

Abstract
Experimental cerebral malaria is a neuroinflammatory condition that results from the host immune response to the parasite. Using intravital microscopy, we investigated leukocyte recruitment in the brain microcirculation and the temporal relationship of this process to the behavioral changes observed in Plasmodium berghei (strain ANKA)-infected C57Bl/6 mice. We found that leukocyte recruitment was increased from day 5 post-infection (p.i.) onwards. Histopathological changes and increased levels of inflammatory cytokines in the brain were also observed. Behavioral performance evaluated by the SHIRPA protocol showed functional impairment from day 6 p.i. onwards. Thus, early leukocyte migration into the brain and associated inflammatory changes may be involved in neurological impairment in parasite-infected C57Bl/6 mice.
Keywords: cerebral malaria, leukocyte recruitment, behavior, cytokines, intravital microscopy
1. Introduction
Malaria infection causes a wide variety of clinical symptoms, ranging from mild unspecific signs to severe forms marked by deep anemia, respiratory distress syndrome and cerebral malaria (CM) (WHO, 2000). CM is the major complication of Plasmodium falciparum infection in humans and, in agreement with WHO clinical criteria, is defined as a potentially reversible diffuse encephalopathy with coma in the absence of other factors that could cause reduced level of arousal (Lou et al., 2001; Medana and Turner, 2006). Long-term neurocognitive impairment has been described in children afflicted by CM (Idro et al., 2006; John et al., 2008a), but the pathogenic mechanisms underlying this impairment remain unclear.
Two main hypotheses have been proposed to explain the pathogenesis of CM: 1) neural injury following direct sequestration of parasite-infected red blood cells in the cerebral microvasculature and endothelia, and 2) neural injury associated with an inflammatory response to the parasite in the central nervous system (CNS). More recently, there is a trend to consider a unified hypothesis in which parasite sequestration and inflammation cooperatively lead to microcirculatory dysfunction and to neurological symptoms (van der Heyde et al., 2006).
Many animal models have been developed to elucidate the inflammatory and/or immunological mechanisms involved in CM (de Souza and Riley, 2002). Experimental CM is characterized by an intravascular accumulation of mononuclear leukocytes and platelets and the presence of perivascular inflammation and parenchymal microhaemorrhages in the CNS (Lackner et al., 2006a). High levels of circulating and cerebral tissue cytokines have also been observed (Grau et al., 1987), including increased expression of CXCL10, CCL2, and CCL5 in mice infected with Plasmodium berghei (strain ANKA) (PbA) (Hanum et al., 2003). These chemokines may represent the signal responsible for the chemoattraction of circulating leukocytes into the CNS and they may also contribute directly to neuronal and glial dysfunction.
Finally, mice infected with PbA may exhibit behavioral symptoms as a result of these neuroinflammatory processes (Lackner et al., 2006b; Desruisseaux et al., 2008). In this study, in order to better understand the timing of processes involved in the behavioral changes that occur during CM, we studied early events in leukocyte migration into the CNS in PbA-infected mice using intravital microscopy.
2. Materials and Methods
2.1. Animals
C57Bl/6 mice (6 to 8-week-old) were obtained from the Animal Care Facilities of the Federal University of Minas Gerais, Brazil. Animals were housed in cages in temperature-controlled rooms and received food and water ad libitum. All procedures described had prior approval from the local animal ethics committee.
2.2. Parasite and experimental infection
Plasmodium berghei (strain ANKA) (PbA) was used in this study. Parasites were maintained in a stabilizer in liquid nitrogen and subjected to at least one in vivo passage prior to use in experimental infection. Mice were infected by intraperitoneal (i.p.) injection of 106 parasitized red blood cells (pRBC) suspended in 0.2 mL PBS (Grau et al., 1986). The level of parasitemia in infected mice was monitored on Giemsa-stained blood films from day 3 onwards and estimated at 1,000 infected RBCs under immersion oil.
