nestler, 2004). CREB is of particular interest in drug addiction because its activation is downstream of the cAMP/PKA signalling pathway, whose up-regulation has been extensively characterized as an adaptation to chronic exposure to drugs of abuse (Chao and Nestler, 2004). Subsequently, however, many other kinases besides PKA have been identified as being capable of phosphorylating CREB in response to various stimuli, such as the mitogen activated protein kinases (MAPK) and protein kinase C (PKC) (Williams et al., 2001).
Precipitated morphine withdrawal has been shown to modify several indices of CREB function within the PVN, including elevated c-Fos expression in rats (Laorden et al., 2002b; Núñez et al. 2007a). Changes in CREB activity may be important for the development and expression of opioid dependence (Carlezon et al. 2005; Martín et al., 2009). The present studies were designed to assess the changes in CREB activation (as measured by pCREB levels) within the PVN during morphine dependence and after naloxone-precipitated morphine withdrawal. Plasma corticosterone was measured as a marker of HPA axis activity. Multiple-label immunohistochemistry and Western blot were used to identify and quantify the expression of pCREB in populations of neuroendocrine cells in general and in CRF neurons, specifically.
The chronic adaptive molecular mechanisms in opioid dependence involve protein kinases, which are implicated in a wide variety of cellular regulatory processes involving protein phosphorylation and gene expression. The MAPK family are known to be transducers of a broad range of responses and one member, ERK, is involved in drug addiction (Eitan et al., 2003). In rat brain, chronic morphine treatment has been shown to regulate ERK1/2 catalytic activity in a region-dependent manner (Muller and Unterwald, 2004). Because phosphorylation of CREB can be associated with distinct signalling pathways, the next objective of the present work was to determine whether activation of PKC and/or the ERK kinase (MEK)-ERK1/2 pathways contributed to morphine withdrawal-induced phosphorylation of CREB in the PVN. The functional neuroendocrine implication of these results was determined by measurement of plasma corticosterone concentration in conscious animals after pretreatment with selective ERK and PKC inhibitors. Furthermore, we examined whether PKCα and PKCγ immunoreactivity in the PVN could be changed following naloxone-induced morphine withdrawal. As we have shown that the blockade of calcium channels attenuates the increase in HPA axis activity observed after naloxone-induced morphine withdrawal, we focused in these two isoforms of PKC because both of them, as classical isoforms, are regulated by calcium, whereas the novel and atypical isoforms are insensitive to calcium (Zarate and Manji, 2009). Moreover, both of these isoforms are expressed in the brain. We also tested the hypothesis that pharmacological inhibition of PKC or MEK would attenuate the somatic signs of morphine withdrawal.
Methods
Animals
All animal care and experimental procedures were in accordance with the European Communities Council Directive of 24 November 1986 (86/609/EEC) and approved by the local Committees for animal research (REGA ES300305440012). Male Sprague-Dawley rats (220–240 g; Harlan, Barcelona, Spain) were housed two-to-three per cage (length, 45 cm; width, 24 cm; height, 20 cm) in a room with controlled temperature (22 ± 2°C) and humidity (50 ± 10%), with free access to water and food. Animals were adapted to a standard 12 h light-dark cycle for 7 days before the beginning of the experiments.
Drug treatment and experimental procedure
Groups of rats were made dependent on morphine by s.c. implantation of pellets of morphine base (75 mg), one on day 1, two on day 3 and three on day 5, under light ether anaesthesia (for about 30 s). Control animals were implanted with placebo pellets containing lactose instead of morphine on the same time schedule. This morphine treatment paradigm has been shown to produce profound states of tolerance and dependence and to result in characteristic biochemical adaptations within the PVN and to induce behavioural alterations (Couceyro and Douglass, 1995; Núñez et al. 2009). On day 8, when rats were morphine dependent, they were injected with saline (s.c.) or naloxone (2 mg·kg−1, s.c.). The weight gain of the rats was checked during treatment to ensure that the morphine was released correctly from the pellets because it is known that chronic morphine treatment induces a decrease in body weight gain due to lower caloric intake (Berhow et al., 1995).
For measurement of the withdrawal syndrome, experiments were carried out in a quiet room. The observer was unaware of the drug combination used. Rats were individually placed into transparent plastic cages 15 min before the naloxone injection and observed continuously for the occurrence of somatic signs of opiate withdrawal up to 30 min after the naloxone injection, at which time many of the acute behavioural effects are displayed (Guitart and Nestler, 1989).
Appearance or non-appearance of the following previously identified behavioural characteristics of the rat opiate abstinence syndrome (Lu et al., 2000) were evaluated (over 30 min): wet-dog shakes, teeth chattering, ptosis, tremor, salivation, rhinorrhoea, lacrimation and irritability. These symptoms are shown as the percentage of animals showing the signs. Weight loss associated with naloxone-precipitated morphine withdrawal is well documented (Funada et al., 2001) and might be associated with increased sympathetic tone, diarrhoea, salivation and breakdown of white adipose tissue depots (Kovacs and Telegdy, 1988). In the present study, body weight loss was determined as the difference between the weight determined immediately before saline or naloxone injection and a second determination made 60 and 90 min later.
In order to determine the effect of inhibiting ERK1/2 activation on the morphine withdrawal-induced changes in pCREB expression at PVN as well as HPA axis activity, pCREB and plasma corticosterone levels were determined in morphine-dependent and control rats treated with SL-327 [a selective inhibitor of MEK (Atkins et al., 1998)] 1 h before the administration of naloxone or saline. This inhibitor was dissolved in dimethyl sulphoxide (DMSO) 100% and injected intraperitoneally (i.p.) at an injection volume of 1 mL·kg−1. On the basis of our initial experiments where the pretreatment with 100 mg·kg−1 of SL-327, but not 50 mg·kg−1, decreased pERK levels in control rats (Núñez et al., 2008), the 100 mg·kg−1 dose was chosen for our experiments.
Furthermore, we tested the hypothesis that pharmacological inhibition of PKC would block morphine withdrawal-induced CREB phosphorylation in the PVN as well as HPA axis activation by infusion of the inhibitors of PKC, calphostin C and chelerythrine. pCREB expression was determined in dependent and control rats pretreated with these inhibitors and compared with that observed in dependent and control animals that had not been so treated. Briefly, animals were continuously infused for 7 days, via s.c. osmotic minipumps (Alzet mod. 2001, 1 µL·h−1), with calphostin C (40 pmol·day−1) or chelerythrine (20 nmol·day−1). The selection of the drug doses used in this study was based on the IC50 values observed in rat brain for PKC inhibitors (Kobayashi et al., 1989; Herbert et al., 1990) and on the dose that has been demonstrated to be effective in inhibiting some hypothalamic biochemical changes induced during morphine withdrawal (Fundytus and Coderre, 1996; Vargas et al., 1997; Cerezo et al., 2002). Pumps were primed for 5 h before implantation at 37°C in sterile saline in order to obtain an optimal flow rate (1 µL·h−1) and then were implanted (under light ether anaesthesia) on the back of animals simultaneously with the first morphine or placebo pellet. Surgical incisions were closed using sterile clamps. On day 8, a withdrawal syndrome was induced by s.c. injection of naloxone (2 mg·kg−1 s.c.). The method of chronic treatment with PKC inhibitors was chosen on the basis of previous experiments where a single dose of PKC inhibitors failed to prevent the development of physical dependence (Gabra et al., 2008) whereas chronic perfusion with calphostin C induced clear inhibition of morphine withdrawal-induced neuroendocrine changes in rats (Cerezo et al. 2002; Benavides et al., 2005).
Western blot analysis
Sixty and 90 min after administration of naloxone or saline, rats were killed by decapitation. The hypothalamic tissue containing the PVN was dissected according to the technique of Palkovits (1973) and the PVN corresponds to those in Plates 25 and 26 in the atlas of Paxinos and Watson (2007). PVN samples were placed in homogenization buffer [phosphate-buffered saline (PBS), 10% sodium dodecyl sulphate (SDS), protease inhibitors plus a phosphatase inhibitor Cocktail Set], homogenized and sonicated for 30 s before centrifugation at 6000×g for 10 min at 4°C. Samples containing 40 µg of protein were loaded on a 10% SDS/polyacrylamide gel, separated by electrophoresis and transferred onto polyvinylidene difluoride membranes (Millipore, Bedford, MA, USA). Non-specific binding of antibodies was prevented by incubating membranes in 1% bovine serum albumin in Tris-buffered saline Tween-20 (TBST; 10 mM Tris HCl, pH 7.6, 150 mM NaCl, 0.15% Tween-20). The blots were incubated with the following primary antibodies: 1:750 polyclonal anti-phospho CREB-123-136 (pCREB; Millipore, Temecula, CA, USA); 1:1000 polyclonal anti-total CREB (Cell Signaling, Beverly, MA, USA); 1:1000 monoclonal anti-pERK1/2 (Santa Cruz Biotechnology, Santa Cruz, CA, USA); 1:6000 polyclonal anti-PKCα (Sigma, St Louis, MO, USA); 1:1000 polyclonal anti-PKCγ (Santa Cruz); and 1:5000 polyclonal anti-α-tubulin (Cell Signaling), in TBST with bovine serum albumin overnight at room temperature (anti-phospho CREB, anti-total CREB and anti-α-tubulin) and 4°C (anti-pERK1/2 and anti-PKCα). After extensive washing with TBST, the membranes were incubated for 1 h at room temperature with peroxidase-labelled secondary antibodies (anti-rabbit sc-2004 at 1:2500 for pCREB, 1:5000 for total CREB and 1:5000 for PKC and α-tubulin; anti-mouse sc-2005 at 1:5000 for pERK1/2). After washing, immunoreactivity was detected with an enhanced chemiluminescence's Western blot detection system (ECL, Amersham Ibérica, Madrid, Spain) and visualized by Amersham Hyper film-ECL. We used α-tubulin (rabbit polyclonal antibody, Cell Signaling #4967) as our loading control for all the experiments. Before re-probing, blots were stripped by incubation with stripping buffer (glycine 25 mM and SDS 1%, pH 2) for 1 h at 37°C. Blots were subsequently reblocked and probed with anti-α-tubulin (1:5000, overnight at room temperature). Quantification of immunoreactivity corresponding to pCREB (43 kDa), total CREB (43 kDa), ERK1 (42 kDa), ERK2 (44 kDa), PKCα (80 kDa), PKCγ (80 kDa) and α-tubulin (52 kDa) bands was carried out by densitometry using Gel Doc (Bio-Rad, Hercules, CA, USA). The integrated optical density of the bands was corrected by subtraction of the background values. The ratios of pCREB/ α-tubulin, total CREB/α-tubulin, pERK1/ α-tubulin, pERK2/ α-tubulin, PKCα/α-tubulin and PKCγ/α-tubulin were calculated and expressed as a percentage of the average of controls in each blot.
Immunohistochemical detection of CREB, p-CREB and CRF
Sixty minutes after administration of saline or naloxone, rats were deeply anesthetized with an overdose of pentobarbital (100 mg·kg−1) and perfused transcardially with PBS (pH 7.4) following by fixative containing paraformaldehyde (4% in PBS). After removal of the perfused brains, they were post-fixed in the same fixative and stored at 4°C overnight, Free-flotation serial coronal brain sections (30 µm thickness) throughout the rostrocaudal extend of the hypothalamus were obtained on a freezing microtome Leica, Nussloch, Germany). Sections were pre-incubated in absolute methanol containing 10% H2O2, rinsed in PBS and treated with normal goat serum (NGS)–PBS (PBS containing 2% swine serum and 0.5% Triton-X-100). Section were then incubated for 60 h at 4°C with rabbit anti-total CREB (1:1500; Cell Signaling), rabbit anti-pCREB antibody (Upstate; 1:750 in NGS–PBS) or rabbit anti-CRF antibody (1:5000 in 2% NGS–PBS; a generous gift from Wylie Vale, The Salk Institute, La Jolla, CA, USA). This was followed by application of a biotinylated anti-rabbit IgG (diluted 1:200 for 1 h or 2 h) in NGS–PBS (Vector, Burlingame, CA), and then with the avidin–biotin complex at room temperature for 1 h. Visualization of the antigen–antibody reaction sites was performed using 0.033% 3,3′-diaminobenzidine (DAB; Sigma, St Louis, MO, USA) and 0.014% H2O2 in 0.05 M Tris–HCl buffer. The reaction was stopped in PBS. The sections were mounted on gelatine coated slides, dehydrated through graded alcohols, cleared in xylene and cover slipped with dibutylphthalate.
