

Summary
Monoclonal antibodies (mAbs) specific for human β2-microglobulin (β2M) have been shown to induce tumour cell apoptosis in haematological and solid tumours via recruiting major histocompatibility complex (MHC) class I molecules into and excluding cytokine receptors from the lipid rafts. Based on these findings, we hypothesized that IgM anti-β2M mAbs might have stronger apoptotic effects because of their pentameric structure. Our results showed that, compared with IgG mAbs, IgM anti-β2M mAbs exhibited stronger tumouricidal activity in vitro against different tumour cells, including myeloma, mantle cell lymphoma, and prostate cancer, and in vivo in a human-like xenografted myeloma mouse model without damaging normal tissues. IgM mAb-induced apoptosis is dependent on the pentameric structure of the mAbs. Disrupting pentameric IgM into monomeric IgM significantly reduced their ability to induce cell apoptosis. Monomeric IgM mAbs were less efficient at recruiting MHC class I molecules into and exclusion of cytokine receptors from lipid rafts, and at activating the intrinsic apoptosis cascade. Thus, we developed and validated the efficacy of anti-β2M IgM mAbs that may be utilized in the clinical setting and showed that IgM anti-β2M mAbs may be more potent than IgG mAbs at inducing tumour apoptosis.
Keywords: Tumour apoptosis, anti-β2 mAbs, IgM pentamer, multiple myeloma, haematological and solid tumours
Introduction
Multiple myeloma (MM), a plasma cell malignant neoplasm, is one of the most common haematological malignancies among people older than 65 years (Jemal, et al 2008). The American Cancer Society estimated that 20,580 new cases of myeloma were diagnosed in 2009, and 10,580 Americans died from this disease. Although several new reagents, such as thalidomide, lenalidomide, and bortezomib, have been reported to prolong myeloma patient survival, MM remains a largely incurable disease and patients are prone to quickly relapse after high-dose chemotherapy (Anderson 2004). So far, monoclonal antibodies (mAbs), such as trastuzumab (Romond, et al 2005), bevacizumab (Hurwitz, et al 2004, Sonpavde 2003), and cetuximab (Bonner, et al 2006, Cunningham, et al 2004), have been commonly used in therapies for breast cancer, renal cancer, colorectal cancer, and squamous-cell carcinoma of the head and neck. This is especially true for haematological malignances because therapeutic efficacy of mAbs can be achieved at low doses, and response can be achieved rapidly. A good example is the anti-CD20 mAb, rituximab, for B-cell malignancies (Coiffier, et al 2002), which is now used as a frontline therapy for diffuse large B-cell lymphoma and other B-cell tumours (Liu, et al 2007, Overman, et al 2008, Spina, et al 2007). To treat MM, several potential target candidates, including CD40 (Tai, et al 2004), CD74 (Stein, et al 2007), interleukin-6 receptor (IL-6R) (Huang and Vitetta 1993), CS1 (Tai, et al 2008) and CD38 (de Weers, et al, 2011), have been identified. Unfortunately, none of these have been approved for the treatment of MM. We have recently demonstrated that humanβ2-microglubulin (β2M) is a potential target for MM treatment (Yang, et al 2006). Our previous studies have shown that anti-β2M mAbs have strong apoptotic effects on myeloma and other haematological malignancies with less toxicity on normal tissues and cells in vitro and in mouse models (Yang, et al 2006), suggesting that anti-β2M mAbs may be a novel therapeutic agent for MM. Furthermore, others have reported similar results by using anti-major histocompatibility complex (MHC) class single-chain Fv diabody or anti-β2M antibodies, respectively, to induce apoptosis in human myeloma (Sekimoto, et al 2007), renal cell carcinoma (Nomura, et al 2007), and prostate cancer (Huang, et al 2006).
Our previous studies have shown that crosslinking anti-β2 M IgG mAbs with surface β2M/MHC class I molecules on myeloma cells leads to recruitment of MHC class I molecules into and exclusion of growth factor receptors from lipid rafts, and activation of apoptosis signalling pathways (Yang, et al 2006, Yang, et al 2007). Because IgM antibodies are polymers, mostly as pentamers with 10 antigenic binding sites, we hypothesized that IgM anti-β2M mAbs have stronger crosslinking ability than IgG mAbs and therefore might have stronger tumouricidal activity. In this study, we generated IgM anti-β2M mAbs and examined their antimyeloma effects. We found that IgM anti-β2M mAbs are more potent than IgG mAbs at inducing tumour apoptosis in vitro and in vivo. Disruption of IgM pentamers by beta-mercaptoethanol (2ME) (Lankester, et al 1994, Shachar, et al 1994) impaired the ability of IgM anti-β2M mAb-induced tumour apoptosis. These results indicate that enhancing the crosslinking ability of anti-β2M mAbs may be a novel approach to further improve mAb-induced tumouricidal efficacy and their therapeutic potential.
Materials and Methods
Cell culture and reagents
Human myeloma cell lines ARP-1 and ARK were established at the Arkansas Cancer Research Center, and the MM.1S cell line was kindly provided by Dr. Steven Rosen of Northwestern University, Chicago, IL. Other tumour cell lines were purchased from American Type Culture Collection (Rockville, MD). Myeloma and mantle cell lymphoma cell lines were cultured in RPMI-1640 medium, and prostate cancer cell lines were cultured in Dulbecco modified Eagle medium (Mediatech, Manassas, VA), containing 10% (vol/vol) heat-inactivated fetal bovine serum (HyClone Laboratories, Logan, UT), 2 mM L-glutamine, 100 u/ml penicillin, and 100 μg/ml streptomycin (Invitrogen, Carlsbad, CA) at 37°C in humidified 95% air and 5% CO2. Primary myeloma cells were isolated from bone marrow aspirates of patients during a routine clinic visit. CD138+ myeloma cells were isolated by magnetic bead sorting (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany). Peripheral blood mononuclear cells (PBMCs) were isolated from blood of healthy donors by centrifugation on Ficoll-Hypaque density gradients. The study was approved by the Institutional Review Board at The University of Texas M. D. Anderson Cancer Center.