2.3. SHIRPA screen
The SmithKline/Harwell/Imperial College/Royal Hospital/Phenotype Assessment (SHIRPA) screen was conceived as a multi-test behavioral battery used for longitudinal studies with standardized guidelines and materials (Rogers et al., 1997). The primary SHIRPA screen consists of a series of observations of reflexes and basic sensorimotor functions and provides a behavioral and functional profile by observational assessment of individual performance (Lalonde et al., 2005).
The SHIRPA protocol was used to evaluate behavioral changes during the course of the infection. After a period of adaptation, the procedure was carried out on day 0 (day of infection) and then from day 3 until death on daily basis. For analysis purpose, the individual parameters assessed by SHIRPA were grouped into five functional categories (neuropsychiatric state; motor behavior; autonomic function; muscle tone and strength, and reflex and sensory function) according to Lackner et al. (2006b), determining an overall score and five domain scores. The reflex and sensory domain involves visual placing, pinna reflex, corneal reflex, toe pinch and righting reflex.
2.4. Intravital microscopy in mouse brain
Intravital microscopy of brain microvasculature was performed as previously described (Vilela et al., 2008, 2009). Briefly, control and infected mice (on day 3 and 5 p.i.) were anesthetized by intraperitoneal (i.p.) injection of a mixture of Ketamine (150 mg/kg, Laboratório Cristália, Brazil) and Xylazine (10 mg/kg, Rompun®, Bayer, Germany) and the tail vein was cannulated. A craniotomy was performed using a high-speed drill (Beltec, Brazil) and the dura matter was removed to expose the underlying pial vasculature. Throughout the experiment, the mouse was maintained at 37°C with a Thermo Plate (TOKAI HIT, Nikon Inc., Japan) and the exposed brain was continuously superfused with artificial cerebrospinal fluid (composition in mM: NaCl 132, KCl 2.95, CaCl2 1.71, MgCl2 0.64, NaHCO3 24.6, dextrose 3.71 and urea 6.7, pH 7.4) held at 37°C.
To observe leukocyte/endothelium interactions, leukocytes were fluorescently labeled by i.v. administration of rhodamine 6G (0.3mg/kg, Sigma-Aldrich, St. Louis, MO) and observed using a microscope (Nikon, Eclipse 501, X10 objective lens) outfitted with a fluorescent light source (epi-ilumination at 510–560 nm, using a 590 nm emission filter). Images were captured by a camera (Nikon, DS-Qi1MC) and projected onto a monitor (LG, FLATRON-W1952TQ). Rolling leukocytes were defined as white cells moving at a velocity less than that of erythrocyte flux. Leucocytes were considered adherent to the venular endothelium (100 μm length) if they remained stationary for 30s or longer.
2.5. Histology
For histological analysis, brains were quickly removed, fixed in 10% formalin, embedded in paraffin and cut into 4 μm sections. The sections were stained with hematoxylin and eosin (H&E) to assess neuronal injury. Slides were examined under a light microscope for qualitative analysis of inflammatory parameters.
2.6. Tissue Extraction and Measurement of NAG Activity
The extent of macrophage sequestration was quantified indirectly by the measuring of N-acetyl-β-D-glucosaminidase (NAG) activity in brain supernatants, as an index of monocyte influx (Barcelos et al., 2005). In brief, the brains of control and infected animals (on day 5 p.i.) were removed, weighed and the tissue was homogenized in extraction solution (100 mg of tissue per mL), containing: 0.4 M NaCl, 0.05% Tween 20, 0.5% BSA, 0.1 mM phenyl methyl sulphonyl fluoride, 0.1 mM benzethonium chloride, 10mM EDTA and 20 KIU aprotinin, using Ultra-Turrax. Brain homogenate was centrifuged at 3,000 g for 10 min at 4° C and supernatants were collected for ELISA and stored at −20° C. The resultant pellet was resuspended in saline/Triton 0.1%. The NAG reaction was run at 37°C for 10 min in a 96-well microplate following the addition of 100 μL p-nitrophenyl-N-acetyl-β-D-glucosaminide (Sigma-Aldrich, St. Louis, MO), dissolved in citrate/phosphate buffer (0.1 M citric acid, 0.1 M Na2HPO4, pH 4.5) at a final concentration of 2.24 mM per 100 μL supernatant derived from tissue sample processing. The reaction was terminated by the addition of 100 μL 0.2 M glycine buffer (pH 10.6). NAG activity was assayed by measuring the change in absorbance (optical density [OD]) at 405nm in a spectrophotometer (Emax, Molecular Devices) and interpolated on a standard curve constructed with p-nitrophenol (0–500 nmol/ml) (Sigma-Aldrich). Results were expressed as change in O.D. per gram of tissue.