Double-labelling immunohistochemistry of pCREB-immunoreactive nuclei and CRF-positive neurons
For pCREB and CRF double-label immunohistochemistry, tissue sections from each rat in each treatment group were processed as follows: pCREB was developed with DAB intensified with nickel, and the CRF revealed with DAB. pCREB immunohistochemistry was performed as described previously, and pCREB antibody–peroxidase complex was visualized by using a mixture of NiSO4.6H2O (33.2 mg·mL−1), DAB (0.033%) and 0.014% H2O2 in 0.175 M sodium acetate solution (pH 7.5). Sections were then incubated with an anti-CRF antibody (diluted 1:1000 in NGS–PBS) 60 h at 4°C. A biotinylated anti-rabbit IgG (diluted 1:200 for 1 h) was used as a secondary antibody. The CRF antibody–peroxidase complex was stained in 0.033% DAB and 0.014% H2O2 in 0.05 M Tris–HCl buffer.
Quantification of pCREB and CRF immunoreactivity
pCREB immunostaining within section of the PVN was quantified bilaterally for each rat and for all treatment groups by an observer unaware of the treatment protocols. The density of pCREB-like immunoreactivity was determined using a computer-assisted image analysis system (Q500MC, Leica, Madrid, Spain). This system consists of a light microscope (DMLB; Leica) connected to a video camera (Sony 151-AP; Sony, Madrid, Spain) and the image analysis computer. A square field (195 µm side) was superimposed upon the captured image (×20 magnification) to use as a reference area.
The CRF immunoreactivity was quantified bilaterally in the medial parvocellular portion of the PVN. The number of CRF-positive neurons was counted in four to five sections from each animal in the PVN, using the same system described above. A square field (195 µm side) was superimposed upon the captured image (×20 magnification) for using as a reference area.
Quantification of pCREB-positive/CRF-positive neurones
Positive nuclei for pCREB immunoreactivity were detected using the same conventional light microscopy described above, and counted at ×20 magnification. pCREB-positive CRF cells were identified as cells with brown cytosolic deposits for CRF-positive staining and blue/dark nuclear staining for pCREB. A square field (195 µm) was superimposed upon captured image to use as reference area. The number of double-labelled pCREB neurons observed bilaterally was counted in four to five sections from each animal in the PVN. The CRF positive cells without a visible nucleus (pCREB-negative CRF cells) were also included in the analysis.
Radioimmunoassay
Sixty minutes after saline or naloxone injection, rats were decapitated. Plasma levels of corticosterone were measured by commercially available kits for rats (125I-corticosterone RIA; MP Biomedicals, Orangeburg, NY, USA). The sensitivity of the assay was 7.7 ng·mL−1.
Statistical analysis
Data are presented as mean ± SEM. Data were analysed using two-way or one-way (when required) analysis of variance (anova) followed by a post hoc Newman–Keuls test. Student's t-test was used when comparisons were restricted to two experimental groups. Differences with a P-value < 0.05 were considered significant.
Materials
Pellets of morphine (75 mg morphine base/pellet; Alcaliber Labs., Madrid, Spain) or lactose (placebo) were prepared by the Department of Pharmacy and Pharmaceutics Technology (School of Pharmacy, Granada, Spain); naloxone HCl was purchased from Sigma Chemical Co. (St Louis, MO, USA), dissolved in sterile 0.9% NaCl (saline) and administered in volumes of 0.1 mL·100 g−1 body weight. SL-327 (Ascent Scientific, North Somerset, UK) was dissolved in DMSO. DMSO was purchased from Sigma. Calphostin C (2-(12-(2-(benzoyloxy) propyl)-3, 10-dihydro-4, 9-dihydroxy-2, 6, 7, 11-tetramethoxy-3, 10-dioxo-1-perylenyl)-1-ethylethyl carbonic acid 4-hydroxyphenyl ester) was purchased from Sigma, dissolved in DMSO and serially diluted in Milli-Q water (final concentration of DMSO was 0.06%). Chelerythrine HCl was purchased from Sigma and diluted in Milli-Q water. Aliquots of the stock solutions were stored at −30°C until used for experimentation. The chronic delivery of calphostin C and chelerythrine was by means of Alzet 2001 osmotic minipumps (Alza, Palo Alto, CA, USA), which deliver at 1 µL·h−1. Drugs were prepared fresh every day. Protease inhibitors (Roche, Mannheim, Germany); phosphatase inhibitor Cocktail Set (Calbiochem, Darmstadt, Germany); goat serum (Sigma); avidin–biotin complex (ABC kits; Vector, Burlingame, CA, USA); nickel sulphate (Sigma). Drug and molecular target nomenclature follows Alexander et al. (2009).
Results
Effects of chronic morphine and precipitated morphine withdrawal on CREB phosphorylation and total CREB as determined by Western blot
To study the effects of chronic morphine and naloxone-precipitated morphine withdrawal on the induction of CREB phosphorylation, we determined pCREB protein levels in cell extracts from the PVN at different time-points (30, 60 and 90 min). No alterations in pCREB levels in the PVN were found at 30 (data not shown) or 90 min (Figure 1B) following naloxone-induced morphine withdrawal. The anova for pCREB at 60 min showed a significant effect of naloxone injection (F1,20= 8.69; P = 0.0080), chronic morphine administration (F1,20= 9.15; P = 0.0067) and an interaction between morphine treatment and naloxone injection (F1,20= 4.84; P = 0.0398). As shown in Figure 1A, there was a significant (P < 0.01) enhancement in pCREB levels after naloxone injection to morphine-dependent rats, compared with control animals also receiving naloxone and with morphine-dependent rats given saline. Acute naloxone injection had no effect in animals chronically treated with placebo, compared with the corresponding control group receiving saline.
Figure 1.
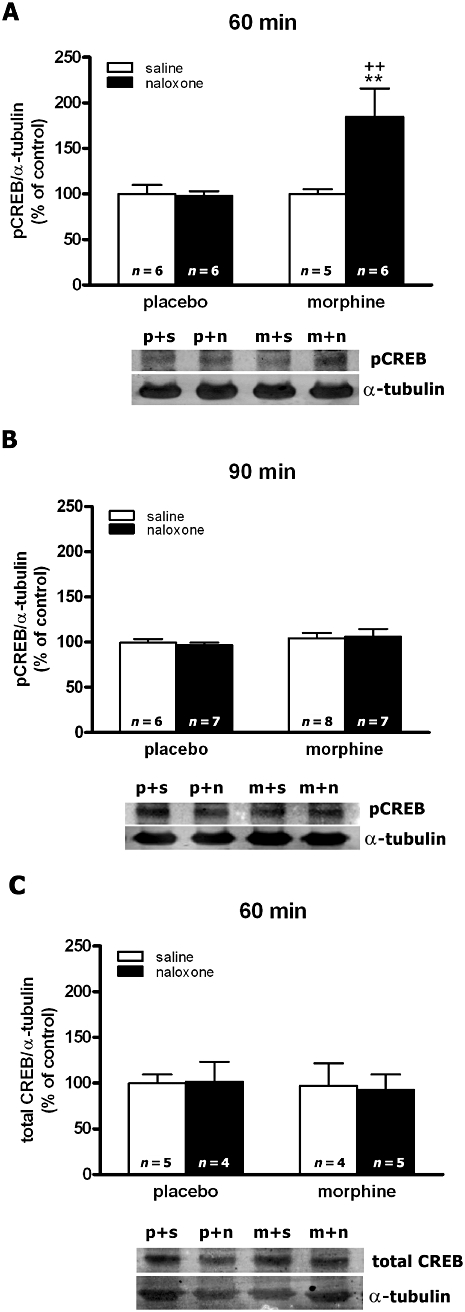
Quantitative analysis of phosphorylated cAMP-response element binding protein (pCREB) (in A,B) and total CREB (in C) in the hypothalamic paraventricular nucleus tissue isolated from placebo (p) or morphine (m)-dependent rats after s.c. administration of saline (s) or naloxone (n), at the times indicated. Levels of pCREB and total CREB were detected by Western blot analysis. Each bar represents the mean optical density ± SEM; values are expressed as the ratio pCREB/α-tubulin and total CREB/α-tubulin (% of controls). Newman–Keuls' post hoc comparison test revealed a significant increase in pCREB in morphine-withdrawn rats at 60 min after naloxone injection. **P < 0.01 versus placebo + naloxone; ++P < 0.01 versus morphine + saline.
The anova for total CREB at 60 min time point showed no main effect of naloxone administration (F1,14= 0.79; P = 0.3878), morphine pretreatment (F1,14= 0.36; P = 0.5600) and no interaction between morphine treatment and naloxone injection (F1,14= 0.17; P = 0.6890; Figure 1C).
pCREB, total CREB and CRF immunoreactivity in the PVN of morphine-dependent and morphine-withdrawn rats
These results were confirmed by immunohistochemical procedures (Figure 2). Antiserum specifically recognizing the Ser133-phosphorylated form of CREB revealed a weak basal expression of pCREB. However, animals killed at 60 min after naloxone-induced morphine withdrawal displayed an increase in the number of positively stained nuclei by neurons of the parvocellular PVN. As shown in Figure 2F–H, in the placebo plus saline- or naloxone-treated rats and in morphine plus saline-injected animals, the cells showed discrete staining for pCREB, whereas native (unphosphorylated) CREB was found to be expressed constitutively in ostensibly neuronal nuclei in the PVN (Figure 2B–D). High levels of pCREB immunoreactivity were observed in the PVN 60 min after naloxone injection to morphine-dependent rats (Figure 2I), whereas total CREB immunoreactivity was similar to the controls (Figure 2E). Two-way anova for pCREB showed an interaction between morphine treatment and naloxone injection (F1,16= 4.54; P = 0.0489), with main effect of naloxone injection (F1,16= 8.85; P = 0.0089) and morphine treatment (F1,16= 18.71; P = 0.0005). Post hoc tests revealed that morphine withdrawal induced a significant elevation in pCREB immunoreactivity compared with the control group also receiving naloxone (P < 0.001; Figure 2J) and with the morphine-dependent rats injected with saline (P < 0.01). However, naloxone injection to placebo-pelleted rats had no effect on CREB phosphorylation compared with the control group receiving saline instead naloxone.
Figure 2.
