

Summary
Background & Objective
The factor VIII (FVIII) B domain shares very little amino acid homology to other known proteins and is not directly necessary for procoagulant activity. Despite this, missense mutations within the B domain have been reported in patients with hemophilia A. Given that the B domain is dispensable for secretion and function of FVIII, we hypothesized that these mutations should not be causative of hemophilia A in these patients.
Methods
Plasmid vectors containing B domain missense mutations that were reported to be associated with moderate/severe hemophilia A (T751S, D826E, V993L, H1047Y, T1353A, N1441K, L1462P, E1579D, A1591S, P1641L and S1669L) were analyzed for their effect on synthesis and secretion compared to FVIII wild-type (WT) following transient transfection into COS-1 and CHO cells in vitro. Further, H1047Y, N1441K and E1579D mutants were expressed in vivo in a hemophilia A mouse model by hydrodynamic tail-vein injection.
Results
FVIII activity and antigen levels for all mutants expressed into the conditioned media of COS-1 and CHO cells were similar to FVIII WT. Also, plasma expression of these mutants was similar to FVIII WT in hemophilia A mice. An in vivo tail clip bleeding assay also demonstrated that blood loss from hemophilia A mice expressing FVIII WT, H1047Y, N1441K and E1579D were similar.
Conclusion
We conclude that most missense mutations within the FVIII B domain would be unlikely to lead to severe hemophilia A and that the majority of such missense mutations represent polymorphisms or non-pathologic mutations.
Keywords: B domain, factor VIII, genotyping, hemophilia A, mutations
Introduction
FVIII is a large plasma glycoprotein that functions as an essential cofactor for the proteolytic activation of factor X by activated factor IX within the intrinsic pathway of blood coagulation [1]. Deficiency or dysfunction of FVIII results in hemophilia A which is phenotypically stratified as mild, moderate, or severe disease based on residual plasma FVIII activity of >5%, 1-5%, or <1% respectively. FVIII is synthesized as a single-chain polypeptide of approximately 280 kDa with the domain structure A1-a1-A2-a2-B-ap-A3-C1-C2 with italics denoting heavy chain acidic regions and a light chain activation peptide [2, 3]. This domain structure is also shared by the homologous coagulation cofactor, factor V (FV). Following synthesis, cleavage within the B domain results in a variably sized heavy chain (A1-a1-A2-a2-B, 90-200 kDa) and a light chain (ap-A3-C1-C2, 80 kDa) that are associated as a heterodimer through a divalent metal ion linkage between the A1 and A3 domains [4]. The A domains share 35% to 40% amino acid identity and are homologous to the A-domains of ceruloplasmin and FV. The C-domains also display 35-40% amino acid identity to each other and to FV and are homologous to proteins that are capable of binding negatively charged phospholipids, suggesting a role in phospholipid interactions [2, 3, 5].
A database of mutations described causing hemophilia A is available on the Internet at The Haemophilia A Mutation, Structure, Test and Resource Site (HAMSTeRS, URL: http://hadb.org.uk/)†. Common gene derangements include gene inversions, insertions and large and small gene deletions, typically associated with a severe phenotype. However, point mutations (missense, nonsense and messenger RNA splice-site mutations) represent ~70% of the described molecular defects in hemophilia A. The study of FVIII missense mutations, in particular, has provided insights on: structure and function of FVIII, mechanisms of hemophilia A, and risk of inhibitors developing to FVIII replacement therapy. Over 900 such missense mutations have now been described and occur throughout the entire coding region for FVIII. Although missense mutations have been shown to interfere with FVIII biosynthesis and secretion, there are a number of hemophilia A missense mutations that affect the functional properties of FVIII. These are clustered in regions known to interact with factor IXa, factor X, von Willebrand factor (VWF), the phospholipid surface or affect the stability of FVIII after activation by thrombin [6]. However, those missense mutations characterized to date have all been located within the A and C domains of FVIII.
The large FVIII B domain is encoded by a single large exon (exon 14) and roughly spans from amino acids 741 to 1648. Unlike the other functional domains of the FVIII molecule, the B domain shares very little amino acid homology with the B domain of FV. FVIII does share with FV the distinctive property of having extensive asparagine (N)-linked glycosylation in its B domain. The B domain alone accounts for 19 of the 25 potential asparagine (N)-linked glycosylation attachment sites found on the FVIII molecule. While the B domain is not directly necessary for the central procoagulant activity of FVIII, more recently it has been shown to play a major role in the intracellular processing and trafficking of FVIII. In addition, there is emerging evidence that portions of the B domain may have functional influences throughout the life cycle of FVIII [7]. However no molecular mechanisms for how missense mutations within the B domain cause hemophilia A have been described.
Figure 1 depicts the reported missense mutations within the FVIII B-domain that have been associated with moderate/severe hemophilia A. None of these reported mutations occurred at sites known to be involved in post translational modifications within the B domain. In this report we have characterized these FVIII B domain missense mutations including in vitro and in vivo expression and functional analysis to elucidate any plausible mechanism for causation for hemophilia A. The list of B domain missense mutations analyzed in this study is shown in Table 1 along with references to their clinical reports.
Figure 1. Missense mutations and sites of key post-translational modifications within the Factor VIII B domain.