2.7. ELISA of proteins in cerebral tissue
Brain tissues were obtained from control and infected mice (on day 3, 5 and 7 p.i.) and the supernatants were collected and stored at −20° C. The concentrations of TNF-α, CXCL1, CXCL9, CCL2, CCL3 and CCL5 were determined by ELISA. Additionally, the systemic concentration of TNF-α and CXCL9 was measured in serum obtained from coagulated blood (15 min at 37° then 30 min a 4°C, stored at −20°C until analysis). Samples at a 1:3 dilution in 0.1% BSA in PBS were assayed by ELISA using commercially available antibodies according to the procedures supplied by the manufacturer (R&D Systems, Minneapolis, MN and Pharmingen, San Diego, CA).
2.8. Statistical analysis
Data are shown as mean ± SEM (except the survival curve). Student t-test was used in NAG analyzes. SHIRPA results, leukocyte-endothelium interaction and ELISA were evaluated by ANOVA, with Newman-Keuls post-test. The survival rate was expressed as the percentage of live animals. A value of p<0.05 was considered significant.
3. Results
3.1. Plasmodium berghei (strain ANKA) infection changes behavioral parameters in C57Bl/6 WT mice
C57Bl/6 WT mice were infected with PbA and monitored daily. Parasitemia progressively increased (Fig. 1A) and mortality peaked on day 7 p.i. (Fig. 1B). There was a marked weight loss in mice during the infection (Fig. 1C).
Fig. 1.

(A) Time course of parasitemia in infected mice (n=8). (B) Time course of survival of mice infected with PbA (n=8). (C) Weight variation in control (n=4) and infected (n=8) mice. Each point of parasitemia and weight loss is expressed as mean ± SEM.
To further analyze the clinical signs of animals, we performed the SHIRPA behavioral battery. SHIRPA analysis confirmed that infected mice developed a wide range of behavioral changes during the course of the disease prior to death (Fig. 2). Neuropsychiatric state was altered in the early course of malaria infection, starting from day 4 p.i. Motor behavior, autonomic function, muscle tone and strength were altered from day 6, while reflex and sensory function did not change until day 7 p.i.
Fig. 2.

Visualisation of performance of control animals and PbA -infected animals in the five distinct functional categories (neuropsychiatric state; motor behavior; autonomic function; muscle tone and strength, and reflex and sensory function) (A–E). Overall scores of the functional categories are shown in groups of at least five mice. Scores of animals at different time points of infection and scores of control animals were compared by One-way ANOVA with Newman-Keuls post-test. *p<0.05; **p<0.01; ***p<0.001.
3.2. PbA-infected mice present several histopathological changes in the brain
Brain samples were evaluated by routine histological techniques in order to determine the progress of the morphological changes in cerebrum and cerebellum of infected mice. Control and infected mice sacrificed on day 3 p.i. had no evidence of morphological changes (Fig. 3A–D). Infected mice on day 5 p.i. showed moderate intravascular infiltrates, consisting primarily of mononuclear cells, and hemorrhagic areas, especially in the cerebellum (Fig. 3E and F). On day 7 p.i., surviving animals exhibited endothelial damage, mild intravascular infiltrates, multifocal hemorrhages (brain parenchyma and cerebellum) and changes consistent with glial activation and neuronal damage (Fig. 3G and H).
Fig. 3.