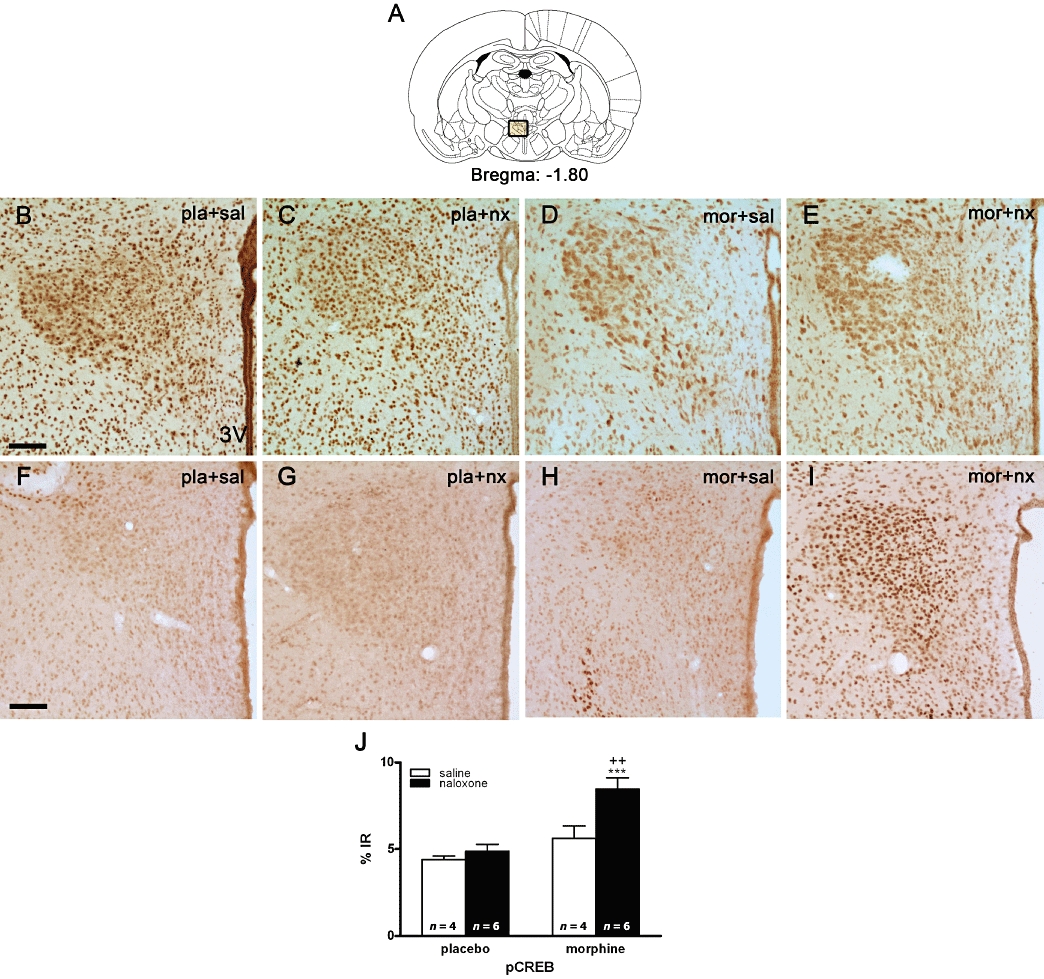
Morphine withdrawal activates cAMP-response element binding protein (CREB) phosphorylation in the hypothalamic paraventricular nucleus (PVN). Rats were made dependent on morphine for 7 days and on day 8 were injected with saline or naloxone. Controls received placebo pellets at the same time schedule and, on day 8, were injected with saline or naloxone. Sixty minutes after injections, rats were perfused, and the PVN was processed for pCREB and total CREB immunohistochemistry. Photomicrographs of sections through a level of the PVN from rats stained with an antiserum against native (unphosphorylated-total) CREB (in B–E) and from animals stained with an antiserum specific to the pCREB (in F–I). CREB antiserum ubiquitously stains neuronal nuclei throughout the PVN whereas, under control conditions, low levels of basal pCREB are evident in the PVN. Morphine withdrawal results in an enhanced level of pCREB-immunoreactivity in the PVN. (A) Schematic diagram illustrating the area measured for quantification of immunostaining signals. (3V) Third ventricle. Scale bar: 100 µm. (J) Quantitative analysis of the % pCREB immunoreactivity (IR) in the PVN. Data shown are means ± SEM. Post hoc analysis revealed a significant increase in pCREB immunoreactivity in morphine-withdrawn rats. ***P < 0.01 versus placebo + naloxone; ++P < 0.01 versus morphine + saline. mor, morphine pellets; nx, naloxone; pla, placebo pellets; sal, saline.
Two-way anova for CRF at the PVN (Figure 3) showed a significant effect of naloxone injection (F1,21= 10.89; P = 0.0034), chronic morphine administration (F1,21= 6.62; P = 0.0177) and an interaction between chronic morphine and naloxone injection (F1,21 = .6.79; P = 0.0165). As shown in Figure 3F, there was a significant (P < 0.01) enhancement in CRF immunoreactivity after naloxone injection to morphine-dependent rats, compared with control animals also receiving naloxone and with morphine-dependent rats given saline. Acute naloxone injection had no effect in animals chronically treated with placebo, compared with the corresponding control group receiving saline.
Figure 3.
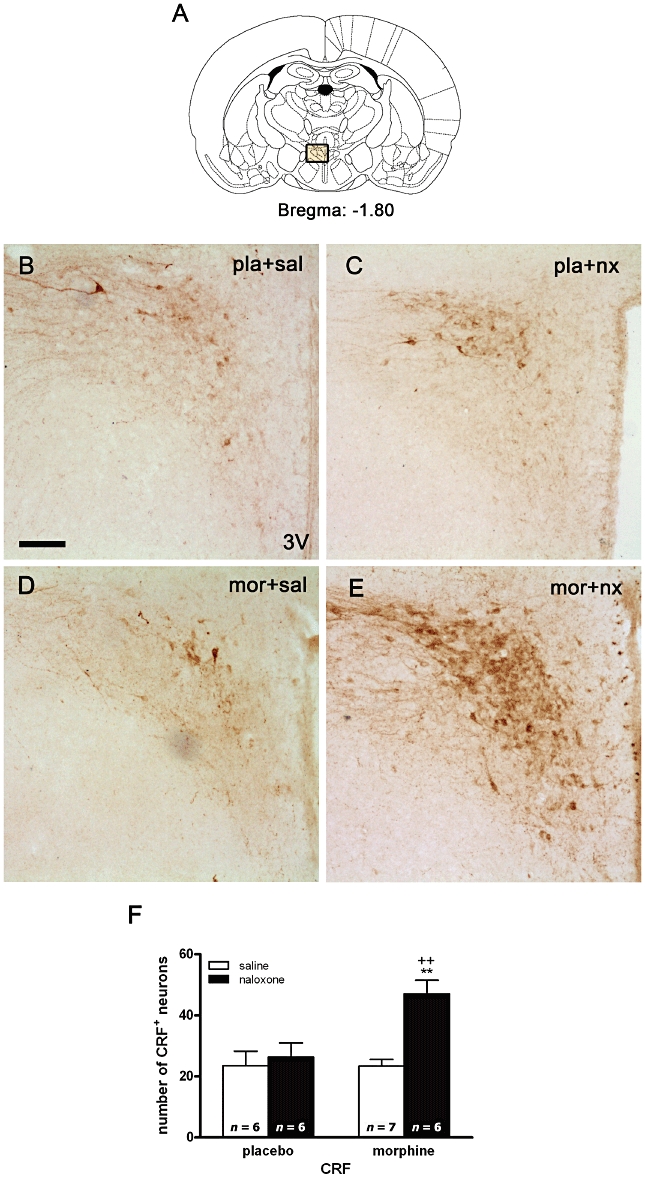
Morphine withdrawal increases corticotrophin-releasing factor (CRF) expression in the hypothalamic paraventricular nucleus (PVN). Rats were made dependent on morphine for 7 days and on day 8 were injected with saline or naloxone. Controls received placebo pellets at the same time schedule and, on day 8, were injected with saline or naloxone. Sixty minutes after injections, rats were perfused, and the PVN was processed for CRF immunohistochemistry. (B–E) Represent immunohistochemical detection of CRF in the PVN from placebo and morphine-treated rats receiving saline or naloxone. (3V) Third ventricle. Scale bar: 100 µm. (F) Quantitative analysis of the number of CRF-positive neurons in the PVN. Data shown are means ± SEM. Post hoc analysis revealed a significant increase in CRF-positive neurons in morphine-withdrawn rats. **P < 0.01 versus placebo + naloxone; ++P < 0.01 versus morphine + saline. mor, morphine pellets; nx, naloxone; pla, placebo pellets; sal, saline.
Induction of CREB phosphorylation in CRF-positive neurons in the PVN
To explore the specificity of morphine withdrawal-induction of CREB phosphorylation observed in the parvocellular part of the PVN, sections from different treatments were immunohistochemically double-labelled for pCREB and CRF (Figure 4). Two-way anova revealed a main effect of chronic morphine (F1,19= 14.85; P = 0.0011) and an interaction between naloxone injection and chronic morphine treatment (F1,19= 10.33; P = 0.0046) for pCREB expression in CRF-positive neurons. Post hoc analysis showed a significant increase in the number of CRF containing neurons expressing pCREB after naloxone-induced morphine withdrawal (Figure 4B), compared with placebo controls receiving naloxone (P < 0.001). However, the number of CRF-positive/pCREB-negative neurons was not altered during morphine dependence or withdrawal (Figure 4C).
Figure 4.
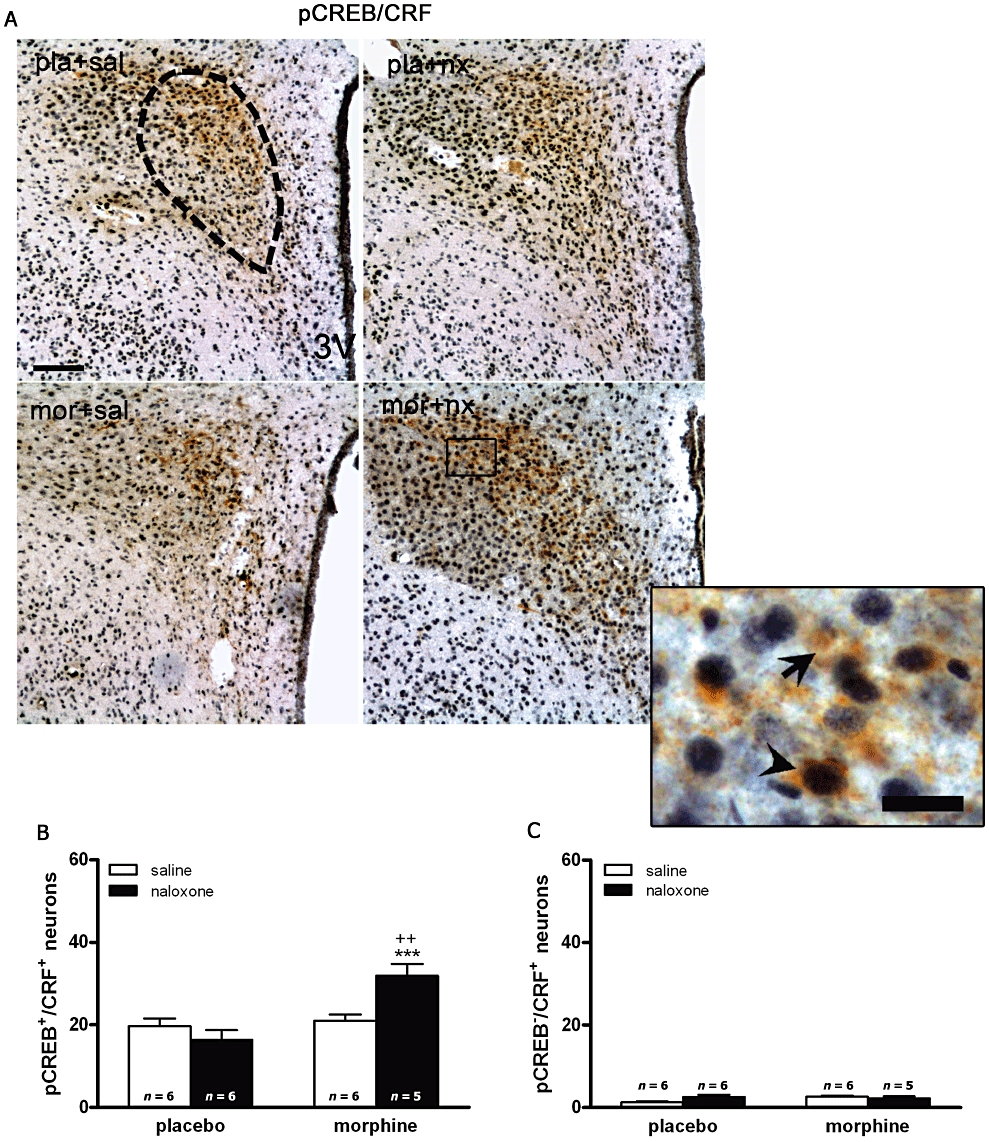
cAMP-response element binding protein (CREB) phosphorylation in the hypothalamic paraventricular nucleus (PVN) is predominantly localized in corticotrophin-releasing factor (CRF)-positive neurons. Rats were made dependent on morphine (mor) and injected with saline (sal) or naloxone (nx). Controls received placebo pellets (pla) and were given saline or naloxone. Sixty minutes after injections, animals were perfused and the PVN was processed for double-labelled (pCREB and CRF) immunohistochemistry. Top panels (A) show immunohistochemical detection of pCREB into CRF neurons after the different treatments. Low and high magnifications images show pCREB-positive (blue-black)/CRF-positive (brown) neurons (arrow head) and CRF-positive/pCREB-negative (arrow) immunoreactivity. Scale bar: 100 µm (low magnification); 20 µm (high magnification). Bottom panels show quantitative analysis of pCREB-positive/CRF-positive (B) and CRF-positive neurons without pCREB (C) neurons in the PVN. Data shown are means ± SEM. Post hoc tests revealed a higher number of pCREB-positive nuclei in CRF immunoreactive neurons. ***P < 0.001 versus placebo + naloxone; ++P < 0.01 versus morphine + saline.