Generation of IgM anti-β2M mAbs
We generated a panel of hybridomas by immunizing Balb/c mice with purified human β2M, fusing spleen lymphocytes with SP2/0 myeloma cells, and screening and expanding hybridomas. After several rounds of fusion and screening, we collected more than twenty clones of hybridomas, which produced significant amounts of mAbs specific for β2M. Among them, three clones, PA41, PA43, and PA51, were IgM isotype and induced tumour cell apoptosis. To produce ascites, 2 × 106 hybridoma cells were intraperitoneally injected into severe combined immunodeficiency (SCID) mice. The mAbs were purified by using protein-M columns (Thermo Scientific, Rockford, IL).
Disruption of IgM pentamer
To disrupt the pentameric structure of IgM mAbs, 2ME (Sigma, St Louis, MO) was used (Lankester, et al 1994, Shachar, et al 1994). Briefly, 2ME was added to IgM mAb solution at 37°C for 1 h, and then incubated with 5 mM Iodo for 15 min on ice. The molecular weight of IgM mAb was determined by using native, 4% to 15% gradient polyacrylamide gel electrophoresis (BN-PAGE Novex Bis-Tris Gels) (Invitrogen, Carlsbad, CA) for 3 h for untreated mAbs and 1 h for 2ME-treated mAbs, followed by Coomassie staining .
Cell apoptosis assays
The fraction of apoptotic cells was determined by staining cells suspended in Annexin-V binding buffer (BD PharMingen, San Diego, CA) with fluorescein isothiocyanate (FITC)-conjugated Annexin-V and propidium iodide (PI), according to the manufacturer’s instructions. After 15 min of incubation at room temperature, samples were analysed by flow cytometry. Apoptotic cells were determined as Annexin V-positive cells, which included both early apoptotic (Annexin V+/PI−) and late apoptotic (Annexin V+/PI+) cells. In addition, cell apoptosis was also analysed with the terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) assay by using a flow cytometry-based cell death detection kit (APO-BrdU; BD PharMingen).
Generation of bone marrow stromal cells (BMSCs) and coculture with myeloma cells
BMSCs were generated from bone marrow mononuclear cells from patients with MM as described previously (Uchiyama, et al 1993). After an adherent cellmonolayer formed, these cells were used for coculture with myeloma cells. At the end of the studies, myeloma cells were detached from BMSCs and subjected to analysis for apoptosis.
Western blotting
Cells were harvested, washed, and lysed with lysis buffer (50 mM Tris, pH 7.5, 140 mM NaCl, 5 mM EDTA, 5 mM NaN3, 1% Triton-X-100, 1% Nonidet P-40, 1 × protease inhibitor cocktail). Cell lysates were subjected to sodium dodecyl sulfate (SDS)-PAGE, transferred to polyvinylidene difluoride membrane, and immunoblotted with antibodies against Bcl-2, Bcl-XL, Bad, and Bax; cleaved or non-cleaved caspase-9, -8, and -3; and PARP (Cell Signaling Technology, Inc., Beverly. MA). The membrane was stripped and reprobed with β-actin specific antibody (Sigma) to ensure equal protein loading. Secondary antibodies conjugated to horseradish peroxidase (HRP) were used, followed by enhanced chemiluminescence (Pierce, Rockford, IL) and autoradiography.
Confocal immunofluorescence
To form lipid raft-antibody capping, 1× 106 cells were treated with or without antibodies or reagents on ice for 30 min, moved to 37°C for another 30 min, and then fixed with 4% paraformaldehyde. Treated cells were stained with Alexa Fluor 674-labelled antibodies against Cholera Toxin Subunit B (CTB; Molecular Probes, Carlsbad, CA) to identify lipid rafts. Cells were also stained with anti-HLA-ABC mAb W6/32 (Serotec Ltd, Oxford, UK) and/or anti-IL-6R antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), followed by staining with Alexa Fluor 488-conjugated second antibody to determine the location of MHC class I and/or IL-6R. DAPI (4',6-diamidino-2-phenylindole) staining (Invitrogen, Carlsbad, CA) was used to visualize nuclei. Immunofluorescent cells were then visualized by Leica confocalmicroscope with plan-apochromat 63×/1.4 NA oil-immersion lens in channels of 405 nm for DAPI, in channels of 647 nm for Alexa Fluor 647, or in channels of 488 nm for Alexa Fluor 488.
Mouse experiments
Six- to 8-week-old NOD.B6-Tg(HLA-A2.1)1Enge/DvsJ, which are non-obese diabetic (NOD)/SCID mice transgenic for human HLA-A2 α-chain (A2-NOD/SCID), were obtained from The Jackson Laboratory (Bar Harbor, ME). All mice were maintained in American Association of Laboratory Animal Care-accredited facilities, and studies were approved by the Institutional Animal Care and Use Committee of The University of Texas M. D. Anderson Cancer Center. Mice were subcutaneously inoculated in the right flank with 1 × 106 myeloma ARP-1 or MM.1S cells. Three to four weeks later, when palpable tumours (5 mm in diameter) developed, mice were treated with intraperitoneal injections of 500μg PA43 mAb every 3 days for a total of 8 injections. Control mice received equal amounts of mouse IgM or phosphate-buffered saline (PBS). Tumour size was measured every 3 days in 2 dimensions by using a caliper, and was calculated by using the formula “tumour size (mm3) = 4π/3 × (tumour width/2)2 × (tumour length/2).” Serum was collected from mice twice a week and used for detection of myeloma-derived M-proteins or their light chains by enzyme-linked immunosorbent assay. Mice were humanely sacrificed when moribund or when subcutaneous tumours reached 15 mm in diameter.
Histological examination and in situ TUNEL assay
Formalin-fixed, paraffin-embedded sections of tumours and murine organs from myeloma-bearing mice were deparaffinized with xylene, and rehydrated through a graded alcohol series. For histology analysis, sections were stained with haematoxylin and eosin, and histological examination was performed by a pathologist. The in situ TUNEL assay was performed for cell apoptosis of tumours or murine tissues according to the manufacturer’s instructions. All slides were observed by light microscopy, and images were captured with a SPOT RT camera (Diagnostic Instruments, Burlingame, CA).