Hemorrhages and intravascular inflammatory infiltrates in the brain of PbA-infected C57Bl/6 mice (H&E). (A and B) Hematoxylin and eosin-stained sections of cerebellum and cerebrum of control mice without morphological changes. (C and D) Cerebellum and cerebrum parenchyma without inflammatory cells and a typical architecture in mice on day 3 p.i. (E) Intravascular inflammatory cells and hemorrhagic areas in the cerebellum on day 5 p.i. (F) Cerebrum parenchyma with intravascular inflammatory infiltrates in the same date. (G) Multifocal hemorrhages in the cerebellum and neuronal damage on day 7 p.i. (H) Vasculat alteration with edema and intravascular inflammatory cells on day 7 p.i. Magnification, ×200.
3.3. Monocytes/macrophages are increased in the brain of PbA-infected mice
To confirm the presence of monocyte/macrophage infiltration in the brain of PbA-infected mice, we measured NAG activity in brain tissue from control and infected mice. In infected mice, on day 5 p.i., the NAG activity was almost two-fold higher than that observed in controls, indicating the presence of monocytes and macrophages (Fig. 4).
Fig. 4.
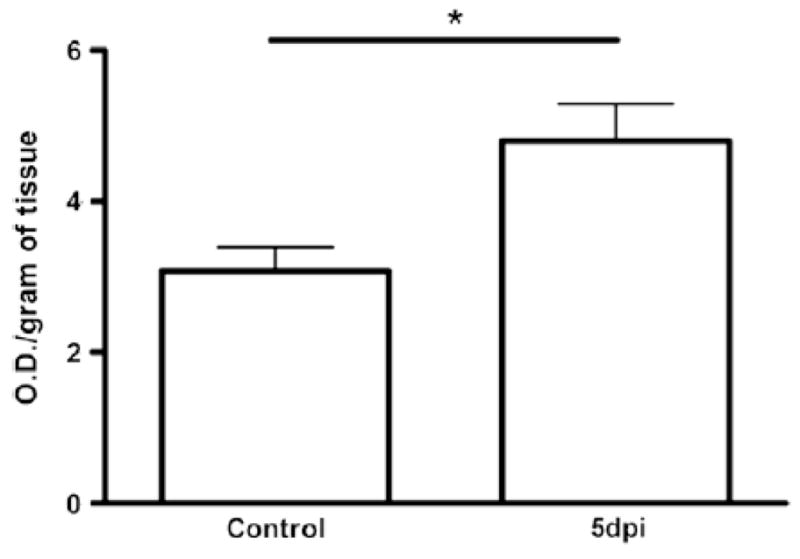
NAG levels in cerebral tissue from control and infected mice. Infected mice on day 5 p.i. showed increased NAG levels when compared with control. Groups of at least five mice and results are expressed as mean ± SEM, where *p<0.05.
3.4. Leukocyte recruitment is increased in the pial microvasculature of PbA-infected mice
To assess the time course of the infiltration of cells into the brain, we performed intravital microscopy in the pia mater vessels. The number of rolling and adherent rhodamine-stained leukocytes was similar in control and infected animals on day 3 p.i., but increased significantly on day 5 p.i. (p<0.001) (Fig. 5), consistent with the heightened monocyte/macrophage infiltration observed by histology and NAG measurements.
Fig. 5.

The study of leukocyte-endothelium interaction was performed by intravital microscopy. The rolling (A) and adhesion (B) of leukocytes in the brain microvasculature were assessed. The protocol included control (sham) and infected animals. Mice, infected by PbA, on day 5 p.i. showed an increase in cell recruitment when compared with control. Groups of at least five mice, results are expressed as mean ± SEM, where **p<0.01; ***p<0.001. ANOVA confirmed the significance.
3.5. CC and CXC chemokines are increased in the brain of PbA-infected mice
Leukocyte activation and recruitment depends on the involvement of cytokines and chemokines. Therefore, we evaluated cerebral levels of TNF-α and the chemokines CCL2, CCL3, CCL5, CXCL1 and CXCL9 (Fig. 6). PbA infection increased cerebral levels of CXCL9 on days 5 and 7 p.i., and CCL2, CCL3 and CCL5 on day 7 p.i., but did not alter CXCL1 levels. Serum levels of TNF-α were increased on days 5 and 7 p.i., and serum CXCL9 levels were increased on days 3, 5 and 7 p.i.
Fig. 6.