PKC activity is required for morphine withdrawal-induced CREB phosphorylation in the PVN
In previous studies, Western blot analysis revealed strong temporal activation (phosphorylation) of ERK1/2 in the PVN after naloxone injection to morphine-dependent rats (Núñez et al., 2008). Because the phosphorylation of CREB can be mediated by several upstream kinases including ERK1/2 (Deak et al., 1998), in the present work we examined whether phosphorylation of ERK1/2 by MEK is also necessary for the morphine withdrawal-induced CREB phosphorylation (Figure 5). Two-way anova for pERK1/2 in rats pretreated with the MEK inhibitor, SL-327, revealed a significant effect of morphine treatment (pERK1: F1,18= 10.94; P = 0.0039; pERK2: F1,18= 12.12; P = 0.0027) and acute SL-327 injection (pERK1: F1,18= 50.68; P < 0.0001; pERK2: F1,18= 75.73; P < 0.0001). No significant interaction for pretreatment-acute treatment was observed. Post hoc analysis revealed that SL-327 effectively prevented the phosphorylation of ERK1/2 in both controls (P < 0.05; P < 0.001) and morphine-withdrawn animals (P < 0.001; Figure 5A). Two-way anova for pCREB in rats pretreated with SL-327 revealed a main effect of SL-327 (F1,17= 12.04; P = 0.0029). As shown in Figure 5B, SL-327 increased (P < 0.01) basal levels of pCREB and did not attenuate the increase in pCREB levels 60 min after administration of naloxone to morphine-dependent rats. From these results, it does not appear that the increased CREB phosphorylation following morphine withdrawal is mediated through the MEK/ERK1/2 pathway.
Figure 5.
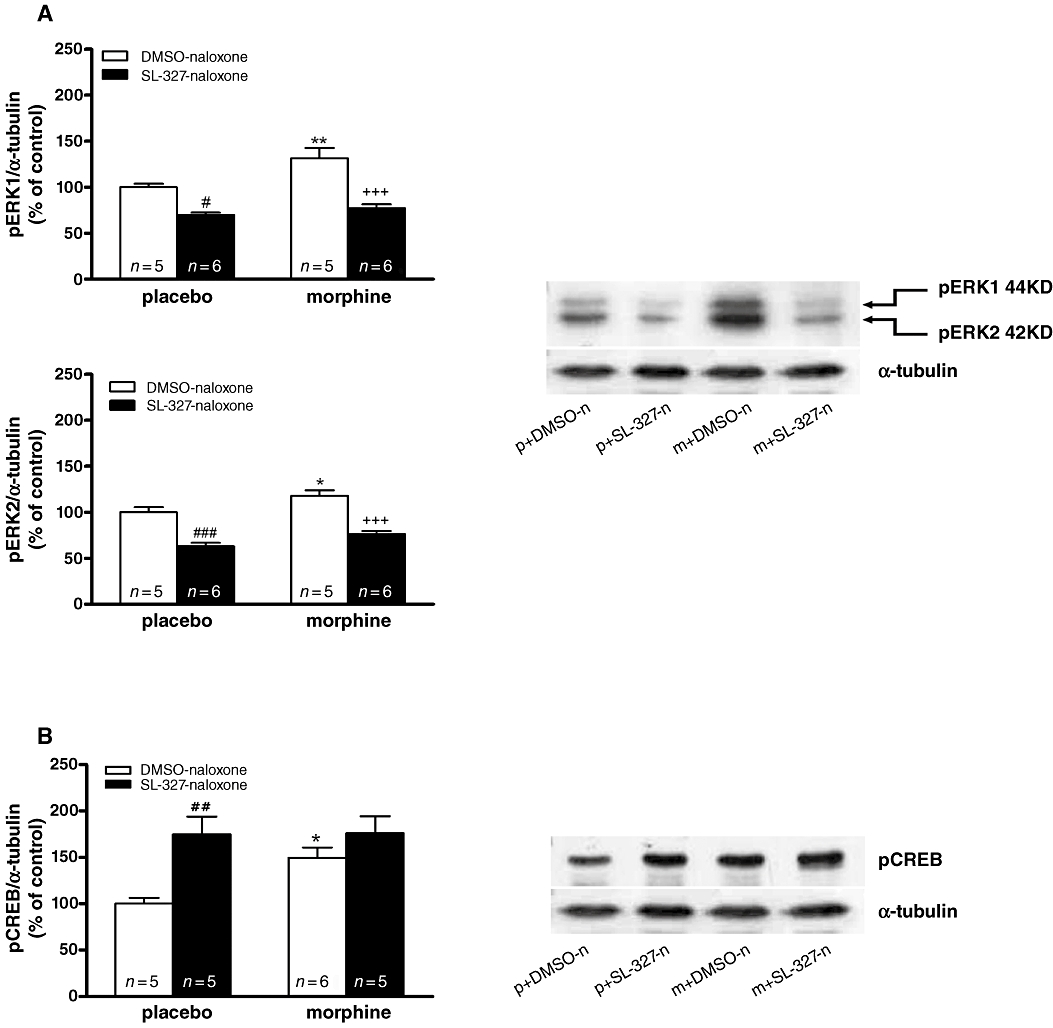
Effects of ERK kinase (MEK) blockade on extracellular signal regulated kinases (ERK) and cAMP-response element binding protein (CREB) phosphorylation. (A,B) Quantitative analysis and representative immunoblots of pERK1/2 and pCREB (respectively) in hypothalamic paraventricular nucleus tissue isolated from placebo or morphine-dependent rats after administration of naloxone in absence or presence of SL-327. Each bar represents mean ± SEM (% of controls). Post hoc analysis revealed that the increase in CREB phosphorylation during morphine withdrawal was not attenuated by SL-327 (B), whereas both ERK1 and ERK2 phosphorylation was attenuated by SL-327 (A). *P < 0.05; **P < 0.01 versus placebo + dimethyl sulphoxide (DMSO) + naloxone; +++P < 0.001 versus morphine + DMSO + naloxone; #P < 0.05; ##P < 0.01; ###P < 0.001 versus placebo + DMSO + naloxone. m, morphine pellets; n, naloxone; p, placebo pellets.
In the present study we also measured the influence of pharmacological inhibition of PKC on morphine withdrawal-induced CREB phosphorylation in the PVN (Figure 6). anova revealed significant differences in pCREB levels (F3,19= 7.042; P = 0.031). As shown in Figure 6A, post hoc comparisons showed that pretreatment with calphostin C significantly (P < 0.05) attenuated the CREB phosphorylation during morphine withdrawal compared with morphine-withdrawn rats pretreated with vehicle. Calphostin C did not modify pCREB levels in placebo-pelleted rats compared with control animals receiving vehicle instead calphostin C. To ensure that the reduction in CREB activation produced by calphostin C was not a non-specific effect, another experiment was performed with the selective PKC antagonist chelerythrine. anova revealed significant differences in pCREB levels (F3,21= 12.77; P < 0.0001).As shown in Figure 6C, chelerythrine significantly (P < 0.001) reduced pCREB levels at the PVN during morphine withdrawal compared with morphine-withdrawn rats receiving vehicle instead of chelerythrine.
Figure 6.
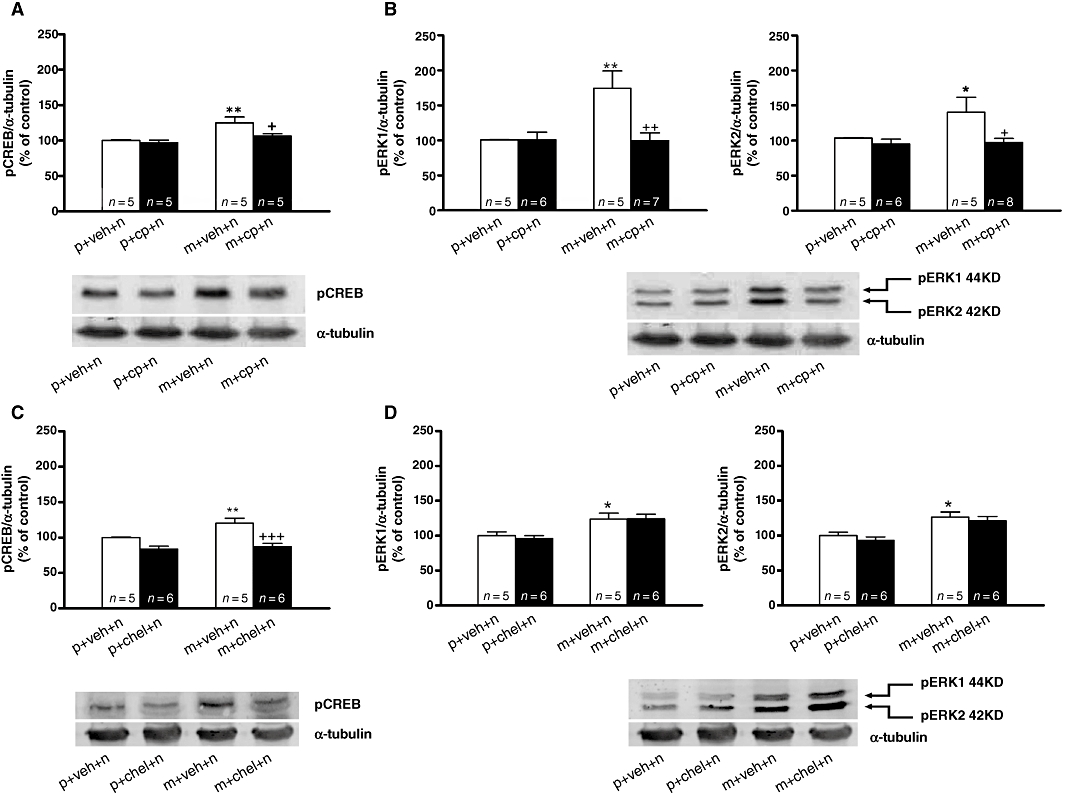
Morphine withdrawal-induced cAMP-response element binding protein (CREB) activation in the hypothalamic paraventricular nucleus (PVN) is protein kinase C dependent. (A,C) Quantitative analysis and representative immunoblots of pCREB in the PVN tissue isolated from placebo or morphine-dependent rats concomitantly treated with calphostin C, chelerythrine or vehicle, 60 min after administration of naloxone. (B,D) Quantitative analysis and representative immunoblots of pERK1/2 expressions in the PVN 60 min after naloxone injection to morphine-dependent rats pretreated with vehicle, calphostin C or chelerythrine. Post hoc analysis revealed that the increase in CREB phosphorylation during morphine withdrawal was attenuated by both calphostin C and chelerythrine, whereas both ERK1 and ERK2 phosphorylation was attenuated by calphostin C (B) but not by chelerythrine (D). Each bar represents mean ± SEM (% of controls). *P < 0.05, **P < 0.01 versus placebo + vehicle + naloxone; +P < 0.05, ++P < 0.01, +++P < 0.001 versus morphine + vehicle + naloxone. chel, chelerythrine; cp, calphostin C; m, morphine pellets; n, naloxone; p, placebo pellets; veh, vehicle.