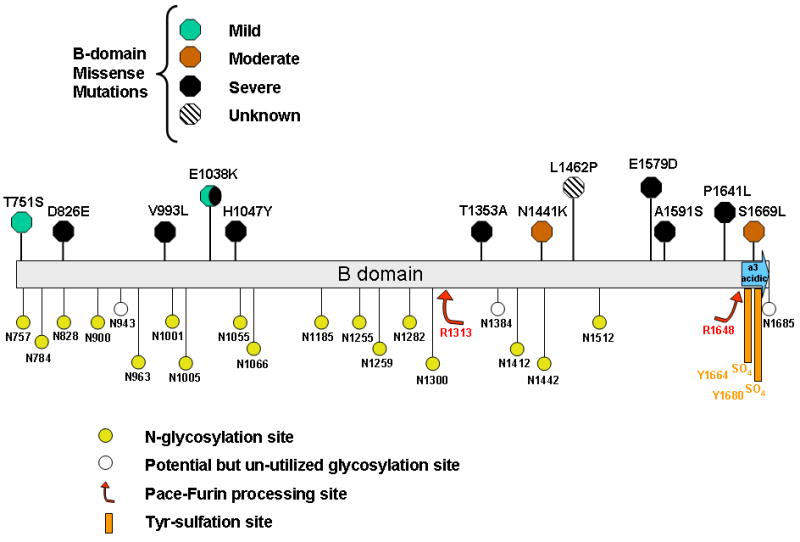
Sites of reported missense mutations within the FVIII B domain and their reported clinical phenotypes are indicated in the upper portion of the figure. Residues that predict sites of asparagine(N)-linked glycosylation, tyrosine(Y)-sulfation and proteolytic modification are indicated in the lower portion of the figure.
Table 1.
FVIII B domain missense mutations analyzed in the present study
| Nucleotide Change* | Amino Acid Change # | Reported Clinical Phenotype | HAMSTeRS | Reference |
|---|---|---|---|---|
| c.2309 C > G | T751S (T770S) | Mild | No | [8] |
| c.2535 C > A | D826E (D845E) | Severe | No | [9] |
| c.3034 G > C | V993L (V1012L) | Severe | Yes | [10] |
| c.3196 C > T | H1047Y (H1066Y) | Severe | Yes | [11] |
| c.4114 A > G | T1353A (T1372A) | Severe | No | [12] |
| c.4380 T > A | N1441K (N1460K) | Cross-reacting material-reduced; Mild/Moderate | Yes | [13] |
| c.4442 T > C | L1462P (L1481P) | UnKnown | Yes | [13] |
| c.4794 G > T | E1579D (E1598D) | Severe | Yes | Unpublished |
| c.4828 G > T | A1591S (A1610S) | Severe | Yes | [14] |
| c.4979 C > T | P1641L (P1660L) | Severe | Yes | [15] |
| c.5063 C > T | S1669L (S1688L) | Moderate | No | [12] |
DNA mutation numbering is based on cDNA sequence (GENBANK # NM_000132), with nucleotide +1 corresponding to A of the ATG translation initiation codon.
Amino acid numbering is for the mature processed protein, with the first alanine numbered as +1 and is representative of the published literature and the FVIII mutation reference database. The corresponding Human Gene Variation Sequence (HGVS) - type numbering is shown in parentheses.
Methods
Materials
FVIII-deficient and normal pooled human plasma were obtained from George King Biomedical (Overland Park, KS). Activated partial thromboplastin (automated aPTT reagent) and CaCl2 were purchased from bioMeriux (Durham, NC). Dulbecco modified Eagle medium (DMEM), alpha-modified essential medium (alpha-MEM) and fetal bovine serum were purchased from Gibco BRL (Gaithersburg, MD). COAMATIC was purchased from DiaPharma (West Chester, OH). Matched-Pair Antibody Set for ELISA of human Factor VIII antigen was purchased from Affinity Biologicals (Ancaster, ON). FuGENE-6 transfection reagent was purchased from Roche Applied Science (Indianapolis, IN). QuikChange XL Site-Directed Mutagenesis Kit was purchased from Stratagene (La Jolla, CA). Male C57BL/6 mice were purchased from Jackson Laboratory. F8 -/- (exon 16 knock-out) hemophilia A mice [16] were housed under pathogen-free conditions at the University of Michigan Laboratory Animal Medicine (ULAM) facility. The University Committee on the Use and Care of Animals (UCUCA) approved the animal protocols.
Plasmid mutagenesis
Mutagenesis was performed within the mammalian expression vector pMT2 [17]containing the full-length human FVIII cDNA (NM_000132). Mutant plasmids were generated through site-directed mutagenesis using the QuikChange XL protocol as described previously [18,19]. The plasmid containing the full-length wild-type human FVIII cDNA sequence was designated FVIII WT. The mutations were confirmed by restriction enzyme digestion and DNA sequencing.
Plasmid transfection and analysis of in vitro expression
Plasmid DNA of FVIII WT, B domain mutants and control mutants were transfected into COS-1 monkey kidney cells by the diethylamino ethanol (DEAE)-dextran method as previously described [20] and into Chinese hamster ovary (CHO) cells using FuGENE-6 transfection reagent per the manufacturer’s recommendations. Conditioned medium was harvested at 60-70 hours post-transfection.