Comparative analysis of TNF-α, CCL2, CXCL1, CXCL9, CCL5 and CCL3 concentration in control and PbA-infected mice by ELISA. Results were expressed as the mean ± SEM from at least five animals per group. Asterisk(s) indicate statistical differences where *p<0.05, **p<0.01 and ***p<0.001.
4. Discussion
In this study, we found a temporal association between behavioral changes and inflammatory parameters in the CNS of PbA-infected mice. The early leukocyte recruitment events observed on day 5 p.i. preceded neurological changes, which were clearly evident from day 6 p.i. until death.
PbA induces a neurological syndrome in susceptible mouse strains that mimics human CM. The concept of experimental CM is traditionally associated with the presence of typical neurological signs, such as ataxia, paralysis, seizures and coma, followed by death (Bagot et al., 2002). However, as the pathogenesis of CM includes different mechanisms with a varying degree of severity, this concept may be too restrictive, not encompassing more subtle neurological signs, including mild to moderate cognitive impairment (Medana et al., 2001). Along this line, Desruisseaux et al. (2008) recently demonstrated that memory dysfunction in experimental malaria correlated with brain inflammation and hemorrhage, as well as microglial activation. Our results corroborate these findings, as a series of neurological signs developed in association with early inflammatory changes in CNS. We found an increase in the rolling and adhesion of rhodamine-stained leukocytes by in vivo analysis of cell recruitment using intravital microscopy. This increase was observed on day 5 p.i., while most neurological changes were observed after 6 days of infection.
Once we had observed these inflammatory changes, we looked at the brain tissue. The major histopathologic findings were hemorrhagic areas and intravascular inflammatory infiltrates composed mainly of mononuclear cells. The increase of NAG in brain supernatants corroborated the histopathologic finding of mononuclear recruitment to brain tissue in infected animals. NAG activity is one of the many parameters that can assess macrophage activity and, hence, is an index of monocyte/macrophage tissue infiltration (Reiner et al., 1981). One limitation of our work is the absence of a more detailed study of the types of leukocytes recruited into CNS. This is an interesting issue as previous studies emphasized the role of determined cell types, such as CD8+ T, in the development of neurological signs in the course of CM (Belnoue et al., 2002). Future studies will address in detail the leukocyte subtypes infiltrating the brain and their putative role in modifying behavioural function.
There was also an increase in the levels of chemokines in the brain. Previous studies showed that chemokines up-regulate the expression of adhesion molecules and facilitate leukocyte migration processes in the brain (Ransohoff, 2002). The specific role of chemokines in determining the clinical severity and/or the outcome of malaria or CM remains to be defined. However, as demonstrated by others (Hanum et al., 2003; Miu et al., 2008; Van den Steen et al., 2008), we found increased cerebral levels of chemokines in the brain of mice infected with PbA. Specifically, we found that CXCL9, an interferon-γ-inducible chemokine (Faber, 1997), was upregulated at day 5 p.i. in the brain and at day 3 p.i. in the serum (Figure 6), predicting the large increase in infiltrating leukocytes in the brain microvasculature (Figures 3 and 5) and the onset of functional deficits (Figure 2). This CXCR3 ligand is associated with macrophage activation (Menke et al., 2008) and leukocyte infiltration in a number of inflammatory disease states associated with parasite infection, including Chagas disease (Cutrullis et al., 2009). Whether CXCL9 in our model was produced by neural cells or by an early wave of infiltrating macrophages and whether the brain CXCL9 promoted additional macrophage recruitment and/or T cell recruitment remains to be determined.
Recently, Miu et al. (2008) showed the up-regulation of CXCL9 and CCL5 in experimental CM at the level of mRNA expression. These two chemokines are directly involved in T cell and monocyte/macrophage recruitment to target tissues (Simpson et al., 2000). Additionally, CXCL9−/− mice (Campanella et al., 2008) and IP-10−/− mice (Nie et al., 2009) were reported to be partially/markedly resistant to CM during PbA infection, and exhibited lower leukocyte migration to the brain. Therefore, the increase in leukocyte recruitment during PbA infection in wild type mice in our model may be a direct consequence of CXCL9 upregulation.