Effects of PKC inhibition on ERK1/2 phosphorylation in the PVN during morphine withdrawal
To determine if PKC mediates the induction of pERK1/2 during naloxone-precipitated morphine withdrawal, placebo- and morphine-pelleted animals were treated with vehicle or the PKC inhibitors calphostin C and chelerythrine and given naloxone on day 8 (Figure 6B,D). anova revealed significant differences in pERK1 (F3,22= 6.459; P = 0.0034 and pERK2 (F3,23= 3.920; P = 0.0237) levels in animals pretreated with calphostin C. As shown in Figure 6B, precipitated withdrawal in rats pretreated with vehicle significantly increased phosphorylation of ERK1 (P < 0.01) and ERK2 (P < 0.05) in the PVN compared with its control group implanted with placebo pellets. Calphostin C blocked morphine withdrawal-induced ERK1 (P < 0.01) and ERK2 (P < 0.05) phosphorylation compared with their respective controls. anova revealed significant differences in pERK1 (F3,21= 6.260; P = 0.0042) and pERK2 (F3,21= 7.732; P = 0.0016) levels in animals pretreated with vehicle or chelerythrine. However, pretreatment with chelerythrine did not modify the increase in pERK1 or pERK2 that was seen in morphine-withdrawn rats pretreated with vehicle (Figure 6D).
Influence of PKC and MEK-inhibition on naloxone-precipitated withdrawal signs in morphine-dependent rats
Before performing the immunodetection assays, we assessed the efficacy of chronic morphine treatment by pellet implantation, which has been previously shown to induce tolerance to and dependence on neuroendocrine effects of morphine at the HPA axis level (Laorden et al., 2000a; 2002a). For this purpose, the weight of animals was recorded on the days of pellet implantation and on the day of killing (day 8), before receiving any injection. Student's t-test showed that rats pretreated with morphine had a significant (t104= 5.761; P < 0.001) lower weight gain (3.80 ± 3.3 g; n = 50) than that observed in animals receiving placebo pellets (25.41 ± 2.2 g; n = 56), as was previously reported (Núñez et al., 2007b), which might be due to the reduced food intake observed during chronic morphine treatment (Núñez et al., 2009).
The body weight loss after saline or naloxone injection to placebo-pelleted and morphine-dependent rats was also recorded as a sign of opiate withdrawal. In agreement with our previous results (Laorden et al., 2002a; Núñez et al., 2007b), two-way anova revealed that morphine pretreatment, naloxone injection, and the interaction between morphine treatment and naloxone injection had a significant effect on body weight loss at 60 (morphine treatment: F1,80= 63.96, P < 0.0001; naloxone injection: F1,80= 71.48, P < 0.0001; interaction: F1,80= 67.75, P < 0.0001) and 90 min (morphine treatment: F1,54= 43.82, P < 0.001; naloxone injection: F1,54= 53.18, P < 0.001; interaction: F1,54= 24.28, P < 0.001; data no shown). Post hoc analysis showed that naloxone injection to morphine-dependent animals significantly increased (P < 0.001) body weight loss when measured 60 min (Figure 7A) after injection when compared with the placebo-pelleted group also receiving naloxone and with morphine-treated rats receiving saline. However, administration of naloxone to control rats resulted in no significant changes in body weight loss when measured 60 or 90 min after drug injection, compared with control rats receiving saline.
Figure 7.
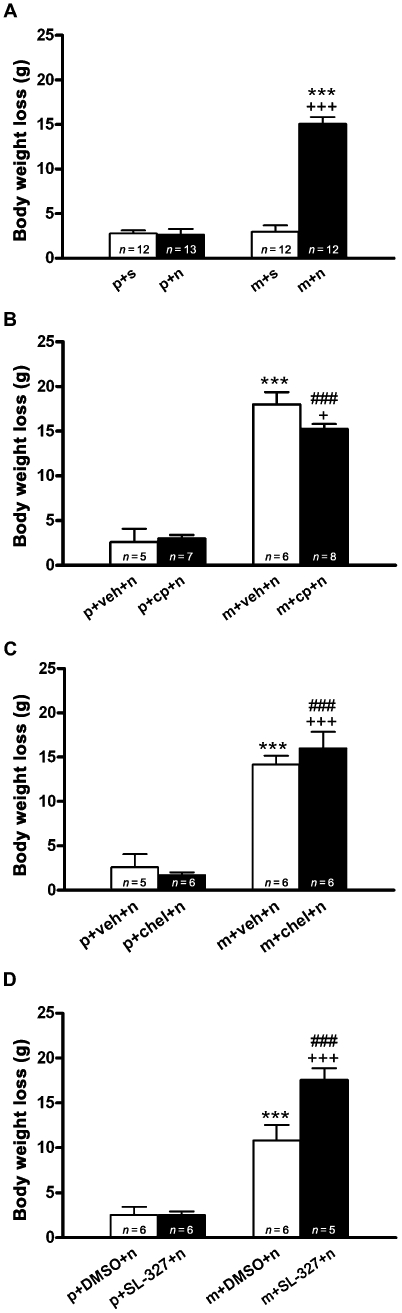
Effects of protein kinase C and ERK kinase (MEK) blockade on body weight loss after naloxone-induced morphine withdrawal. Rats were implanted with morphine or placebo pellets for 7 days and pretreated with vehicle, calphostin C, chelerythrine or SL-327. Weight loss was checked immediately before and 60 min after naloxone injection. Data shown are means ± SEM. Post hoc tests revealed that calphostin C significantly attenuated the increase in body weight loss that was seen during morphine withdrawal. (A) ***P < 0.001 versus placebo + naloxone; +++P < 0.001 versus morphine + saline. (B,C) ***P < 0.001 versus placebo + vehicle + naloxone; +P < 0.05, +++P < 0.001 versus morphine + vehicle + naloxone. (D) +++P < 0.001 versus morphine + dimethyl sulphoxide (DMSO) + naloxone. (B–D) ###P < 0.001 versus placebo + calphostin + naloxone, placebo + chelerythrine + naloxone or placebo + SL-327 + naloxone respectively. chel, chelerythrine; cp, calphostin C; m, morphine pellets; n, naloxone; p, placebo pellets; veh, vehicle.
In animals pretreated with calphostin C, one-way anova revealed significant differences in body weight loss (F3,25= 71.79; P < 0.0001). As shown in Figure 7B, there was a significant (P < 0.05) decrease in body weight loss during morphine withdrawal in animals receiving calphostin C compared with morphine-withdrawn rats receiving vehicle. However, pretreatment with chelerythrine did not attenuate the increase in body weight (Figure 7C). Two-way anova for body weight loss in animals pretreated with SL-327 revealed that morphine pretreatment, SL-327 injection, and the interaction between morphine pretreatment and acute SL-327 injection had a significant effect on body weight loss at 60 (morphine treatment: F1,19= 96.14, P < 0.0001; SL-327 injection: F1,19= 8.02, P = 0.0107; interaction: F1,19= 8.02, P = 0.0107). As shown in Figure 7D, post hoc analysis shows that SL-327 did not block the increase (P < 0.001) in body weight loss induced in morphine-withdrawn rats. By contrast, an increase (P < 0.001) in body weight loss was seen in animals pretreated with the MEK antagonist.
Seven days after the implantation of morphine or placebo pellets, rats were challenged with naloxone and immediately tested for the occurrence of somatic signs of opiate withdrawal. The following somatic signs were observed in morphine-treated groups when compared with placebo-treated groups: irritability, wet dog shakes, teeth chattering, rhinorrhoea, lacrimation, salivation, body tremor and ptosis (Table 1). A lower frequency of irritability was noted in the dependent group pretreated with calphostin C. Signs of withdrawal were not observed in the control group receiving placebo and naloxone. In general, when naloxone was injected to morphine-dependent rats, animals were very irritable. Instead, rats receiving calphostin showed decreased irritability during 30 min of observation. anova revealed significant differences (F3,62= 62.92; P < 0.001) in the number of animals showing irritability in rat pretreated with calphostin C. As shown in Table 1, irritability was significantly (P < 0.001) decreased in rats receiving calphostin C, whereas pretreatment with chelerythrine did not modify physical signs of withdrawal. Treatment with the MEK inhibitor SL-327 significantly attenuated rhinorrhoea (P < 0.05).
Table 1.
Effects of protein kinase C and ERK kinase (MEK) blockade on the incidence of physical signs of opiate withdrawal
| Signs | ||||||||
|---|---|---|---|---|---|---|---|---|
| Treatment | Irritability | Wet dog shakes | Teeth chattering | Rhinorrhoea | Lacrimation | Salivation | Tremor | Ptosis |
| p + n (23) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| m + n (27) | 92.6*** | 44.4*** | 85.2*** | 62.9*** | 40.7** | 88.9*** | 88.9*** | 66.7*** |
| m + veh + n (6) | 100 | 50 | 83.3 | 33.3 | 33.3 | 83.3 | 100 | 83.3 |
| m + cp + n (8) | 37.5*** | 100* | 100 | 0 | 12.5 | 100 | 87.5 | 62.5 |
| m + veh + n (6) | 85.7 | 57.1 | 71.4 | 42.9 | 28.6 | 85.7 | 100 | 85.7 |
| m + chel + n (6) | 100 | 83.3 | 100 | 16.7 | 33.3 | 100 | 83.3 | 33.3 |
| m + DMSO + n (5) | 100 | 83.3 | 83.3 | 66.7 | 50 | 50 | 66.7 | 66.7 |
| m + SL-327 + n (6) | 100 | 40 | 100 | 0* | 20 | 40 | 80 | 100 |
P < 0.05
P < 0.01
P < 0.001 versus its respective control group; one-way anova.
Rats were treated with pellets of placebo (p) or morphine (m) to induce dependence and withdrawal precipitated by naloxone (n). Some groups also received vehicle (veh), dimethyl sulphoxide (DMSO), calphostin C (cp), chelerythrine (chel) or SL-327. The withdrawal symptoms were checked for 30 min after naloxone injection. Data show the percentage of animals (means ± SEM; numbers in each group are shown in brackets) showing the signs of withdrawal as indicated.
Influence of naloxone-induced morphine withdrawal on PKCα,γ-isoforms
The influence of morphine withdrawal on the immunoreactivity of PKCα and PKCγ isoforms was examined by Western blot analysis in the PVN after injection of naloxone to morphine-dependent rats and to control placebo-pelleted animals (Figure 8). As previously reported (Benavides et al., 2005) there was a decrease (t6= 4.59; P < 0.01) of PKCα isoform immunoreactivity 60 min after naloxone administration to morphine-dependent rats, compared with the control group (Figure 8A). However, PKCγ isoform immunoreactivity was not modified (t9= 1.08; P = 0.3215) in the PVN during morphine withdrawal (Figure 8B).
Figure 8.
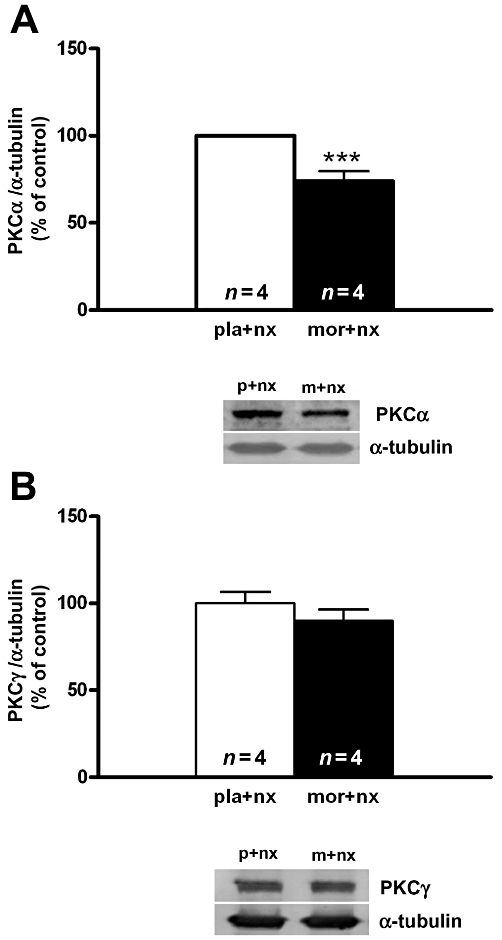
Western blot analysis of protein kinase C PKC α (A) and PKC γ (B) isoforms immunoreactivity levels in the hypothalamic paraventricular nucleus. Control animals received a 7 day chronic administration of placebo pellets. Opiate withdrawal was induced by administration of naloxone to morphine dependent rats. Animals were killed 60 min after naloxone administration. The immunoreactivity of PKC is expressed as the percentage of that in the control group (placebo plus naloxone). Each bar represents mean ± SEM (% of controls). ***P < 0.001 versus placebo + naloxone. nx, naloxone; mor, m, morphine pellets; pla, p, placebo pellets.