Statistical analysis
All data are shown as means ± standard deviation (SD). The Student t test was used to compare various experimental groups. Significance was set at P < 0.05.
Results
IgM anti-β2M mAbs are more potent at inducing tumour cell apoptosis
In this study, we generated a panel of IgM anti-β2M mAbs. Among them, three clones, PA41, PA43, and PA51, were shown to induce myeloma cell apoptosis (data not shown). We selected clone PA43 for this study because it displayed the strongest antimyeloma effects in vitro (data not shown). Our first experiment was to compare the tumouricidal effects of PA43 and IgG anti-β2M mAb D1 (Yang, et al 2006). As shown in Figure 1, although both mAbs induced myeloma cell apoptosis in a dose- (Figure 1A) and time(Figure 1B) dependent manner, significantly stronger tumouricidal effects were observed with PA43 mAb (P < 0.05 to P < 0.01), compared with D1 mAb at the same doses or time points. About 70% of ARP-1 cells treated with 50 μg/ml of PA43 were apoptotic at 12 h. The maximum apoptosis of ARP-1 cells occurred with the treatment of 50 μg/ml PA43 for 24 h. Treatment with purified mouse IgM or IgG, or medium had no effects. Tumour cell apoptosis was confirmed by TUNEL assay (data not shown).
Figure 1.

IgM anti-β2M mAbs are stronger inducers of tumour cell apoptosis than IgG mAbs. Shown is Annexin V-staining for detection of apoptosis in myeloma and other tumour cells in cultures with addition of IgM (clone PA43) or IgG (clone D1) anti-β2M mAbs or equal amounts of mouse IgM (mIgM) or IgG (mIgG). (A) Dose-dependent (in a 24-hour culture) or (B) Time-dependent response of mAb-induced apoptosis (50 μg/ml mAbs) in myeloma cell line ARP-1. Results of four independent experiments using the cell line are shown. Similar results were also obtained with other myeloma cell lines. (C) Percentages of apoptotic myeloma (ARK, ARP-1, MM.1S, U266, and RPMI8226), MCL (Jeko-1, MINO, SP53, and Grant519), and prostate cancer (PC; LN3 and LN4) cells in cultures with medium or with addition of 50 μg/ml IgM PA43 mAb or mouse IgM (mIgM) for 24 h. To examine the toxicity of the mAbs on normal cells, peripheral blood mononuclear cells (PBMC) from four blood donors (PBMC1-PBMC4) were used. (D) Percentages of apoptotic primary myeloma cells from 4 patients (Pt1–Pt4) in cultures with medium or with addition of 50 μg/ml IgM PA43 mAb or mouse IgM (mIgM) for 24 h. Results of three experiments are shown. Similar results were obtained with TUNEL assay. *P < 0.05; **P < 0.01.
PA43 IgM anti-β2M mAb was also effective at inducing apoptosis in other myeloma and tumour cells. As shown in Figure 1C, treatment with 50 μg/ml PA43 mAb exhibited strong tumouricidal activity on all five myeloma cell lines ARK, ARP-1, MM.1S, U266, and RPMI-8226. The mAb also killed four mantle cell lymphoma cell lines MINO, SP53, Jeko-1, and Grant519, and two prostate cancer cell lines LN3 and LN4. Treatment with purified mouse IgM or medium did not induce cell apoptosis. In contrast, similar numbers of apoptotic cells were detected in normal PBMCs in cultures with PA43 mAb compared with cultures with medium or mouse IgM alone, indicating that the anti-β2M mAbs were selective against tumour cells, as we reported previously (Yang, et al 2006). Furthermore, we investigated whether the mAb would also be active on freshly isolated primary myeloma cells from patients. As shown in Figure 1D, purified myeloma plasma cells were uniformly killed by PA43 mAb. At 24 h of culture, 55% to 82% of primary tumour cells cultured with 50 μg/ml PA43 mAb were apoptotic , while the percentages of apoptotic primary myeloma in control medium were 10 to 25%. The IgM mAbs were specific for surfaceβ2M because treatment with the mAbs did not induce apoptosis of Daudi cells, which are a β2M-negative cell line (data not shown). These results indicate that IgM anti-β2M mAbs indeed exhibit stronger antitumour activity than IgG mAbs on haematological and solid tumour cell lines and on primary myeloma cells.
As IL-6 and IGF-I are the most important cytokines for myeloma growth and survival, we investigated whether the cytokines would compromise IgM anti-β2M mAb-induced tumour apoptosis. As shown in Figure 2, treatment with PA43 mAb effectively reduced the viability of myeloma cells. However, the addition of 10 ng/ml IL-6 or 50 ng/ml IGF-I had no effect on PA43 mAb-induced myeloma cell apoptosis (Figure 2A), whereas both cytokines significantly reduced the percentages of apoptotic cells induced by dexamethasone (Figure 2B). These results were reproduced in experiments with four other myeloma cell lines, ARK, MM.1S, RPMI-8266, and U266 (Figure 2C). Additionally, we examined whether BMSCs, which are well known to protect myeloma cells from chemotherapy drug-induced apoptosis, would protect myeloma cells from IgM mAb-induced apoptosis. As shown in Figure 2D, PA43 mAb effectively killed myeloma cells cultured alone or cocultured with BMSCs. These results suggest that the presence of growth factors and BMSCs may not compromise PA43 IgM mAb-induced tumouricidal activity.
Figure 2.

Tumouricidal activity of IgM anti-β2M mAbs is not compromised by myeloma growth factors or bone marrow stromal cells. Shown are percentages of apoptotic myeloma ARP-1 cells in 24-h cultures with (A) IgM PA43 mAb (50 μg/ml) or (B) dexamethasone (10 μM) with or without IL-6 (10 ng/ml) or IGF-I (50 ng/ml). Figures in the left quadrant represent the percentage of live cells. (C) Pooled data of apoptotic cells of five myeloma cell lines induced by IgM anti-β2M mAb with or without IL-6 or IGF-I. (D) Percentage of apoptotic cells from six myeloma cell lines in the cocultures with bone marrow stromal cells (BMSCs) with addition of IgM PA43 mAb (50 μg/ml) for 24 h. Myeloma cells cultured with medium or with the mAb without BMSCs served as controls. After coculture, myeloma cells were recovered and apoptotic cells were detected by using Annexin V-binding assay. Results of three experiments are shown.