Factor VIII activity and antigen analysis
FVIII activity was measured by a: (1) 1-stage aPTT clotting assay on an MLA Electra 750 fibrinometer (Medical Laboratory Automation, Pleasantville, NY) by reconstitution of human FVIII-deficient plasma or (2) 2-stage assay using the COAMATIC chromogenic assay according to the manufacturer’s instructions. The FVIII plasma standard was FACT plasma (normal pooled plasma) from George King Biomedical for the one-stage aPTT clotting assay. The calibration standard included with the COAMATIC chromogenic assay is assayed according to the Fourth International WHO standard. FVIII antigen was quantified by an anti-FVIII light chain sandwich ELISA according to the manufacturer’s instructions.
Plasmid expression in vivo by hydrodynamic tail-vein injection into the hemophilia A (exon 16 -/-) mouse model
A F8 exon 16 knock-out mouse model of hemophilia A was used to analyze the in vivo expression of the FVIII B domain mutants. Plasmid DNA (100ug) was diluted in 2.0 mL Lactated Ringers and infused over 10 seconds into the tail vein [21, 22]. Peripheral blood was collected from the retro-orbital venous plexus at 24 hours and mixed in 10% volume of 3.8% sodium citrate. Plasma was prepared by centrifugation of the blood at 3000 rpm for 15 min. FVIII activity and antigen levels were analyzed by COAMATIC chromogenic assay and human FVIII-specific ELISA.
Murine Tail Clip Bleeding assay
A quantitative in vivo hemostasis assay was performed at between 24 to 48 hours after hydrodynamic tail vein injection. Mice were anesthetized (ketamine 80 mg/kg; xylazine 5 mg/kg IP) and the tail was placed on a heat pad at 37°C for 5 min. Next, the tail was transected with a sterile razor blade over a lateral tail vein at a position where the diameter of the tail was approximately 1.5 mm. After transection, the tail was immediately placed in a 15-mL falcon tube filled with 0.9 % NaCl warmed to 37°C. The weight of lost blood was measured by a micro balance within a 10 minute window. Immediately after the conclusion of the bleeding assay, peripheral blood was collected from the mouse retro-orbital venous plexus and mixed in 10% volume of 3.8% sodium citrate. Plasma was prepared by centrifugation of the blood at 3000 rpm for 15 min. FVIII activity was analyzed by COAMATIC chromogenic assay.
Statistical Analysis
Data are expressed as mean values ± S.D. Statistical significance was determined by a 2-sided student t test and established at P < 0.05.
Results
Analysis of FVIII B domain mutants in silico
To compare amino acid sequence variation between species at sites of FVIII B domain hemophilia-associated missense mutations, we utilized the Basic Local Alignment Search Tool (BLAST, http://blast.ncbi.nlm.nih.gov/Blast.cgi). Amino acid alignments were performed by MacVector (Symantec). Although there was relatively high conservation of the residues between species at the homologous sites for the B domain missense mutation, especially at position 1047, the greatest divergence was at amino acid 1241 (Table 2). Interestingly, some of the reported hemophilia A mutations appear as native sequence in other species.
Table 2.
Conservation across species and in silico prediction of reported FVIII B domain sequence variations
| Amino Acid Change | Species Conservation1 H/CM/D/M/P/B/Rb/R |
In Silico Prediction
|
||
|---|---|---|---|---|
| Polyphen | SIFT | Align GVGD (likely to affect function) | ||
| T751S | T/T/T/A/T/T/T/S | Benign | Tolerated | Likely |
| D826E | D/D/E/D/E/E/D/H | Benign | Tolerated | Less Likely |
| V993L | V/V/V/D/V/V/V/- | Benign | Tolerated | Least Likely |
| E1038K | E/E/E/E/-/E/E/E | Benign | Affects Function* | Likely |
| H1047Y | H/H/H/H/-/H/Y/H | Possibly Damaging | Tolerated | Most Likely |
| D1241E | D/E/E/Y/G/E/-/E | Benign | Tolerated | Less Likely |
| T1353A | T/T/T/K/-/T/T/K | Benign | Tolerated | Likely |
| N1441K | N/N/N/N/N/N/N/D | Possibly Damaging | Affects Function* | Most Likely |
| L1462P | L/V/L/P/L/L/L/P | Benign | Tolerated | Most Likely |
| E1579D | E/E/E/M/E/E/E/Q | Benign | Tolerated | Less Likely |
| A1591S | A/A/A/S/A/A/D/R | Benign | Tolerated | Most Likely |
| P1641L | P/P/P/P/P/P/S/L | Benign | Tolerated | Most Likely |
| S1669L | S/S/S/T/S/S/S/V | Benign | Tolerated | Most Likely |
|
| ||||
| A284E | A/A/A/A/A/A/A/A | Benign | Tolerated | Most Likely |
| R593C | R/R/R/R/R/R/R/R | Probably Damaging | Affects Function | Most Likely |
| R2150C | R/R/R/R/R/R/R/R | Probably Damaging | Affects Function | Most Likely |
FVIII Conservation across species: H, human; CM, common marmoset; D, dog; M, mouse; P, pig; B, bat; Rb, rabbit; R, rat. Non-conserved amino acids are in bold; Divergent amino acids are italicized; reported mutations appearing as native sequences in other species are underlined.