Effects of MEK- and PKC-inhibition on morphine withdrawal-induced HPA axis hyper activation
To evaluate whether a causal link exists between ERK phosphorylation by MEK and/or PKC and HPA axis activation during morphine withdrawal, we measured plasma corticosterone concentration in animals made dependent on morphine and pretreated with SL-327, calphostin C or chelerythrine (Figure 9). Two-way anova for corticosterone in rats pretreated with SL-327 revealed a main effect of morphine pretreatment (F1,19= 8.59; P = 0.0086), and acute SL-327 treatment (F1,19= 8.78; P = 0.0080). As shown in Figure 9A, pretreatment with SL-327 C did not modify plasma corticosterone levels in morphine-withdrawn rats. On the other hand, control rats receiving SL-327 showed a slight but significant (P < 0.05) increase in corticosterone secretion. However, anova revealed significant differences (F3,24= 52.43; P < 0.001) in corticosterone secretion in animals pretreated with calphostin. Post hoc analysis showed that calphostin C significantly (P < 0.05) attenuated the increase (P < 0.001) in plasma corticosterone levels during morphine withdrawal (Figure 9B). Similar results were obtained with chelerythrine. anova for corticosterone revealed significant effects of chelerythrine pretreatment (F3,21= 48.29; P < 0.001). Post hoc tests showed that rats pretreatment with chelerythrine significantly (P < 0.01) antagonized corticosterone release in morphine-withdrawn rats (Figure 9C).
Figure 9.
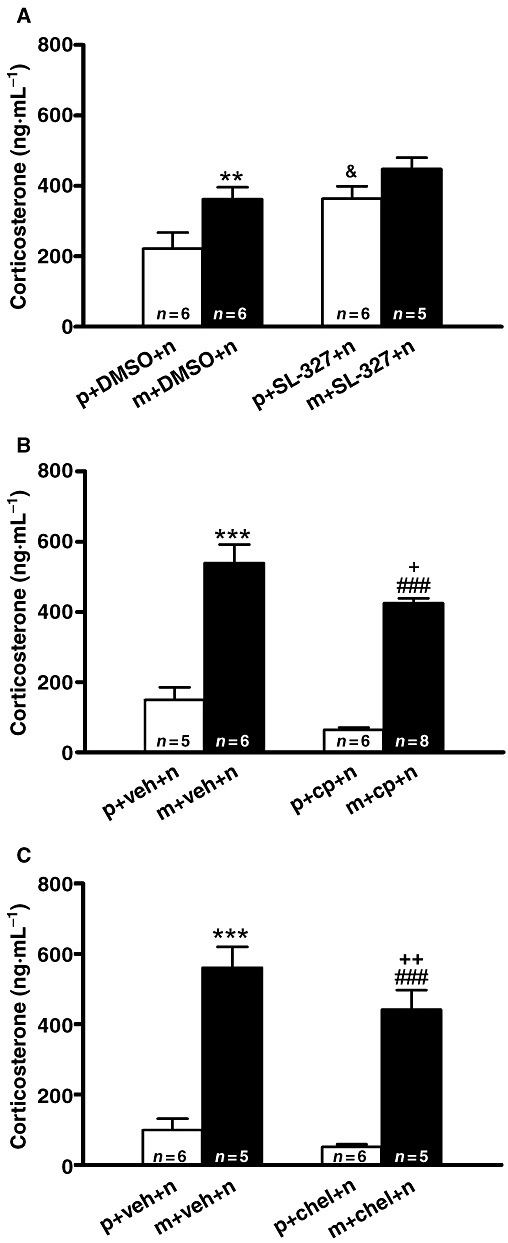
Activation of the hypothalamic-pituitary-adrenocortical axis during morphine withdrawal is attenuated by PKC inhibition. Placebo or morphine-dependent rats were pretreated with dimethyl sulphoxide (DMSO), SL-327, calphostin C or chelerythrine and plasma levels of corticosterone (a marker of hypothalamic-pituitary-adrenocortical axis activity) were determined 60 min after naloxone injection. Each bar represents mean ± SEM. Post hoc analysis revealed a significant increase in plasma corticosterone concentration after naloxone-induced morphine withdrawal, which was attenuated in calphostin C-pretreated rats (B) and chelerythrine-pretreated rats (C) but not in SL-327-pretreated rats. (A) **P < 0.01 versus placebo + DMSO + naloxone; &P < 0.05 versus placebo + DMSO + naloxone. (B) ***P < 0.001 versus placebo + vehicle + naloxone; +P < 0.05 versus morphine + vehicle + naloxone; ###P < 0.001 versus placebo + calphostin C + naloxone. (C) ***P < 0.001 versus placebo + vehicle + naloxone; ++P < 0.01 versus morphine + vehicle + naloxone; ###P < 0.001 versus placebo + chelerythrine + naloxone. chel, chelerythrine; cp, calphostin C; m, morphine pellets; n, naloxone; p, placebo pellets.
Discussion
The principal findings of the present study are as follows: (i) naloxone-induced morphine withdrawal increased CREB phosphorylation in the PVN; (ii) the induction of CREB activity in the PVN occurred predominantly in CRF-positive neurons of the parvocellular part of the PVN; (iii) pretreatment with PKC antagonists blocked CREB phosphorylation during morphine withdrawal but not the physical signs of dependence; blockade of the MEK/ERK pathway did not modify phosphorylation of CREB; and (iv) PKC inhibition but not MEK inhibition reduced the morphine withdrawal-induced corticosterone release.
The link between CREB function and drug addiction has been characterized in numerous works (Blendy and Maldonado, 1998; Carlezon et al., 2005; Briand and Blendy, 2010). Such role for CREB has most been studied in the brain's reward regions. However, the functional impact of these adaptations on the transcriptional activity of CREB in brain stress-related areas during chronic opiate administration and opioid withdrawal is not well known. Present results show that, upon naloxone administration to morphine-dependent rats, higher pCREB levels were found in the PVN. We also observed that morphine withdrawal did not change the expression of total CREB in the PVN, suggesting that the effects of morphine abstinence are mediated through the activation (phosphorylation) of CREB, but not through the up-regulation of its expression. The results obtained by both Western blot and immunohistochemistry were in good agreement.
It has been shown that acute and chronic psycho-stimulants, such as amphetamine and cocaine lead to an increase in CREB activity in a variety of brain areas related to drug addiction (see Briand and Blendy, 2010). In contrast to these drugs that lead to increases in CREB activation, it has been shown that acute and chronic opiate administration leads to a decrease in CREB phosphorylation in the nucleus accumbens and hippocampus (Yang and Pu, 2009). By contrast, opiate withdrawal has the opposite effect. Thus, it has been shown an increase in pCREB levels in the amygdala, the nucleus accumbens and the dorsal striatum during morphine and heroin withdrawal (Chartoff et al., 2006; Edwards et al., 2009), which is in agreement with present findings. In contrast to present results, an increase in CREB activation has been reported in the PVN after chronic morphine treatment (Espinosa et al., 2008). These conflicting data may be due to the different dose of morphine used for inducing dependence. Thus, Espinosa et al. used four pellets of 75 mg, whereas in the present work it has been used six pellets of 75 mg. So, the methods of inducing morphine dependence may lead to different results.
The HPA axis and the brain stress system (both mediated by CRF) are dysregulated by chronic administration of drugs of abuse, with a common response of elevated ACTH and corticosterone and extended amygdala CRF immunoreactivity during acute withdrawal from almost all drugs of abuse (Koob and Moal, 2005; Koob, 2009). Using probes complementary to intronic sequences of genes encoding neuropeptides in parvocellular neurosecretory neurons of the PVN, we found robust increases in the precursor mRNA (hnRNA) for CRF and AVP in morphine-dependent rats during naloxone-precipitated withdrawal (Núñez et al., 2007a). The present work showed that the induction of CREB activity that occurred during morphine withdrawal, was seen predominantly in CRF-positive neurons of the parvocellular part of the PVN, consistent with transcriptional regulation of CRF neurons from the PVN by chronic morphine treatment. In addition, immunohistochemical analysis of CREB phosphorylation in the PVN CRF neurons during morphine withdrawal indicates that morphine withdrawal-induced CREB phosphorylation is associated with HPA axis hyperactivity, as shown by increased plasma corticosterone levels. Ongoing drug use in opiate-addicted individuals could be reinforced by withdrawal reactions that occur when drug use is halted abruptly (Koob and Moal, 2001). Activation of the brain stress system may contribute not only to the negative motivational state associated with acute abstinence, but also to the vulnerability to stressors observed during protracted abstinence (Koob, 2008). Hyperactivity of CRF in the brain stress system has been implicated in opiate dependence (Milanés et al., 1998; Koob and Kreek, 2007; Núñez et al., 2007a). CREB is a member of the basic leucine zipper family of transcription factors that has been proposed as one candidate that is critical for both the response to stress and drug exposure (Briand and Blendy, 2010). Transcriptional activity of CREB is regulated by phosphorylation at serine residues. Phosphorylation has been shown to increase CREB DNA binding activity without changing the total CREB protein level in vivo (Yao and Denver, 2007). CREB regulates the transcription of over 10 000 genes, including those implicated in stress and addiction, such as CRF (Itoi et al., 1996). A highly conserved cAMP response element (CRE) is present in the CRF promoter, and it has been shown that this element in the CRF gene promoters is responsible for gene activation (Yao et al., 2007). Several studies have shown that in the rat, CREB is phosphorylated in CRF neurons in response to various stressors, preceding elevation of CRF expression in those cells (Kovács and Sawchenko, 1996). In the present study, we have showed that morphine withdrawal, a kind of stress, increased phosphorylated CREB immunoreactivity in the PVN and that pCREB immunoreactivity co-localized with CRF neurons. In parallel, an increased CRF immunoreactivity was seen, which was associated with the increase in corticosterone secretion (a marker of HPA axis activity), according to previous findings (Núñez et al., 2007a). In line with present data, studies from Kovács and Sawchenko (1996) showed that in animals killed after stress pCREB nuclei in the medial parvocellular part of the PVN overlaid by a positive hybridization signal for CRF hnRNA. Furthermore, it has been shown that phosphorylated CREB is essential for activation of CRF transcription (Liu et al., 2008). Although indirectly, these results might suggest that activation of CREB could contribute to increased transcription of CRF gene during morphine withdrawal response. Supporting this hypothesis are the findings of Itoi et al. (1996) showing that injection of antisense oligodeoxynucleotides to CREB blocked the increase in CRF mRNA caused by stress. Still lacking, however, is the evidence that shows CREB to be essential for the transcriptional activation of the CRF gene.
A variety of signal transduction pathways regulate CREB phosphorylation, suggesting that this transcription factor may integrate signalling information from multiple sources (Nestler, 2001). The activation of stimulatory G-protein coupled receptors activates adenylyl cyclase, which in turn leads to an accumulation of cAMP in the cytosol. Cyclic AMP then activates PKA, which phosphorylates CREB at Ser133 (Gonzalez et al., 1991). In addition, CREB can be activated by other kinases including ERK1/2 and PKC, which induce its transcriptional activity (Liu and Anand, 2001; Johannessen et al. 2004). Previous findings have shown that activated PKC may be critical for the activation of the HPA axis in response to morphine withdrawal (Benavides et al., 2005). From present results, a straightforward explanation for enhanced CREB activation in the PVN is that morphine withdrawal stimulated PKC, which then phosphorylated CREB. Indeed, inhibition of PKC activity with both calphostin C or chelerythrine, selective inhibitors of PKC at either the catalytic or the regulatory site (with chelerythrine or calphostin respectively), attenuated and totally blocked, respectively, the induction of pCREB in the PVN after naloxone-induced morphine withdrawal, which suggests that PKC could be one of the transduction pathways that mediate the regulation of CREB. In agreement with the findings in the present study, it has been shown that the PKC activator phorbol 12-myristate 13-acetate (PMA) increased CREB phosphorylation. In addition, chelerythrine inhibited PMA-induced increases in pCREB-labelled neurons in striatal cultures (Mao et al., 2007).