Disruption of pentameric structure reduces IgM mAb-induced tumour apoptosis
To prove that the pentameric IgM was responsible for the observed stronger tumouricidal effects, we generated monomeric IgM mAbs by disrupting the pentameric IgM with 2ME, a specific agent that irreversibly breaks the J-chain of IgM antibody (Lankester, et al 1994, Shachar, et al 1994). The change of the mAb structure was visualized by native gel electrophoresis. As shown in Figure 3A, pentameric mAb PA43 was shown as one band with a molecular weight of 900 kDa. Treatment of the mAb with 3 mM of 2ME led to disappearance of the 900-kDa band and appearance of a strong 180-kDa band, which is the same size as a monomeric IgM (Fig. 3B). This indicates that treatment with 2ME completely disrupted the pentameric structure of IgM mAbs, leading to generation of IgM monomers.
Figure 3.

Disruption of pentameric structure of IgM anti-β2M mAb reduces their ability to induce myeloma cell apoptosis. Gel electrophoresis using 4–15% gradient gels to show (A) Pentameric IgM, and (B) Monomeric IgM, monomers induced by 2ME. 2ME was added to PA43 mAb solution at 37°C for 1 h, and then incubated with 5 mM Iodo for 15 min on ice. Native IgM PA43 mAb is shown as pentamers with one band of 900 kDa. After treatment with 3 mM 2ME, IgM mAbs displayed a strong band at 175 kDa, which corresponds to IgM monomer. M: marker. (C) Representative histograms showing apoptotic ARP-1 cells in 24-h cultures with medium or with addition of IgM PA43 mAb (50 μg/ml) pretreated with or without 2ME, and (D) Pooled data of apoptotic cells in three myeloma cell lines. Apoptotic cells were detected by Annexin V-binding assay. Representative results of three independent experiments are shown. **P < 0.01. (E) Representative staining (mean fluorescence intensity) for detection of mAb binding to cell surface β2M on ARP-1 myeloma cells. The mAbs included native PA43 or 2ME-treated PA43. IgG anti-β2M mAb D1 was used as a control. The binding of the mAbs was visualized by FITC-conjugated antibody against mouse IgG or IgM. Similar results were obtained from other myeloma cell lines.
We next examined the effects of 2ME-treated IgM mAbs on tumour cell apoptosis. Tumour cells were incubated with 50 μg/ml of untreated or 2ME-treated PA43 for 24 h. As shown in Figure 3C, untreated PA43 mAb induced 77% of myeloma cells into apoptosis, whereas 2ME-pretreated PA43 mAb had reduced activity of inducing tumour cell apoptosis at the same doses (P < 0.01). These results were confirmed with other myeloma cells (P < 0.05 to P < 0.01; Figure 3D). Similar results were also obtained from mantle cell lymphoma and prostate cancer cells (data not shown). To exclude the possibility that 2ME-treated anti-β2M IgM mAb may bind poorly with surface β2M, ARP-1 cells were incubated with either PA43 mAb pretreated with 3 mM 2ME or untreated PA43 mAb, followed by staining with FITC-conjugated anti-mouse IgM antibody. As shown in Figure 3E, 2ME-treated PA43 mAb bound to the surface of myeloma cells, similar to anti-β2M IgG mAb D1, indicating that the reduced apoptotic ability of monomeric PA43 mAb was not due to its ability to bind with cell surface β2M. These results confirm that the enhanced activity of IgM mAbs in tumour apoptosis depends on the pentameric structure of the mAb.
IgM anti-β2M mAbs are efficient at redistributing MHC class I molecules into and IL-6 receptor from lipid rafts
We investigated whether IgM mAbs are more efficient at recruiting MHC class I molecules into the lipid rafts. To induce PA43-mediated β2M/MHC class I capping, ARP-1 myeloma cells were incubated with 50 μg/ml PA43 mAb, either pretreated with 2ME or untreated, or with equal amounts of mouse IgM. Cells were subsequently stained with Alexa Fluor 674-conjugated antibody specific for Cholera Toxin Subunit B (CTB, red colour), a lipid raft-associated protein, to visualize lipid rafts, and with Alexa Fluor 488-conjugated anti-HLA-ABC antibody (green colour) to detect MHC class I molecules. Stained cells were analysed by confocal microscopy. In cells treated with mouse IgM, MHC class I and lipid rafts (CTB positive) were evenly distributed (Figure 4A, upper panels). Treatment of the cells with pentameric PA43 mAb induced formation of large aggregation or capping of MHC class I and lipid rafts, which also co-localized (Figure 4A, middle panels). Treatment with 2ME-pretreated PA43 mAb could also induce capping, but a large portion of surface MHC class I molecules remained outside of lipid rafts (Figure 4A, lower panels).
Figure 4.