Low confidence prediction
We also investigated whether missense mutations within the FVIII B domain associated with hemophilia A were reported in normal population studies by searching the National Center for Biotechnology Information (NCBI) database of SNPs (http://www.ncbi.nlm.nih.gov/sites/entrez?db=snp)‡. Out of ten SNPs also reported to result in missense mutations of FVIII, four were found in the B domain (data not shown).
We utilized three software tools available online, namely, PolyPhen (http://genetics.bwh.harvard.edu/pph/), SIFT (http://sift.jcvi.org/) and Align GVGD (http://agvgd.iarc.fr/) to predict whether these missense substitutions were likely to be damaging. Of all the B domain mutations, Polyphen predicted only H1047Y and N1441K to be “possibly damaging”, while all the others were considered to be “benign”. Similarly, SIFT analysis indicated that only E1038K and N1441K might affect FVIII function and it is to be noted here that both of these were low confidence predictions. Align GVGD analysis suggested that H1047Y, N1441K, L1462P, A1591S, P1641L and S1669L were most likely to affect function while the rest were benign. The utility of these in silico prediction tools was further underscored by the fact that all three of them predicted the A and C domain mutations, namely, R593C and R2150C, included as controls in this study, to be deleterious.
Expression of FVIII B-domain mutants is similar to WT in COS-1 and CHO cells
FVIII WT and B-domain mutants were compared after transient DNA transfection into COS-1 monkey kidney cells in vitro. FVIII activity and antigen levels were measured by a 1-stage clotting assay and FVIII-specific ELISA, respectively, from the conditioned media 60-70 hrs after transfection. As controls for our experimental approach, previously well characterized FVIII A and C domain mutants, namely, A284E, R593C and R2150C were included in our assays. All B-domain mutants exhibited FVIII activity and antigen levels similar to FVIII WT(Figure 2A). To determine if the observations were cell-line specific, the same experiments were repeated in CHO cells and no difference was observed for the B domain mutants compared to FVIII WT (Figure 2B).Of the controls, A284E displayed a mild impairment of FVIII activity while FVIII antigen levels were similar to that of FVIII WT. In the case of R593C, a mild to moderate impairment of FVIII activity and a moderate to severe impairment of antigen levels were observed. R2150C on the other hand, displayed a mild phenotype with both FVIII activity and antigen levels.
Figure 2. Expression of FVIII B domain missense mutants is similar to WT in COS-1 and CHO cells.
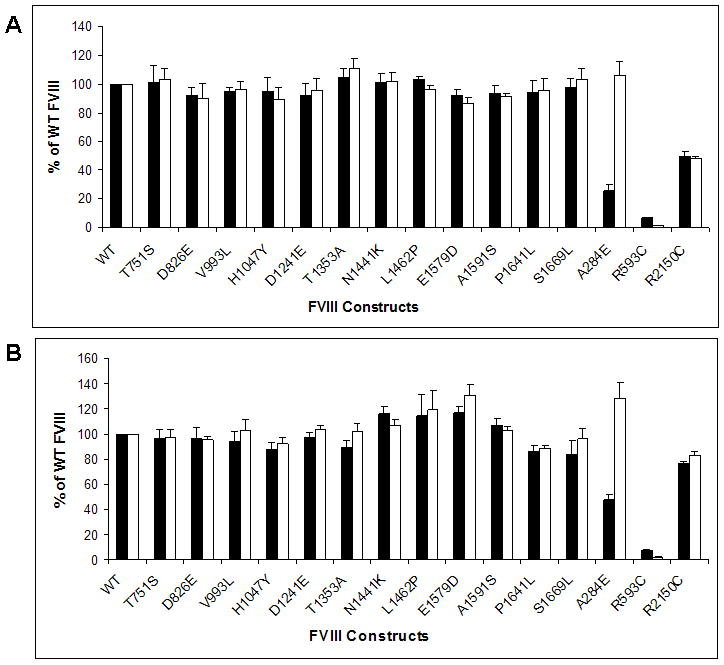
FVIII activity (■) and antigen levels (□) in the conditioned media of COS-1 (A) and CHO (B) cells were measured by APTT and ELISA respectively, at 60-70 hours following transfection with the indicated FVIII constructs. Data are reported as percentages of FVIII WT expression. Data shown is representative of several independent experiments (N ≥ 6), and the error bars represent the standard deviation (SD).
B domain mutants express efficiently in vivo in the hemophilia A mouse model
We next compared the in vivo expression for select B domain mutants with FVIII WT in the hemophilia A mouse model. We chose H1047Y and N1441K since they were considered “possibly damaging” by the different prediction tools. In addition, we also chose the known polymorphism D1241E and E1579D which is an unpublished entry in HAMSTeRS database with a severe phenotype. We also included A284E and R2150C as control mutants. We induced transient expression in the liver of hemophilia A mice using a hydrodynamic tail vein injection of plasmid DNA. No statistically significant difference in mean plasma levels was found between FVIII WT, H1047Y, D1241E, N1441K and E1579D measured at 265.1 mU/mL (range 10.8-734.1), 203.7 mU/mL (96.1-368.3), 208.4 mU/mL (25.51-608.1), 335.5 mU/mL (150-485) and 212 mU/mL (40.5-720) respectively (Figure 3A). The mean plasma levels of the controls A284E and R2150C were 12 mU/mL (9.4-38.3) and 84.1 mU/mL (18.4-143) respectively and were significantly lower than FVIII WT (p<0.05).