Finally, systemic inflammation, as suggested by the increase of serum CXCL9 levels on day 3 p.i., could explain the early alteration in neuropsychiatric state starting at day 4 p.i. However, whether chemokine expression directly results in cognitive dysfunction or whether recruited leukocytes are primarily responsible remains unclear. The association of inflammatory chemokine and cytokine production in the CNS with mild cognitive impairment (John et al., 2008b) and under conditions of CNS viral infection (Sui et al., 2007) suggests that these factors may directly impair neuronal function.
In the present study, we found that deficits in cognitive function associated with cerebral malaria in mice triggered by infection with Plasmodium berghei (strain ANKA) follows an early wave of leukocyte recruitment to the CNS. This recruitment may be driven predominantly by CXCL9, as this was the chemokine that we observed to be increased in the brain prior to severe cognitive impairment. The upregulation of CCL2, CCL3, and CCL5 in the brain at later timepoints just prior to death suggests that these chemokines are not responsible for the initial inflammatory infiltration into the malarial CNS. Further studies are necessary to define leukocyte subsets recruited into the CNS and possibly involved in behavioral changes.
Acknowledgments
The authors thank Carlos Henrique da Silva and Catherine Kelly Martins for their technical assistance. This work was financially supported by Fundação de Amparo à Pesquisa do Estado de Minas Gerais (Fapemig), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Rede Instituto Brasileiro de Neurociência (Rede IBN-Net), Brazil.
References
- Bagot S, Idrissa Boubou M, Campino S, Behrschmidt C, Gorgette O, Guenet JL, Penha-Goncalves C, Mazier D, Pied S, Cazenave PA. Susceptibility to experimental cerebral malaria induced by Plasmodium berghei ANKA in inbred mouse strains recently derived from wild stock. Infection and Immunity. 2002;70:2049–2056. doi: 10.1128/IAI.70.4.2049-2056.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcelos LS, Talvani A, Teixeira AS, Vieira LQ, Cassali GD, Andrade SP, Teixeira MM. Impaired inflammatory angiogenesis, but not leukocyte influx, in mice lacking TNFR1. Journal of Leukocyte Biology. 2005;78:352–358. doi: 10.1189/jlb.1104682. [DOI] [PubMed] [Google Scholar]
- Belnoue E, Kayibanda M, Vigario AM, Deschemin JC, van Rooijen N, Viguier M, Snounou G, Rénia L. On the pathogenic role of brain-sequestered alphabeta CD8+ T cells in experimental cerebral malaria. Journal of Immunology. 2002;169:6369–6375. doi: 10.4049/jimmunol.169.11.6369. [DOI] [PubMed] [Google Scholar]
- Campanella GS, Tager AM, El Khoury JK, Thomas SY, Abrazinski TA, Manice LA, Colvin RA, Luster AD. Chemokine receptor CXCR3 and its ligands CXCL9 and CXCL10 are required for the development of murine cerebral malaria. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:4814–4819. doi: 10.1073/pnas.0801544105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutrullis RA, Postan M, Petray PB, Corral RS. Timing of expression of inflammatory mediators in skeletal muscles from mice acutely infected with the RA strain of Trypanosoma cruzi. Pathobiology. 2009;76:170–180. doi: 10.1159/000218333. [DOI] [PubMed] [Google Scholar]
- Desruisseaux MS, Gulinello M, Smith DN, Lee SC, Tsuji M, Weiss LM, Spray DC, Tanowitz HB. Cognitive dysfunction in mice infected with Plasmodium berghei strain ANKA. The Journal of Infectious Diseases. 2008;197:1621–1627. doi: 10.1086/587908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Souza JB, Riley EM. Cerebral malaria: the contribution of studies in animal models to our understanding of immunopathogenesis. Microbes and Infection. 2002;4:291–300. doi: 10.1016/s1286-4579(02)01541-1. [DOI] [PubMed] [Google Scholar]