The results of the present study show that naloxone-precipitated morphine withdrawal exerts a down-regulation of the PKCα isoform, which agree with previous results from our laboratory (Benavides et al., 2005). Opioids are capable of activating PKC, as shown by enhanced translocation of the enzyme to the cell membrane (Kramer and Simon, 1999). Furthermore, it has been confirmed that PKC is markedly activated in different brain areas in morphine-withdrawn rats (Tokuyama et al., 1995). Translocation of PKC to the membrane is believed to be its primary mode of phosphorylation (activation). Once phosphorylated, PKC can respond to second messengers and phosphorylate downstream targets. In addition, after translocation PKC is known to be rapidly proteolysed and subject to down-regulation. In fact, activation by treatment of cells with phorbol esters results in down-regulation of PKC (Newton, 2003). Therefore, the results of the present study showing a decreased PKCα immunoreactivity may suggest that naloxone-induced morphine withdrawal would induce an increase in PKC activity at the PVN. Because PKCγ isoform immunoreactivity levels did not change during morphine withdrawal, it is therefore reasonable to propose that activation of the PKCα isoform may have a function mediating CREB activation in the PVN during morphine withdrawal.
Our present results showed that pretreatment with calphostin C attenuated the increase in body weight loss and the anxiety-like behaviour during morphine withdrawal. However, pretreatment with chelerythrine did not modify somatic signs of withdrawal, although chelerythrine blocked CREB phosphorylation. In addition, chronic chelerythrine slightly increased the frequency of several counted symptoms, such as wet dog shakes and jumps. The physical dependence observed when an opioid antagonist is given, is manifested as withdrawal symptoms. Previous studies demonstrated that mutant mice with a genetic disruption of the α and Δ isoforms of CREB showed attenuation of several signs of morphine withdrawal, such as piloerection and teeth chattering, but not wet dog shakes or jumping, which were increased (Maldonado et al., 1996). On the other hand, it has also been reported that the aversive properties of morphine withdrawal are present in CREB mutant mice (Walters and Blendy, 2001). Previous findings have shown that chronic administration of chelerythrine concurrently with morphine produced a significant reduction in time spent in withdrawal, whereas a single dose of this PKC inhibitor was ineffective in reducing withdrawal behaviour (Fundytus and Coderre, 1996). Taken together, these results would suggest that PKC is not clearly involved in the somatic expression of morphine withdrawal.
Consistent with previous results (Cerezo et al., 2002; Martín et al., 2009) was the enhanced corticosterone release in response to morphine withdrawal, attenuated by PKC inhibition with both calphostin C and chelerythrine. These findings are compatible with the observation that PKC is activated in the PVN after naloxone-induced morphine withdrawal and may suggest that PKC activity is necessary for the increased HPA axis activity during morphine withdrawal (Benavides et al., 2005). Previous findings have indicated that the secretor activity of the HPA axis after morphine withdrawal results from an increase in the activity of noradrenergic pathways innervating the PVN that is dependent on α1-adrenoceptor activation (Laorden et al., 2000b). PKC is activated in response to enhancement of intracellular diacylglycerol as second messenger, activated, among the others by stimulation of receptors positively coupled to phospholipase C, like α1-adrenoceptors. Taken with all these findings, our present results strongly suggest the involvement of the PKC pathway in mediating the naloxone-induced morphine withdrawal activation of the HPA axis activity.
It is not known which components of signal transduction pathways downstream of PKC activation are involved in CREB induction in the CRF neurons. In the present study, we investigated whether ERK activation was responsible for CREB phosphorylation at the PVN and for HPA axis hyperactivity following morphine withdrawal. Present results show that although ERK1/2 were activated in response to morphine withdrawal, the phosphorylation of ERK by MEK is most probably not a necessary event for the morphine withdrawal-induced CREB activation, because the MEK inhibitor SL-327 at a dose that inhibits ERK phosphorylation (Núñez et al., 2008; Martín et al., 2009) did not prevent either the morphine withdrawal-evoked CREB phosphorylation or the corticosterone hypersecretion. In contrast to MEK inhibition, PKC inhibition (with both calphostin C and chelerythrine) greatly reduced the withdrawal-triggered phosphorylation of CREB as well as corticosterone release. These results are consistent with previous data showing that activation of CRF neurons in the PVN depends in part on PKC activation (Benavides et al., 2005; Núñez et al., 2007b). In addition, it has been found that PKC, at least in vitro, can directly phosphorylate CREB (Thonberg et al., 2002), whereas inhibitors of ERK1/2 failed to decrease CREB phosphorylation, in agreement with the present results.
The PKC regulates expression of MAPK in many cell types (Zheng and Keifer, 2008). Our results showed that calphostin C but not chelerythrine significantly attenuated the phosphorylation of ERK1/2 60 min after morphine withdrawal. Therefore, PKC would seem to have a minor function mediating the activation of the MEK/ERK pathway during morphine withdrawal. On the other hand, the divergent results observed with chelerythrine and calphostin C on ERK1/2 phosphorylation and withdrawal behaviour could be due to the inhibition of protein kinase C activity at either the catalytic or the regulatory site with chelerythrine or calphostin C respectively.
In conclusion, the present data show an activation of CREB in the CRF neurons of the PVN during morphine withdrawal, which was attenuated by PKC inhibitors. Present data, together with previous findings suggest that morphine withdrawal may activate PKC, which could be among the protein kinases that can positively modulate CREB phosphorylation in the CRF neurons of the PVN.
Acknowledgments
This work was supported by the Ministerio de Ciencia e Innovación (Grants SAF/FEDER 2006-00331, 2007-62758 and 2009-07178) and Red de Trastornos Adictivos (RTA; RD06/0001/1006). Fatima Martin was supported by a fellowship from the Ministerio de Ciencia e Innovación.
Glossary
Abbreviations
- AVP
arginine-vasopressin
- CREB
cAMP-response element binding protein
- CRF
corticotrophin-releasing factor
- ERK
extracellular signal regulated kinase
- HPA
hypothalamic-pituitary-adrenocortical
- MAPK
mitogen-activated protein kinase
- PKC
protein kinase C
- PVN
hypothalamic paraventricular nucleus
Conflict of interest
Authors have none to declare.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD. The MAPK cascade is required for mammalian associative learning. Nat Neurosci. 1998;1:602–609. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- Benavides M, Laorden ML, Marin MT, Milanes MV. Role of PKC-alpha,gamma isoforms in regulation of c-Fos and TH expression after naloxone-induced morphine withdrawal in the hypothalamic PVN and medulla oblongata catecholaminergic cell groups. J Neurochem. 2005;95:1249–1258. doi: 10.1111/j.1471-4159.2005.03445.x. [DOI] [PubMed] [Google Scholar]
- Berhow MT, Russel DS, Terwilliger RZ, Beitner-Johnson D, Self DW, Lindsay RM, et al. Influence of neurotrophic factors on morphine- and cocaine-induced biochemical changes in the mesolimbic dopamine system. Neuroscience. 1995;68:969–979. doi: 10.1016/0306-4522(95)00207-y. [DOI] [PubMed] [Google Scholar]
- Blendy JA, Maldonado R. Genetic analysis of drug addiction: the role of cAMP response element binding protein. J Mol Med. 1998;76:104–110. doi: 10.1007/s001090050197. [DOI] [PubMed] [Google Scholar]
- Briand LA, Blendy JA. Molecular and genetic substrates linking stress and addiction. Brain Res. 2010;1314:219–234. doi: 10.1016/j.brainres.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlezon JR, Duman RS, Nestler EJ. The many faces of CREB. Trends Neurosci. 2005;28:436–445. doi: 10.1016/j.tins.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Cerezo M, Laorden ML, Milanes MV. Inhibition of protein kinase C but not protein kinase A attenuates morphine withdrawal excitation of rat hypothalamus-pituitary-adrenal axis. Eur J Pharmacol. 2002;452:57–66. doi: 10.1016/s0014-2999(02)02245-8. [DOI] [PubMed] [Google Scholar]
- Chao J, Nestler EJ. Molecular Neurobiology of Drug Addiction. Annu Rev Med. 2004;55:113–132. doi: 10.1146/annurev.med.55.091902.103730. [DOI] [PubMed] [Google Scholar]
- Chartoff EH, Mague SD, Barhight MF, Smith AM, Carlezon WA., Jr Behavioral and molecular effects of dopamine D1 receptor stimulation during naloxone-precipitated morphine withdrawal. J Neurosci. 2006;26:6450–6457. doi: 10.1523/JNEUROSCI.0491-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleck JN, Blendy JA. Making a bad thing worse: adverse effects of stress on drug addiction. J Clin Invest. 2008;118:454–461. doi: 10.1172/JCI33946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couceyro P, Douglass J. Precipitated morphine withdrawal stimulates multiple activator protein-1 signaling pathways in rat brain. Mol Pharmacol. 1995;47:29–39. [PubMed] [Google Scholar]
- Deak M, Clifton AD, Lucocq LM, Alessi DR. Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J. 1998;17:4426–4441. doi: 10.1093/emboj/17.15.4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards S, Graham DL, Whisler KN, Self DW. Phosphorylation of GluR1, ERK, and CREB during spontaneous withdrawal from chronic heroin self-administration. Synapse. 2009;63:224–235. doi: 10.1002/syn.20601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eitan S, Bryant CD, Saliminejad N, Yang YC, Vojdani E, Keith D, et al. Brain region-specific mechanisms for acute morphine-induced mitogen-activated protein kinase modulation and distinct patterns of activation during analgesic tolerance and locomotor sensitization. J Neurosci. 2003;23:8360–8369. doi: 10.1523/JNEUROSCI.23-23-08360.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa VP, Liu Y, Ferrini M, Anghel A, Nie Y, Tripathi PV, et al. Differential regulation of PC1/3, PC2 and phosphorylated CREB by short-term and long-term morphine treatment: implication for understanding the ‘switch’ to opiate addiction. Neuroscience. 2008;156:788–799. doi: 10.1016/j.neuroscience.2008.07.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funada M, Hara C, Wada K. Involvement of corticotropin-releasing factor receptor subtype 1 in morphine withdrawal regulation of the brain noradrenergic system. Eur J Pharmacol. 2001;430:277–281. doi: 10.1016/s0014-2999(01)01402-9. [DOI] [PubMed] [Google Scholar]
- Fundytus ME, Coderre TJ. Chronic inhibition of intracellular Ca++ release or protein kinase C activation significantly reduces the development of morphine dependence. Eur J Pharmacol. 1996;300:173–181. doi: 10.1016/0014-2999(95)00871-3. [DOI] [PubMed] [Google Scholar]