Figure 3. In vivo expression and hemostatic efficacy of Factor VIII B domain mutants in a hemophilia A mouse model.
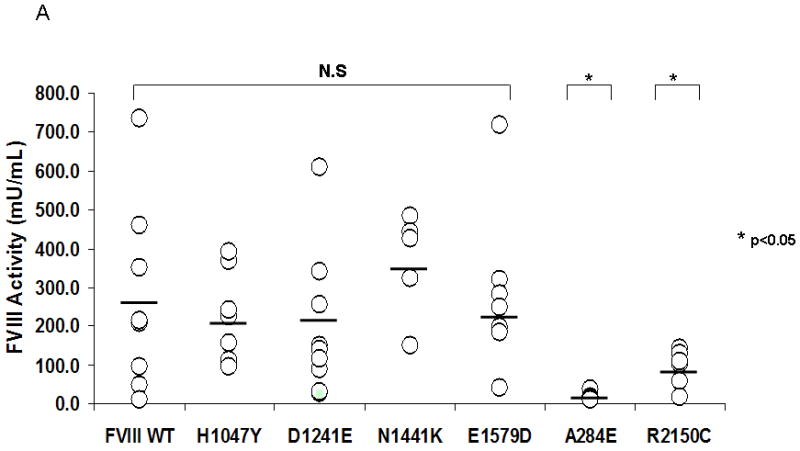
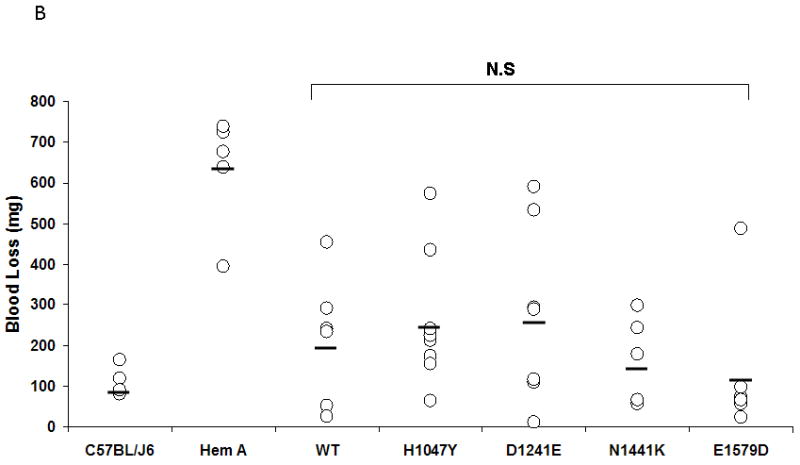
A. FVIII activity in the plasma of hemophilia A mice was measured by a COAMATIC chromogenic assay following transient expression of the indicated FVIII constructs by hydrodynamic tail vein injection as described under Methods. Data presented (○) represent multiple independent expression studies (N ≥ 5) with the mean indicated by a bar. No statistical difference was observed between FVIII WT and the B domain mutants, H1047Y, D1241E, N1441K and E1579D. The FVIII activity of the controls, A284E and R2150C, however, was significantly lower than FVIII WT (p<0.05) B. Hemophilia A mice were subjected to a tail clip bleeding assay following in vivo expression of FVIII WT and B domain mutants and blood loss over 10 min was quantified. C57BL/6J and Hem A mice were included as positive and negative controls, respectively. Data presented (○) are from several independent expression studies (N ≥ 5) with the mean indicated by a bar. No significant difference was observed between FVIII WT and the B domain mutants.
B domain mutants exhibit effective hemostasis in vivo in the hemophilia A mouse model
To investigate the functional effect of select FVIII B domain mutants, we utilized an in vivo tail clip bleeding assay in the hemophilia A mouse model (n≥5). The mean quantitation of blood loss in untreated HA mice was 633.6 mg (range 395-737; n=5) while in C57BL/6J control mice, it was 115 mg (82-165). The mean blood loss measured for FVIII WT, H1047Y, D1241E, N1441K and E1579D was 216.2 mg (26-453), 260 mg (65-573), 278.14 mg (12-532), 169.4 mg (57-299) and 134.33 mg (25-488) respectively. No statistically significant difference was observed between FVIII WT and the B domain mutants (p>0.05).
Discussion
Hemophilia A results from varied genetic alterations of the F8 gene including gene inversions, deletions and insertions. Intron 22 or intron 1 gene inversions account for nearly half of the families with severe hemophilia A [23]. In mild/moderate hemophilia A, missense mutations within the exon coding for the three A domains or the two C domains account for most of the mutations detected [24]. Missense mutations occur in fewer than 20% of individuals with severe hemophilia A but nearly in all of those with mild or moderate hemophilia [23] due to defects in (1) protein and intracellular trafficking, (2) VWF interaction, (3) thrombin activation, (4) stability of FVIIIa, (5) phospholipid binding, and (6) interaction with FIXa. Point mutations in the nucleotide sequence resulting in splicing errors, rare small rearrangements and promoter mutations also account for hemophilia with varying phenotypes [6].