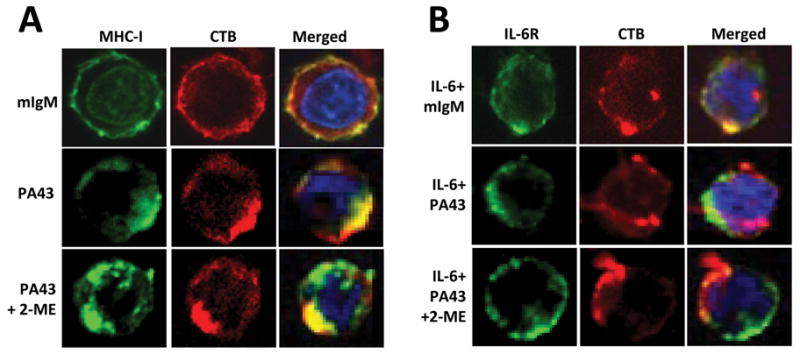
Pentameric IgM anti-β2M mAbs are more efficient than monomeric mAbs at redistributing MHC class I and IL-6R in lipid rafts. Visualization by confocal microscopy shows the distribution and localization of: (A) MHC class I (MHC-I) and (B) IL-6R in relationship with lipid rafts on myeloma cells ARP-1 treated with 50 μg/ml of IgM PA43 mAb or 2ME-pretreated PA43 mAb in presence or absence of IL-6 (10 ng/ml) for 1 h. Cells treated with mouse IgM (mIgM) served as controls. After the treatment, cells were fixed with 4% paraformaldehyde and stained with anti-MHC class I mAb W6/32 (left panels), or with anti-gp130 antibody for IL-6R (right panels), followed by incubation with Alexa Fluor 488-conjugated secondary antibody (green). Cells were then stained with anti-CTB antibody (red) for detection of lipid rafts and DAPI staining (blue) to visualize nuclei. Representative results of three independent experiments are shown.
We also wanted to know whether IgM anti-β2M mAbs might be more effective at excluding IL-6R from lipid rafts. ARP-1 myeloma cells were treated with 50 μg/ml of PA43 mAb, either pretreated with 3 mM 2ME or untreated, or equal amounts of mouse IgM in the presence of 10 ng/ml human recombinant IL-6. The distribution of IL-6R in the cell membrane was determined with Alexa Fluor 488-conjugated anti-IL-6 antibody (green colour). As shown in Figure 4B, the addition of IL-6 recruited IL-6R into lipid rafts on control (mouse IgM-treated) cells (upper panels), determined by Alexa Fluor 674-conjugated anti-CTB antibody (red colour). However, treatment of the cells with pentameric PA43 mAb excluded IL-6R from lipid rafts, and almost all IL-6R was found in non-lipid raft fraction in cells treated with PA43 mAb even in the presence of IL-6 (middle panels). In contrast, 2ME-pretreated PA43 mAb was less effective at excluding IL-6R from lipid rafts, because a part of IL-6R was still localized within the lipid raft fraction (lower panels). Similar results were also obtained from other myeloma cell lines treated with PA43 mAbs (data not shown). Taken together, these results indicate that intact PA43 mAb was more effective than 2ME-pretreated PA43 at relocating MHC class I molecules into and excluding IL-6R from lipid raft domains.
To examine whether IgM anti-β2M mAb-mediated redistribution of MHC class I and IL-6R in lipid rafts would activate apoptosis signalling pathways in myeloma cells, we examined the activation and cleavage of caspase cascades. As shown in Figure 5A, treatment with PA43 mAb led to cleavage of caspase-9, -3 and PARP as early as 12 h after the treatment. Additionally, our data showed that myeloma cell apoptosis induced by IgM PA43 mAb was caspase-9 dependent, because the addition of the pan-caspase inhibitor Z-VAD and caspase-9 inhibitor Z-LEHD significantly inhibited PA43 mAb-induced tumour apoptosis (Figure 5B). Although caspase-8 was also cleaved 48 h after treatment, caspase-8 inhibitor did not abrogate the apoptotic effects of PA43 mAb on tumour apoptosis (data not shown). These results suggest that the intrinsic apoptotic pathway might be a major pathway activated by the mAbs. To confirm these results, we examined the expression of pro-apoptotic proteins, Bax and Bad, or anti-apoptotic proteins, Bcl-2 and Bcl-XL, in PA43 mAb-treated myeloma cells. The results showed that treatment with PA43 mAb downregulated Bcl-2 and Bcl-XL expression, whereas the levels of Bad and Bax were unchanged (Figure 5C). Furthermore, we found that 2ME-pretreated PA43 mAb was less effective at activating caspase-9, -3 and PARP (Figure 5D). These results indicate that the pentameric structure of the PA43 mAb contributes to the mAb-induced crosslinking and redistribution of MHC class I and IL-6R in lipid rafts and activation of the apoptosis signalling pathway.
Figure 5.

Pentameric IgM anti-β2M mAbs induce strong caspase activation in myeloma cells. (A) Western blot analysis shows the levels of pro and cleaved caspase-9, -8, -3 and PARP in myeloma cells treated with 50 μg/ml IgM mAbs for different times. (B) Inhibition of apoptosis in IgM mAb-treated ARP-1 cells by the pan-caspase inhibitor (Z-VAD) or caspase-9 inhibitor (Z-LEHD). Culture with medium or dimethyl sulfoxide (DMSO) served as controls. (C) Western blot analysis shows protein levels of mitochondria-related anti-apoptotic proteins Bcl-2 and Bcl-XL, and pro-apoptotic proteins Bad and Bax in myeloma cells treated with different doses of IgM mAbs for 24 h. (D) Western blot analysis shows the levels of pro- and cleaved caspase-9, -3 and PARP in myeloma cells treated with different doses of IgM mAbs pretreated with or without 2ME for 24 h. Arrows indicate cleaved (c) caspases and PARP. The levels of β-actin served as loading controls. Results obtained with IgM PA43 mAb on ARP-1 from one representative experiment out of three performed are shown. Similar results were obtained with other tumour cell lines.
IgM anti-β2M mAbs are therapeutic against established myeloma in vivo
As IgM mAbs are large molecules, we examined whether IgM anti-β2M mAbs would be therapeutic on established myeloma in vivo in a myeloma mouse model. In this study, we used our recently described, human-like myeloma mouse model (A2-NOD/SCID mice), in which mature and functional human MHC class I (HLA-A2 and human β2M) is expressed on murine organs and high levels of human β2M derived from human myeloma cells circulate. Our results showed that treatment with PA43 mAb significantly reduced myeloma tumour volumes (Figure 6A) and decreased the levels of circulating M-proteins (Figure 6B) in treated myeloma-bearing A2-NOD/SCID mice. Treatment with PA43 mAb was efficient at reducing tumour growth of established myeloma (P < 0.01, compared with mouse IgM controls). Similar results were also obtained from MM.1S-bearing mice treated with PA43 mAb (data not shown).
Figure 6.