- Gabra BH, Bailey CP, Kelly E, Smith FL, Henderson G, Dewey WL. Pre-treatment with a PKC or PKA inhibitor prevents the development of morphine tolerance but not physical dependence in mice. Brain Res. 2008;1217:70–77. doi: 10.1016/j.brainres.2008.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez GA, Menzel P, Leonard J, Fischer WH, Montminy MR. Characterization of motifs which are critical for activity of the cyclic AMP-responsive transcription factor CREB. Mol Cell Biol. 1991;11:1306–1312. doi: 10.1128/mcb.11.3.1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guitart X, Nestler EJ. Identification of morphine- and cyclic AMP-regulated phosphoprotein (MARPPs) in the locus coeruleus and other regions of rat brain: regulation by acute and chronic morphine. J Neurosci. 1989;9:4371–4387. doi: 10.1523/JNEUROSCI.09-12-04371.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert JM, Augereau JM, Gleye J, Maffrand JP. Chelerythrine is a potent and specific inhibitor of protein kinase C. Biochem Biophy Res Commun. 1990;172:993–999. doi: 10.1016/0006-291x(90)91544-3. [DOI] [PubMed] [Google Scholar]
- Itoi K, Horiba N, Tozawa F, Sakai Y, Sakai K, Demura H, et al. Major role of 3′,5′-cyclic adenosine monophosphate-dependent protein kinase A pathways in corticotropin-releasing factor gene expression in the rat hypothalamus in vivo. Endocrinology. 1996;137:2389–2396. doi: 10.1210/endo.137.6.8641191. [DOI] [PubMed] [Google Scholar]
- Johannessen M, Delghandi MP, Moens U. What turns CREB on? Cell Signal. 2004;16:1211–1227. doi: 10.1016/j.cellsig.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Kobayashi E, Nakano H, Morimoto M, Tamaoki T. Calphostin C (UCN-1028C), a novel microbial compound, is a highly potent and specific inhibitor of protein kinase C. Biochem Biophys Res Commun. 1989;159:548–553. doi: 10.1016/0006-291x(89)90028-4. [DOI] [PubMed] [Google Scholar]
- Koob GF. A role for brain stress system in addiction. Neuron. 2008;59:11–34. doi: 10.1016/j.neuron.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF. Dynamics of neuronal circuits in addiction: reward, antireward, and emotional memory. Pharmacopsychiatry. 2009;42:S32–S41. doi: 10.1055/s-0029-1216356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob G, Kreek MJ. Stress, dysregulation of drug reward pathways, and the transition to drug dependence. Am J Psychiatry. 2007;164:1149–1159. doi: 10.1176/appi.ajp.2007.05030503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Drug addiction, dysregulation of reward and allostasis. Neuropsychopharmacology. 2001;24:97–129. doi: 10.1016/S0893-133X(00)00195-0. [DOI] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Plasticity of reward neurocircuitry and the ‘dark side’ of drug addiction. Nat Neurosci. 2005;8:1442–1444. doi: 10.1038/nn1105-1442. [DOI] [PubMed] [Google Scholar]
- Kovács KJ, Sawchenko PE. Regulation of stress-induced transcriptional changes in the hypothalamic neurosecretory neurons. J Mol Neurosci. 1996;7:125–133. doi: 10.1007/BF02736792. [DOI] [PubMed] [Google Scholar]
- Kovacs GL, Telegdy G. Hypothalamo-neurohypophyseal neuropeptides and experimental drug addiction. Brain Res Bull. 1988;20:893–895. doi: 10.1016/0361-9230(88)90107-4. [DOI] [PubMed] [Google Scholar]
- Kramer HK, Simon EJ. Role of protein kinase C (PKC) in agonist-induced m-opioid receptor down-regulation. I. PKC translocation to the membrane of SH-SY5Y neuroblastoma cells is induced by m-opioid agonists. J Neurochem. 1999;72:585–593. doi: 10.1046/j.1471-4159.1999.0720585.x. [DOI] [PubMed] [Google Scholar]
- Laorden ML, Castells MT, Martínez MD, Martínez PJ, Milanés MV. Activation of c-fos expression in hypothalamic nuclei by mu- and kappa-receptor agonists. Correlation with catecholaminergic activity in the hypothalamic paraventricular nucleus. Endocrinology. 2000a;141:1366–1376. doi: 10.1210/endo.141.4.7407. [DOI] [PubMed] [Google Scholar]
- Laorden ML, Fuertes G, González-Cuello A, Milanés MV. Changes in catecholaminergic pathways innervating paraventricular nucleus and pituitary-adrenal axis response during morphine dependence: implication of a1- and a2-adrenoceptors. J Pharmacol Exp Ther. 2000b;293:578–584. [PubMed] [Google Scholar]
- Laorden ML, Castells MT, Milanés MV. Effects of morphine and morphine withdrawal on brainstem neurons innervating hypothalamic nuclei that control the pituitary-adrenocortical axis in rats. Br J Pharmacol. 2002a;136:67–75. doi: 10.1038/sj.bjp.0704684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laorden ML, Núñez C, Almela P, Milanés MV. Morphine withdrawal-induced c-fos expression in the hypothalamic paraventricular nucleus is dependent on the activation of catecholaminergic neurones. J Neurochem. 2002b;83:132–140. doi: 10.1046/j.1471-4159.2002.01123.x. [DOI] [PubMed] [Google Scholar]
- Liu JG, Anand KJ. Protein kinases modulate the cellular adaptations associated with opioid tolerance and dependence. Brain Res Brain Res Rev. 2001;38:1–19. doi: 10.1016/s0165-0173(01)00057-1. [DOI] [PubMed] [Google Scholar]
- Liu Y, Kamitakahara A, Kim AJ, Aguilera G. Cyclic adenosine 3′, 5′-monophoaphate responsive element binding protein phosphorylation is required but not sufficient for activation of corticotropin-releasing hormone transcription. Endocrinology. 2008;149:3512–3520. doi: 10.1210/en.2008-0052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Liu D, Ceng X, Ma L. Differential role of corticotropin-releasing factor receptor subtypes 1 and 2 in opiate withdrawal and in relapse to opiate dependence. Eur J Neurosci. 2000;12:4398–4404. [PubMed] [Google Scholar]
- Maldonado R, Blendy JA, Tzavara E, Gass P, Roques BP, Haoune J, et al. Reduction of morphine abstinence in mice with a mutation in the gene encoding CREB. Science. 1996;273:657–659. doi: 10.1126/science.273.5275.657. [DOI] [PubMed] [Google Scholar]
- Mao LM, Tang Q, Wang JQ. Protein kinase C-regulated cAMP response element-binding protein phosphorylation in cultured rat striatal neurons. Brain Res Bull. 2007;72:302–308. doi: 10.1016/j.brainresbull.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín F, Laorden ML, Milanes MV. Morphine withdrawal regulates phosphorylation of cAMP response element binding protein (CREB) through PKC in the nucleus tractus solitarius-A2 catecholaminergic neurons. J Neurochem. 2009;110:1422–1432. doi: 10.1111/j.1471-4159.2009.06234.x. [DOI] [PubMed] [Google Scholar]
- McClung CA, Nestler EJ. Neuroplasticity Mediated by Altered Gene Expression. Neuropsychopharmacology. 2008;33:3–17. doi: 10.1038/sj.npp.1301544. [DOI] [PubMed] [Google Scholar]
- Milanés MV, Laorden ML, Chapleur-Chateau M, Burlet A. Alterations in corticotropin-releasing factor and vasopressin content in rat brain during morphine withdrawal. Correlation with hypothalamic noradrenergic activity and pituitary-adrenal response. J Pharmacol Exp Ther. 1998;285:700–706. [PubMed] [Google Scholar]
- Muller DL, Unterwald EM. In vivo regulation of extracellular signal-regulated protein kinase (ERK) and protein kinase B (Akt) phosphorylation by acute and chronic morphine. J Pharmacol Exp Ther. 2004;3010:774–782. doi: 10.1124/jpet.104.066548. [DOI] [PubMed] [Google Scholar]
- Nestler EJ. Molecular basis of long-term plasticity underlying addiction. Nat Rev Neurosci. 2001;2:119–128. doi: 10.1038/35053570. [DOI] [PubMed] [Google Scholar]
- Newton AC. Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. Biochem J. 2003;370:361–371. doi: 10.1042/BJ20021626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Núñez C, Foldes A, Laorden ML, Milanes MV, Kovács KJ. Activation of stress-related hypothalamic neuropeptide gene expression during morphine withdrawal. J Neurochem. 2007a;101:1060–1071. doi: 10.1111/j.1471-4159.2006.04421.x. [DOI] [PubMed] [Google Scholar]
- Núñez C, Laorden ML, Milanes MV. Regulation of serine (Ser)-31 and Ser40 tyrosine hydroxylase phosphorylation during morphine withdrawal in the hypothalamic paraventricular nucleus and nucleus tractus solitarius-A2 cell group. Role of ERK1/2. Endocrinology. 2007b;148:5780–5793. doi: 10.1210/en.2007-0510. [DOI] [PubMed] [Google Scholar]
- Núñez C, Castells MT, Laorden ML, Milanes MV. Regulation of extracellular signal-regulated kinases (ERKs) by naloxone-induced morphine withdrawal in the brain stress system. Naunyn Schmiedebergs Arch Pharmacol. 2008;378:407–420. doi: 10.1007/s00210-008-0304-9. [DOI] [PubMed] [Google Scholar]
- Núñez C, Földes A, Pérez-Flores D, García-Borrón JC, Laorden ML, Kovács KJ, et al. Elevated glucocorticoid levels are responsible for induction of tyrosine hydroxylase (TH) mRNA expression, phosphorylation and enzyme activity in the nucleus of the solitary tract (NTS-A2) during morphine withdrawal. Endocrinology. 2009;150:3118–3127. doi: 10.1210/en.2008-1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palkovits M. Isolated removal of hypothalamic or other brain nuclei of the rat. Brain Res. 1973;59:449–450. doi: 10.1016/0006-8993(73)90290-4. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Amsterdam: Academic Press; 2007. [Google Scholar]
- Swanson LW, Sawchenko PE, Rivier J, Vale WW. Organization of ovine corticotropin-releasing factor immunoreactive cells and fibers in the rat brain: an immunocitochemical study. Neuroendocrinology. 1983;36:165–186. doi: 10.1159/000123454. [DOI] [PubMed] [Google Scholar]
- Thonberg H, Frediksson M, Nedergaard J, Cannon B. A novel pathway for adrenergic stimulation of cAMP-response-element-binding protein (CREB) phosphorylation: mediation via a1-adrenoceptors and protein kinase C activation. Biochem J. 2002;364:73–79. doi: 10.1042/bj3640073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuyama S, Feng Y, Wakabayashi H, Ho IK. Possible involvement of protein kinases in physical dependence on opioids: studies using protein kinase inhibitors, H-7 and H-8. Eur J Pharmacol. 1995;284:101–107. doi: 10.1016/0014-2999(95)00370-z. [DOI] [PubMed] [Google Scholar]
- Vargas ML, Martínez-Piñero MG, Milanés MV. Neurochemical activity of noradrenergic neurons and pituitary-adrenal response after naloxone-induced withdrawal: the role of calcium channels. Naunyn Schmiedebergs Arch Pharmacol. 1997;355:501–506. doi: 10.1007/pl00004975. [DOI] [PubMed] [Google Scholar]
- Walters CL, Blendy JA. Different Requirements for cAMP Response Element Binding Protein in Positive and Negative Reinforcing Properties of Drugs of Abuse. J Neurosci. 2001;21:9438–9444. doi: 10.1523/JNEUROSCI.21-23-09438.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JT, Christie MJ, Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiological Rev. 2001;81:299–343. doi: 10.1152/physrev.2001.81.1.299. [DOI] [PubMed] [Google Scholar]
- Yang HY, Pu XP. Chronic morphine administration induces over-expression of aldolase C with reduction of CREB phosphorylation in the mouse hippocampus. Eur J Pharmacol. 2009;609:51–57. doi: 10.1016/j.ejphar.2009.03.028. [DOI] [PubMed] [Google Scholar]
- Yao M, Denver RJ. Regulation of vertebrate corticotropin-releasing factor genes. Gen Comp Endocrinol. 2007;153:200–216. doi: 10.1016/j.ygcen.2007.01.046. [DOI] [PubMed] [Google Scholar]
- Yao M, Stenzel-Poore M, Denver RJ. Structural and functional conservation of vertebrate corticotropin-releasing factor genes: evidence for a critical role for a conserved cyclic AMP response element. Endocrinology. 2007;148:2518–2531. doi: 10.1210/en.2006-1413. [DOI] [PubMed] [Google Scholar]
- Zarate CA, Manji HK. Protein kinase C inhibitors: rationale for use and potential in the treatment of bipolar disorder. CNS Drugs. 2009;23:569–582. doi: 10.2165/00023210-200923070-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Z, Keifer J. Protein kinase C-dependent and independent signaling pathways regulate synaptic GluR1 and GluR4 AMPAR subunits during in vitro classical conditioning. Neuroscience. 2008;156:872–884. doi: 10.1016/j.neuroscience.2008.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]