Detection and identification of F8 gene mutations has been demonstrated to predict disease severity, the likelihood of inhibitor development and the success of immune tolerance induction (ITI) therapy for FVIII inhibitor patients [6, 25-27]. Since the first sequences of the human F8 gene were published, various studies have been conducted on the mechanisms of hemophilia A. Although investigation into the structural and functional roles of the FVIII A and C domains have progressed [28], there has not been extensive characterization of the the FVIII B-domain [7]. FVIII and factor V (FV) comprise the same basic domain structure and therefore share structural and functional features. Despite these similarities, there is a marked difference in rates of secretion between the two proteins in vitro, with FVIII being less efficiently secreted. Removal of the B domain of FVIII does not impair FVIII activity and results in an increased expression of mRNA [29]. However, this does not result in more efficient secretion due to intracellular retention of the primary translation product. Nevertheless, the net balance is improved expression of BDD FVIII compared to FVIII WT in vitro [20]. A significant portion of the FVIII primary translation product is misfolded, resulting in its retention within the endoplasmic reticulum (ER). FVIII is co-translationally translocated into the lumen of the ER where it folds and assembles into its tertiary structure. Within the ER, FVIII acquires N-linked oligosaccharide structures. Productive secretion of FVIII requires interaction with and subsequent release from several ER chaperones including immunoglobulin binding protein, calnexin (CNX), and calreticulin (CRT). CNX and CRT both display substrate specificity for glycoproteins containing partially glycosylated N-linked core oligosaccharides. Interaction of FVIII with CNX and CRT is mediated in part by interaction with N-linked oligosaccharides within the B-domain. Properly folded FVIII is released from these chaperones but requires interaction with the mannose-binding lectin complex LMAN1/MCFD2 for efficient transport from the ER to Golgi apparatus. Recent work has also demonstrated that LMAN1 directly interacts with FVIII and that high mannose-containing oligosaccharides, mostly clustered within the B-domain, provide a significant contribution to this interaction. Thus, the N-linked oligosaccharides within the B-domain can participate in the folding interactions within the ER as well as potentially facilitate ER-Golgi transport[7]. Thus, it could be postulated that missense mutations in the B domain, particularly those that may disrupt post-translational modifications to FVIII could affect its secretion efficiency.
However, none of the B domain mutants in our study would be predicted to interfere with N-glycosylation. Two mutations, P1641L and S1669L, are close to the intracellular cleavage site at residue R1648 or tyrosine-sulfation sites at residues Y1664 and Y1680, respectively. Nevertheless, we did not recognize any effect on expression or functional activity.
D1241E was previously widely reported as a missense mutation associated with mild or severe hemophilia but recent observations have firmly established it as a non-synonymous single-nucleotide substitution polymorphism [30-32]. The expression results in our study demonstrated that both FVIII antigen levels and activity in conditioned media from cell lines and in plasma within the hemophilia A mouse were similar to FVIII WT. Additionally, in a tail clip bleeding assay, D1241E exhibited hemostatic efficacy similar to FVIII WT. Similar observations were obtained with H1047Y, N1441K and E1579D. Accordingly, these results suggest that none of the FVIII B domain missense mutations we investigated would be likely to be causative of severe hemophilia A.
There are several examples where patients have been identified with FVIII B domain missense mutations coincident with other FVIII gene derangements. E1038K has a reported phenotype ranging from mild to severe [14,33,34]. However, in one of these cases [14], E1038K was found associated with the intron 22 inversion, which is a known causal mutation for severe hemophilia A. Therefore, it is quite unlikely that the E1038K B domain missense variant is responsible for the hemophilia A phenotype, but rather represents a polymorphism. Such misattribution of causation to a missense variant highlights the potential hazards when genotyping is adopted broadly, particularly when such information might be used to identify carriers or utilized in prenatal decision-making. A recent paper by Schneppenheim et al (35) clearly emphasizes this point wherein a novel rare missense variant was mistakenly attributed to be causative of hemophilia A leading to prenatal diagnosis and peripartal management until the subsequent identification of the actual causative mutation in the family’s index case.
In conclusion, most missense mutations within the FVIII B-domain would be unlikely to lead to severe hemophilia A. Possibly additional causative mutations in these patients are outside the targeted sequencing regions. When sequencing patients with hemophilia A, if a B-domain missense mutation is identified, another causative mutation should continue to be sought. It would also be important to perform expression studies of these novel sequence variants to prove their causative nature.A search for other potential pathological mutations would be recommended within the non-analyzed sequences, such as within introns, in the promoter region, or the 3’-untranslated region or specialized studies to identify duplications.
Acknowledgments
We thank Arno Scheller for help with construction of the FVIII constructs. This work was supported by National Institutes of Health grant HL82619 to S.W.P. and by the Goerlich Foundation.