In vivo therapeutic effects of IgM anti-β2M mAbs on established myeloma in a human-like, A2-NOD/SCID mouse model. A2-NOD/SCID mice were xenografted subcutaneously with 2 × 106 ARP-1 per mouse. When subcutaneous tumours reached 5 mm in diameter, mice were injected intraperitoneally with 500 μg IgM PA43 mAb, 500 μg mouse IgM (mIgM), or 500 μl phosphate-buffered saline (PBS) every 3 days for a total of 8 injections. Tumour burdens were determined by (A) tumour volumes measured every 3 days, and (B) levels of circulating human M-proteins or their light chains secreted by the tumour cells by ELISA. **P < .01. After treatment, mice were sacrificed, and tumours and mouse organs were removed for (C) histological examination and (D) detection of apoptotic cells by in situ TUNEL assay. Significant tissue damage and cell apoptosis were found in tumours but not in normal mouse tissues, such as liver and kidney, of mice treated with IgM anti-β2M mAb P43. Similar results were also obtained from established myeloma MM.1S and other mouse organs. Scale bar 20 μM.
To further investigate the potential toxicity of PA43 mAb on normal tissues in vivo, myeloma-bearing A2-NOD/SCID mice were sacrificed after mAb treatment, and tumours and murine organs were removed for histological examination. As shown in Figure 6C, kidney and liver tissues from myeloma-bearing mice treated with PA43 mAb (lower panels) displayed normal histology similar to myeloma-bearing mice treated with mouse IgM (upper panels). Similar results were also obtained by examining other mouse tissues including heart, lung, spleen, and bone marrow from mice treated with PA43 mAb or untreated mice (data not shown). Furthermore, cell damage was only observed in the tumour from mice treated with PA43 mAb (lower panels) but not from mice treated with mouse IgM (upper panels). We also used in situ TUNEL assay to visualize apoptotic cells in normal tissues and tumours of the mice. As shown in Figure 6D, large numbers of apoptotic cells were found in tumours from mice treated with PA43 mAb (lower panels), but not in mice treated with mouse IgM (upper panels). The treatments did not induce cell apoptosis in mouse normal tissues, including the kidney, liver, and heart, lung, spleen, and bone marrow (data not shown). As these mice expressed mature and functional human MHC class I (HLA-A2 and human β2M) on murine organs (Yang, et al 2009), these results indicate that IgM anti-β2M mAbs were therapeutic against established myeloma without damaging normal tissues and cells in vivo.
Discussion
This study examined whether it is possible to improve anti-β2M mAb-induced tumour apoptosis through enhancing the crosslinking ofβ2M/MHC class I molecules by using pentameric IgM mAbs. Our results showed that, compared with monomeric mAbs, IgM mAbs exhibit stronger tumouricidal activity on tumours in vitro and in myeloma-bearing A2-NOD/SCID mice. Thus, we developed and validated the efficacy of anti-β2M IgM mAbs that may be utilized in a clinical setting and showed that IgM anti-β2M mAbs may be more potent than IgG mAbs at inducing tumour apoptosis.
Thus far, the majority of mAbs applied in cancer therapy are IgG isotype, whereas only 5% of them are IgM mAbs (Reichert and Valge-Archer 2007). One reason is that IgM mAbs, which are large molecules with molecular weight of approximately 900 kDa, contain five identical IgG-like subunits linked by J-chain disulfides to form circular pentamers. IgM may have low tissue penetration capacity (Jain and Baxter 1988), which would limit their utility in the clinical setting, particularly for treatment of solid tumours. However, recent studies have demonstrated that alterations in glycosylation or other modifications in size, and in antibody-antigen binding affinity could increase the ability of IgM mAbs to penetrate solid tumours and enhance the tumouricidal efficacy of the mAbs (Adams and Weiner 2005). IgM mAbs, which are more heavily glycosylated than IgG mAbs (Tchoudakova, et al 2009), are reported to be effective in the treatment of melanoma (Azuma, et al 2007, Irie, et al 2004). In line with these findings, our results show that IgM anti-β2M mAbs are more efficient at killing myeloma, mantle cell lymphoma, and prostate cancer cells, as compared with monomeric mAbs. This is because IgM mAbs, which contain ten antigenic binding sites, are more potent at recruiting MHC class I molecules into and excluding IL-6R and IGF-IR from lipid rafts and activating caspase cascades, as shown in Figures 4 and 5. Moreover, IgM anti-β2M mAb-induced tumour apoptosis is stronger and earlier than those induced by IgG mAbs at the same antibody doses (Figure 1). Despite their large size, IgM anti-β2M mAbs were also therapeutic in the human-like A2-NOD/SCID myeloma-mouse model. Consistent with our previous results (Yang, et al 2009), the mAbs did not induce normal cell apoptosis or cause tissue necrosis even though the murine tissues in A2-NOD/SCID mice expressed functional human β2M/MHC class I molecules. These results suggest that the mAbs, in the forms of humanized or chimeric, may be used in the clinical setting because of their strong ability to induce myeloma cell apoptosis.
Our results also clearly show that the efficacy of IgM anti-β2M mAbs depend on their pentameric structure. Treatment with 2ME, which irreversibly disrupts the IgM pentamer, reduced IgM pentamers to IgM monomers and significantly compromised their ability to induce tumour cell apoptosis. As 2ME-pretreated IgM monomeric mAbs bound well to myeloma cell surface, similar to IgG mAbs, we believe that the compromised tumouricidal ability of the IgM monomer may be attributed to its weaker ability to crosslink their targeted molecules, compared with IgM pentamers. Indeed, we found that 2ME-pretreated IgM mAbs were much less efficient at redistributing MHC class I molecules and IL-6R in lipid rafts on myeloma cells and, as a consequence, induced much weaker caspase-9, -8, -3, and PARP activation and less tumour apoptosis. These findings support the notion that the pentameric structure of IgM contributes to the enhanced mAb-mediated crosslinking of MHC class I molecules and to improved mAb-induced tumour apoptosis.