- Farber JM. Mig and IP-10: CXC chemokines that target lymphocytes. Journal of Leukocyte Biology. 1997;61:246–257. [PubMed] [Google Scholar]
- Grau GE, Piguet PF, Engers HD, Louis JÁ, Vassali P, Lambert PH. L3T4+ T lymphocytes play a major role in the pathogenesis of murine cerebral malaria. Journal of Immunology. 1986;137:2348–2354. [PubMed] [Google Scholar]
- Grau GE, Fajardo LF, Piguet PF, Allet B, Lambert PH, Vassalli P. Tumor necrosis factor (cachectin) as an essential mediator in murine cerebral malaria. Science. 1987;237:1220–1212. doi: 10.1126/science.3306918. [DOI] [PubMed] [Google Scholar]
- Idro R, Carter JA, Fegan G, Neville BG, Newton CR. Risk factors for persisting neurological and cognitive impairments following cerebral malaria. Archives of Disease in Childhood. 2006;91:142–148. doi: 10.1136/adc.2005.077784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanum PS, Hayano M, Kojima S. Cytokine and chemokine responses in a cerebral malaria-susceptible or -resistant strain of mice to Plasmodium berghei ANKA infection: early chemokine expression in the brain. International Immunology. 2003;15:633–640. doi: 10.1093/intimm/dxg065. [DOI] [PubMed] [Google Scholar]
- John CC, Bangirana P, Byarugaba J, Opoka RO, Idro R, Jurek AM, Wu B, Boivin MJ. Cerebral malaria in children is associated with long-term cognitive impairment. Pediatrics. 2008a;122:92–99. doi: 10.1542/peds.2007-3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John CC, Panoskaltsis-Mortari A, Opoka RO, Park GS, Orchard PJ, Jurek AM, Idro R, Byarugaba J, Boivin MJ. Cerebrospinal fluid cytokine levels and cognitive impairment in cerebral malaria. The American Journal of Tropical Medicine and Hygiene. 2008b;78:198–205. [PMC free article] [PubMed] [Google Scholar]
- Lackner P, Beer R, Helbok R, Broessner G, Engelhardt K, Brenneis C, Schmutzhard E, Pfaller K. Scanning electron microscopy of the neuropathology of murine cerebral malaria. Malaria Journal. 2006a;5:116. doi: 10.1186/1475-2875-5-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackner P, Beer R, Heussler V, Goebel G, Rudzki D, Helbok R, Tannich E, Schmutzhard E. Behavioural and histopathological alterations in mice with cerebral malaria. Neuropathology and Applied Neurobiology. 2006b;32:177–188. doi: 10.1111/j.1365-2990.2006.00706.x. [DOI] [PubMed] [Google Scholar]
- Lalonde R, Dumont M, Staufenbiel M, Strazielle C. Neurobehavioral characterization of APP23 transgenic mice with the SHIRPA primary screen. Behavioural Brain Research. 2005;157:91–98. doi: 10.1016/j.bbr.2004.06.020. [DOI] [PubMed] [Google Scholar]
- Lou J, Lucas R, Grau GE. Pathogenesis of cerebral malaria: recent experimental data and possible applications for humans. Clinical Microbiology Reviews. 2001;14:810–820. doi: 10.1128/CMR.14.4.810-820.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medana IM, Chaudhri G, Chan-Ling T, Hunt NH. Central nervous system in cerebral malaria: ‘Innocent bystander’ or active participant in the induction of immunopathology? Immunology and Cell Biology. 2001;79:101–120. doi: 10.1046/j.1440-1711.2001.00995.x. [DOI] [PubMed] [Google Scholar]