Footnotes
The Haemophilia A Mutation, Structure, Test and Resource Site (HAMSTeRS, URL: http://hadb.org.uk/) Accessed on 12/10/2010
National Center for Biotechnology Information (NCBI) database of SNPs (http://www.ncbi.nlm.nih.gov/sites/entrez?db=snp)Accessed on 12/10/2010
References
- 1.Mann KG. Biochemistry and physiology of blood coagulation. Thromb Haemost. 1999;82:165–74. [PubMed] [Google Scholar]
- 2.Toole JJ, Knopf JL, Wozney JM, Sultzman LA, Buecker JL, Pittman DD, Kaufman RJ, Brown E, Shoemaker C, Orr EC, Amphlett GW, Foster WB, Coe ML, Knutson GJ, Fass DN. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature. 1984;312:342–7. doi: 10.1038/312342a0. [DOI] [PubMed] [Google Scholar]
- 3.Vehar GA, Keyt B, Eaton D, Rodriguez H, O’Brien DP, Rotblat F, Oppermann H, Keck R, Wood WI, Harkins RN. Structure of human factor VIII. Nature. 1984;312:337–42. doi: 10.1038/312337a0. [DOI] [PubMed] [Google Scholar]
- 4.Kaufman RJ, Wasley LC, Dorner AJ. Synthesis, processing, and secretion of recombinant human factor VIII expressed in mammalian cells. J Biol Chem. 1988;263:6352–62. [PubMed] [Google Scholar]
- 5.Stubbs JD, Lekutis C, Singer KL, Bui A, Yuzuki D, Srinivasan U, Parry G. cDNA cloning of a mouse mammary epithelial cell surface protein reveals the existence of epidermal growth factor-like domains linked to factor VIII-like sequences. ProcNatlAcadSciUSA. 1990;87:8417–21. doi: 10.1073/pnas.87.21.8417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.d’Oiron R, Pipe SW, Jacquemin M. Mild/moderate haemophilia A: new insights into molecular mechanisms and inhibitor development. Haemophilia. 2008;14(Suppl 3):138–46. doi: 10.1111/j.1365-2516.2008.01730.x. [DOI] [PubMed] [Google Scholar]
- 7.Pipe SW. Functional roles of the factor VIII B domain. Haemophilia. 2009;15:1187–96. doi: 10.1111/j.1365-2516.2009.02026.x. [DOI] [PubMed] [Google Scholar]
- 8.Bogdanova N, Markoff A, Eisert R, Wermes C, Pollmann H, Todorova A, Chlystun M, Nowak-Gottl U, Horst J. Spectrum of molecular defects and mutation detection rate in patients with mild and moderate hemophilia A. Human mutation. 2007;28:54–60. doi: 10.1002/humu.20403. [DOI] [PubMed] [Google Scholar]
- 9.Liu J, Zhang Y, Wang H. A novel missense mutation in the B domain of factor VIII causes severe hemophilia A. Zhonghua Xue Ye Xue Za Zhi. 1998;19:458–60. [PubMed] [Google Scholar]
- 10.Becker J, Schwaab R, Moller-Taube A, Schwaab U, Schmidt W, Brackmann HH, Grimm T, Olek K, Oldenburg J. Characterization of the factor VIII defect in 147 patients with sporadic hemophilia A: family studies indicate a mutation type-dependent sex ratio of mutation frequencies. Am J Hum Genet. 1996;58:657–70. [PMC free article] [PubMed] [Google Scholar]
- 11.Habart D, Kalabova D, Novotny M, Vorlova Z. Thirty-four novel mutations detected in factor VIII gene by multiplex CSGE: modeling of 13 novel amino acid substitutions. J Thromb Haemost. 2003;1:773–81. doi: 10.1046/j.1538-7836.2003.00149.x. [DOI] [PubMed] [Google Scholar]
- 12.Santacroce R, Acquila M, Belvini D, Castaldo G, Garagiola I, Giacomelli SH, Lombardi AM, Minuti B, Riccardi F, Salviato R, Tagliabue L, Grandone E, Margaglione M. Identification of 217 unreported mutations in the F8 gene in a group of 1,410 unselected Italian patients with hemophilia A. J Hum Genet. 2008;53:275–84. doi: 10.1007/s10038-007-0238-y. [DOI] [PubMed] [Google Scholar]
- 13.McGinniss MJ, Kazazian HH, Jr, Hoyer LW, Bi L, Inaba H, Antonarakis SE. Spectrum of mutations in CRM-positive and CRM-reduced hemophilia A. Genomics. 1993;15:392–8. doi: 10.1006/geno.1993.1073. [DOI] [PubMed] [Google Scholar]
- 14.Chan V, Pang A, Chan TP, Chan VW, Chan TK. Molecular characterization of haemophilia A in southern Chinese. Br J Haematol. 1996;93:451–6. doi: 10.1046/j.1365-2141.1996.4981042.x. [DOI] [PubMed] [Google Scholar]
- 15.Bogdanova N, Markoff A, Pollmann H, Nowak-Gottl U, Eisert R, Wermes C, Todorova A, Eigel A, Dworniczak B, Horst J. Spectrum of molecular defects and mutation detection rate in patients with severe hemophilia A. Human mutation. 2005;26:249–54. doi: 10.1002/humu.20208. [DOI] [PubMed] [Google Scholar]
- 16.Bi L, Lawler AM, Antonarakis SE, High KA, Gearhart JD, Kazazian HH., Jr Targeted disruption of the mouse factor VIII gene produces a model of haemophilia A. Nat Genet. 1995;10:119–21. doi: 10.1038/ng0595-119. [DOI] [PubMed] [Google Scholar]
- 17.Kaufman RJ. Vectors used for expression in mammalian cells. Methods Enzymol. 1990;185:487–511. doi: 10.1016/0076-6879(90)85041-l. [DOI] [PubMed] [Google Scholar]