In conclusion, this study shows that the effects of anti-β2M mAbs on tumour apoptosis can be improved through enhancing mAb-mediated crosslinking of MHC class I molecules, which in turn recruits more MHC class I molecules into and excludes more IL-6R from lipid rafts, leading to stronger activation of the intrinsic caspase cascades. Although IgM mAbs are large molecules, they still have in vivo tumouricidal efficacy on established myeloma. Our study suggests that enhancing the crosslinking ability of IgG anti-β2M mAbs may be a novel approach to further improve mAb-induced tumouricidal efficacy and their therapeutic potential.
Acknowledgments
We thank Ms. Alison Woo for providing editorial assistance and our Departmental Myeloma Tissue Bank for providing patient samples.
Grant Support
This work was supported by National Cancer Institute R01 CA138402, R01 CA138398, and P50 CA142509 (Q. Yi), the Leukemia and Lymphoma Society Translational Research Grants (Q. Yi), Multiple Myeloma Research Foundation (Q. Yi), Commonwealth Foundation for Cancer Research (Q. Yi), National Cancer Institute K99/R00 CA137158 (J. Yang), International Myeloma Foundation (J. Yang), Lymphoma Research Foundation (J. Yang), American Society of Hematology (J. Yang), and by funds from the University Cancer Foundation and the Center for Targeted Therapy of The University of Texas M. D. Anderson Cancer Center (Q. Yi).
Footnotes
Disclosure of Potential Conflicts of Interest
The authors have no potential conflicts of interest.
Contribution: Y.C., YL, JY, and Q.Y. initiated the work, designed the experiments, and wrote the paper. Y.C., YL, JQ, YZ, SH, HL performed the experiments and statistical analysis. MW, L.K. and DL provided patients’ samples and critical suggestions to this study.
References
- Adams GP, Weiner LM. Monoclonal antibody therapy of cancer. Nat Biotechnol. 2005;23:1147–1157. doi: 10.1038/nbt1137. [DOI] [PubMed] [Google Scholar]
- Anderson KC. Clinical update: novel targets in multiple myeloma. Semin Oncol. 2004;31:27–32. doi: 10.1053/j.seminoncol.2004.10.016. discussion 33. [DOI] [PubMed] [Google Scholar]
- Azuma Y, Ishikawa Y, Kawai S, Tsunenari T, Tsunoda H, Igawa T, Iida S, Nanami M, Suzuki M, Irie RF, Tsuchiya M, Yamada-Okabe H. Recombinant human hexamer-dominant IgM monoclonal antibody to ganglioside GM3 for treatment of melanoma. Clin Cancer Res. 2007;13:2745–2750. doi: 10.1158/1078-0432.CCR-06-2919. [DOI] [PubMed] [Google Scholar]
- Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB, Jones CU, Sur R, Raben D, Jassem J, Ove R, Kies MS, Baselga J, Youssoufian H, Amellal N, Rowinsky EK, Ang KK. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354:567–578. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- Coiffier B, Lepage E, Briere J, Herbrecht R, Tilly H, Bouabdallah R, Morel P, Van Den Neste E, Salles G, Gaulard P, Reyes F, Lederlin P, Gisselbrecht C. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N Engl J Med. 2002;346:235–242. doi: 10.1056/NEJMoa011795. [DOI] [PubMed] [Google Scholar]
- Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Verslype C, Chau I, Van Cutsem E. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351:337–345. doi: 10.1056/NEJMoa033025. [DOI] [PubMed] [Google Scholar]
- de Weers M, Tai YT, van der Veer MS, Bakker JM, Vink T, Jacobs DC, Oomen LA, Peipp M, Valerius T, Slootstra JW, Mutis T, Bleeker WK, Anderson KC, Lokhorst HM, van de Winkel JG, Parren PW. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J Immunol. 2011;186:1840–1848. doi: 10.4049/jimmunol.1003032. [DOI] [PubMed] [Google Scholar]
- Huang WC, Wu D, Xie Z, Zhau HE, Nomura T, Zayzafoon M, Pohl J, Hsieh CL, Weitzmann MN, Farach-Carson MC, Chung LW. beta2-microglobulin is a signaling and growth-promoting factor for human prostate cancer bone metastasis. Cancer Res. 2006;66:9108–9116. doi: 10.1158/0008-5472.CAN-06-1996. [DOI] [PubMed] [Google Scholar]
- Huang YW, Vitetta ES. A monoclonal anti-human IL-6 receptor antibody inhibits the proliferation of human myeloma cells. Hybridoma. 1993;12:621–630. doi: 10.1089/hyb.1993.12.621. [DOI] [PubMed] [Google Scholar]
- Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, Ferrara N, Fyfe G, Rogers B, Ross R, Kabbinavar F. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–2342. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- Irie RF, Ollila DW, O'Day S, Morton DL. Phase I pilot clinical trial of human IgM monoclonal antibody to ganglioside GM3 in patients with metastatic melanoma. Cancer Immunol Immunother. 2004;53:110–117. doi: 10.1007/s00262-003-0436-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain RK, Baxter LT. Mechanisms of heterogeneous distribution of monoclonal antibodies and other macromolecules in tumors: significance of elevated interstitial pressure. Cancer Res. 1988;48:7022–7032. [PubMed] [Google Scholar]
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- Lankester AC, van Schijndel GM, Fromme J, Cordell JL, van Lier RA, van Noesel CJ. Evidence for a direct physical interaction of membrane IgM, IgD, and IgG with the B29 gene product. J Immunol. 1994;152:2157–2162. [PubMed] [Google Scholar]
- Liu YY, Leboeuf C, Shi JY, Li JM, Wang L, Shen Y, Garcia JF, Shen ZX, Chen Z, Janin A, Chen SJ, Zhao WL. Rituximab plus CHOP (R-CHOP) overcomes PRDM1-associated resistance to chemotherapy in patients with diffuse large B-cell lymphoma. Blood. 2007;110:339–344. doi: 10.1182/blood-2006-09-049189. [DOI] [PubMed] [Google Scholar]
- Nomura T, Huang WC, Seo S, Zhau HE, Mimata H, Chung LW. Targeting beta2-microglobulin mediated signaling as a novel therapeutic approach for human renal cell carcinoma. J Urol. 2007;178:292–300. doi: 10.1016/j.juro.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Overman MJ, Feng L, Pro B, McLaughlin P, Hess M, Samaniego F, Younes A, Romaguera JE, Hagemeister FB, Kwak L, Cabanillas F, Rodriguez MA, Fayad LE. The addition of rituximab to CHOP chemotherapy improves overall and failure-free survival for follicular grade 3 lymphoma. Ann Oncol. 2008;19:553–559. doi: 10.1093/annonc/mdm511. [DOI] [PubMed] [Google Scholar]
- Reichert JM, Valge-Archer VE. Development trends for monoclonal antibody cancer therapeutics. Nat Rev Drug Discov. 2007;6:349–356. doi: 10.1038/nrd2241. [DOI] [PubMed] [Google Scholar]
- Romond EH, Perez EA, Bryant J, Suman VJ, Geyer CE, Jr, Davidson NE, Tan-Chiu E, Martino S, Paik S, Kaufman PA, Swain SM, Pisansky TM, Fehrenbacher L, Kutteh LA, Vogel VG, Visscher DW, Yothers G, Jenkins RB, Brown AM, Dakhil SR, Mamounas EP, Lingle WL, Klein PM, Ingle JN, Wolmark N. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med. 2005;353:1673–1684. doi: 10.1056/NEJMoa052122. [DOI] [PubMed] [Google Scholar]
- Sekimoto E, Ozaki S, Ohshima T, Shibata H, Hashimoto T, Abe M, Kimura N, Hattori K, Kawai S, Kinoshita Y, Yamada-Okabe H, Tsuchiya M, Matsumoto T. A single-chain Fv diabody against human leukocyte antigen-A molecules specifically induces myeloma cell death in the bone marrow environment. Cancer Res. 2007;67:1184–1192. doi: 10.1158/0008-5472.CAN-06-2236. [DOI] [PubMed] [Google Scholar]
- Shachar I, Rabinovich E, Kerem A, Bar-Nun S. Thiol-reducing agents and calcium perturbants alter intracellular sorting of immunoglobulin M. J Biol Chem. 1994;269:27344–27350. [PubMed] [Google Scholar]
- Sonpavde G. Bevacizumab in renal-cell cancer. N Engl J Med. 2003;349:1674. doi: 10.1056/NEJM200310233491719. [DOI] [PubMed] [Google Scholar]
- Spina M, Simonelli C, Tirelli U. Phase II trial of CHOP plus rituximab in patients with HIV-associated non-Hodgkin's lymphoma. J Clin Oncol. 2007;25:e7. doi: 10.1200/JCO.2006.09.0407. [DOI] [PubMed] [Google Scholar]
- Stein R, Mattes MJ, Cardillo TM, Hansen HJ, Chang CH, Burton J, Govindan S, Goldenberg DM. CD74: a new candidate target for the immunotherapy of B-cell neoplasms. Clin Cancer Res. 2007;13:5556s–5563s. doi: 10.1158/1078-0432.CCR-07-1167. [DOI] [PubMed] [Google Scholar]
- Tai YT, Catley LP, Mitsiades CS, Burger R, Podar K, Shringpaure R, Hideshima T, Chauhan D, Hamasaki M, Ishitsuka K, Richardson P, Treon SP, Munshi NC, Anderson KC. Mechanisms by which SGN-40, a humanized anti-CD40 antibody, induces cytotoxicity in human multiple myeloma cells: clinical implications. Cancer Res. 2004;64:2846–2852. doi: 10.1158/0008-5472.can-03-3630. [DOI] [PubMed] [Google Scholar]
- Tai YT, Dillon M, Song W, Leiba M, Li XF, Burger P, Lee AI, Podar K, Hideshima T, Rice AG, van Abbema A, Jesaitis L, Caras I, Law D, Weller E, Xie W, Richardson P, Munshi NC, Mathiot C, Avet-Loiseau H, Afar DE, Anderson KC. Anti-CS1 humanized monoclonal antibody HuLuc63 inhibits myeloma cell adhesion and induces antibody-dependent cellular cytotoxicity in the bone marrow milieu. Blood. 2008;112:1329–1337. doi: 10.1182/blood-2007-08-107292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchoudakova A, Hensel F, Murillo A, Eng B, Foley M, Smith L, Schoenen F, Hildebrand A, Kelter AR, Ilag LL, Vollmers HP, Brandlein S, McIninch J, Chon J, Lee G, Cacciuttolo M. High level expression of functional human IgMs in human PER.C6 cells. MAbs. 2009;1:163–171. doi: 10.4161/mabs.1.2.7945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchiyama H, Barut BA, Mohrbacher AF, Chauhan D, Anderson KC. Adhesion of human myeloma-derived cell lines to bone marrow stromal cells stimulates interleukin-6 secretion. Blood. 1993;82:3712–3720. [PubMed] [Google Scholar]
- Yang J, Qian J, Wezeman M, Wang S, Lin P, Wang M, Yaccoby S, Kwak LW, Barlogie B, Yi Q. Targeting beta(2)-microglobulin for induction of tumor apoptosis in human hematological malignancies. Cancer Cell. 2006;10:295–307. doi: 10.1016/j.ccr.2006.08.025. [DOI] [PubMed] [Google Scholar]
- Yang J, Zhang X, Wang J, Qian J, Zhang L, Wang M, Kwak LW, Yi Q. Anti beta2-microglobulin monoclonal antibodies induce apoptosis in myeloma cells by recruiting MHC class I to and excluding growth and survival cytokine receptors from lipid rafts. Blood. 2007;110:3028–3035. doi: 10.1182/blood-2007-06-094417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Cao Y, Hong S, Li H, Qian J, Kwak LW, Yi Q. Human-like mouse models for testing the efficacy and safety of anti-beta2-microglobulin monoclonal antibodies to treat myeloma. Clin Cancer Res. 2009;15:951–959. doi: 10.1158/1078-0432.CCR-08-1823. [DOI] [PMC free article] [PubMed] [Google Scholar]