- 18.Qi D, Scholthof KB. A one-step PCR-based method for rapid and efficient site-directed fragment deletion, insertion, and substitution mutagenesis. J Virol Methods. 2008;149:85–90. doi: 10.1016/j.jviromet.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 19.Zheng L, Baumann U, Reymond JL. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 2004;32:e115. doi: 10.1093/nar/gnh110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pittman DD, Alderman EM, Tomkinson KN, Wang JH, Giles AR, Kaufman RJ. Biochemical, immunological, and in vivo functional characterization of B-domain-deleted factor VIII. Blood. 1993;81:2925–35. [PubMed] [Google Scholar]
- 21.Liu F, Song Y, Liu D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999;6:1258–66. doi: 10.1038/sj.gt.3300947. [DOI] [PubMed] [Google Scholar]
- 22.Zhang G, Budker V, Wolff JA. High levels of foreign gene expression in hepatocytes after tail vein injections of naked plasmid DNA. Hum Gene Ther. 1999;10:1735–7. doi: 10.1089/10430349950017734. [DOI] [PubMed] [Google Scholar]
- 23.Kaufman RJ, Antonarakis SE, Fay PJ. Hemostasis and Thrombosis: Basic Principles and Clinical Practice. 5. Philadelphia: J B Linpincott Company; 2006. Factor VIII and hemophilia A; pp. 151–75. [Google Scholar]
- 24.Kemball-Cook G, Tuddenham EGD, Wacey AI. The factor VIII Structure and Mutation Resource Site: HAMSTeRS version 4. Nucleic Acids Res. 1998;26:216–9. doi: 10.1093/nar/26.1.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goodeve AC, Peake IR. The molecular basis of hemophilia A: genotype-phenotype relationships and inhibitor development. Semin Thromb Hemost. 2003;29:23–30. doi: 10.1055/s-2003-37936. [DOI] [PubMed] [Google Scholar]
- 26.Gouw SC, van den Berg HM. The multifactorial etiology of inhibitor development in hemophilia: genetics and environment. Semin Thromb Hemost. 2009;35:723–34. doi: 10.1055/s-0029-1245105. [DOI] [PubMed] [Google Scholar]
- 27.Coppola A, Margaglione M, Santagostino E, Rocino A, Grandone E, Mannucci PM, Di Minno G. Factor VIII gene (F8) mutations as predictors of outcome in immune tolerance induction of hemophilia A patients with high-responding inhibitors. J Thromb Haemost. 2009;7:1809–15. doi: 10.1111/j.1538-7836.2009.03615.x. [DOI] [PubMed] [Google Scholar]
- 28.Markoff A, Gerke V, Bogdanova N. Combined homology modelling and evolutionary significance evaluation of missense mutations in blood clotting factor VIII to highlight aspects of structure and function. Haemophilia. 2009;15:932–41. doi: 10.1111/j.1365-2516.2009.02009.x. [DOI] [PubMed] [Google Scholar]
- 29.Dorner AJ, Bole DG, Kaufman RJ. The relationship of N-linked glycosylation and heavy chain-binding protein association with the secretion of glycoproteins. J Cell Biol. 1987;105:2665–74. doi: 10.1083/jcb.105.6.2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Viel KR, Machiah DK, Warren DM, Khachidze M, Buil A, Fernstrom K, Souto JC, Peralta JM, Smith T, Blangero J, Porter S, Warren ST, Fontcuberta J, Soria JM, Flanders WD, Almasy L, Howard TE. A sequence variation scan of the coagulation factor VIII (FVIII) structural gene and associations with plasma FVIII activity levels. Blood. 2007;109:3713–24. doi: 10.1182/blood-2006-06-026104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scanavini D, Legnani C, Lunghi B, Mingozzi F, Palareti G, Bernardi F. The factor VIII D1241E polymorphism is associated with decreased factor VIII activity and not with activated protein C resistance levels. Thromb Haemost. 2005;93:453–6. doi: 10.1160/TH04-09-0629. [DOI] [PubMed] [Google Scholar]
- 32.Nossent AY, Eikenboom JC, Vos HL, Bakker E, Tanis BC, Doggen CJ, Bertina RM, Rosendaal FR. Haplotypes encoding the factor VIII 1241 Glu variation, factor VIII levels and the risk of venous thrombosis. Thromb Haemost. 2006;95:942–8. doi: 10.1160/TH06-01-0024. [DOI] [PubMed] [Google Scholar]
- 33.Higuchi M, Antonarakis SE, Kasch L, Oldenburg J, Economou-Petersen E, Olek K, Arai M, Inaba H, Kazazian HH., Jr Molecular characterization of mild-to-moderate hemophilia A: detection of the mutation in 25 of 29 patients by denaturing gradient gel electrophoresis. Proc Natl Acad Sci U S A. 1991;88:8307–11. doi: 10.1073/pnas.88.19.8307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hwang SH, Kim MJ, Lim JA, Kim HC, Kim HS. Profiling of factor VIII mutations in Korean haemophilia A. Haemophilia. 2009 doi: 10.1111/j.1365-2516.2009.02086.x. [DOI] [PubMed] [Google Scholar]
- 35.Schneppenheim R, Schroder J, Obser T, Oyen F, Schneppenheim S, Oldenburg J. The problem of novel FVIII missense mutations for haemophilia A genetic counseling. Hamostaseologie. 2009;29:158–60. [PubMed] [Google Scholar]