- Medana IM, Turner GD. Human cerebral malaria and the blood-brain barrier. International Journal for Parasitology. 2006;36:555–568. doi: 10.1016/j.ijpara.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Menke J, Zeller GC, Kikawada E, Means TK, Huang XR, Lan HY, Lu B, Farber J, Luster AD, Kelley VR. CXCL9, but not CXCL10, promotes CXCR3-dependent immune-mediated kidney disease. Journal of the American Society of Nephrology: JASN. 2008;19:1177–1189. doi: 10.1681/ASN.2007111179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miu J, Mitchell AJ, Müller M, Carter SL, Manders PM, McQuillan JA, Saunders BM, Ball HJ, Lu B, Campbell IL, Hunt NH. Chemokine gene expression during fatal murine cerebral malaria and protection due to CXCR3 deficiency. Journal of Immunology. 2008;180:1217–1230. doi: 10.4049/jimmunol.180.2.1217. [DOI] [PubMed] [Google Scholar]
- Nie CQ, Bernard NJ, Norman MU, Amante FH, Lundie RJ, Crabb BS, Heath WR, Engwerda CR, Hickey MJ, Schofield L, Hansen DS. IP-10-mediated T cell homing promotes cerebral inflammation over splenic immunity to malaria infection. PLoS Pathogens. 2009;5:e1000369. doi: 10.1371/journal.ppat.1000369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM. The chemokine system in neuroinflammation: an update. The Journal of Infectious Diseases. 2002;186:152–156. doi: 10.1086/344266. [DOI] [PubMed] [Google Scholar]
- Reiner RG, Tanner AR, Keyhani AH, Wright R. A comparative study of lysosomal enzyme activity in monocytes and Kupffer cells isolated simultaneously in a rat model of liver injury. Clinical and Experimental Immunology. 1981;43:376–380. [PMC free article] [PubMed] [Google Scholar]
- Rogers DC, Fisher EM, Brown SD, Peters J, Hunter AJ, Martin JE. Behavioral and functional analysis of mouse phenotype: SHIRPA, a proposed protocol for comprehensive phenotype assessment. Mammalian Genome. 1997;8:711–713. doi: 10.1007/s003359900551. [DOI] [PubMed] [Google Scholar]
- Simpson JE, Newcombe J, Cuzner ML, Woodroofe MN. Expression of the interferon-γ-inducible chemokines IP-10 and Mig and their receptor, CXCR3, in multiple sclerosis lesions. Neuropathology and Applied Neurobiology. 2000;26:133–142. doi: 10.1046/j.1365-2990.2000.026002133.x. [DOI] [PubMed] [Google Scholar]
- Sui Z, Sniderhan LF, Schifitto G, Phipps RP, Gelbard HA, Dewhurst S, Maggirwar SB. Functional synergy between CD40 ligand and HIV-1 Tat contributes to inflammation: implications in HIV type 1 dementia. Journal of Immunology. 2007;178:3226–3236. doi: 10.4049/jimmunol.178.5.3226. [DOI] [PubMed] [Google Scholar]
- Van den Steen PE, Deroost K, Van Aelst I, Geurts N, Martens E, Struyf S, Nie CQ, Hansen DS, Matthys P, Van Damme J, Opdenakker G. CXCR3 determines strain susceptibility to murine cerebral malaria by mediating T lymphocyte migration toward IFN-gamma-induced chemokines. European Journal of Immunology. 2008;38:1082–1095. doi: 10.1002/eji.200737906. [DOI] [PubMed] [Google Scholar]
- van der Heyde HC, Nolan J, Combes V, Gramaglia I, Grau GE. A unified hypothesis for the genesis of cerebral malaria: sequestration, inflammation and hemostasis leading to microcirculatory dysfunction. Trends in Parasitology. 2006;22:503–508. doi: 10.1016/j.pt.2006.09.002. [DOI] [PubMed] [Google Scholar]
- Vilela MC, Mansur DS, Lacerda-Queiroz N, Rodrigues DH, Arantes RM, Kroon EG, Campos MA, Teixeira MM, Teixeira AL. Traffic of leukocytes in the central nervous system is associated with chemokine up-regulation in a severe model of herpes simplex encephalitis: An intravital microscopy study. Neuroscience Letters. 2008;445:18–22. doi: 10.1016/j.neulet.2008.08.072. [DOI] [PubMed] [Google Scholar]
- Vilela MC, Mansur DS, Lacerda-Queiroz N, Rodrigues DH, Lima GK, Arantes RM, Kroon EG, da Silva Campos MA, Teixeira MM, Teixeira AL. The chemokine CCL5 is essential for leukocyte recruitment in a model of severe Herpes simplex encephalitis. Annals of the New York Academy of Sciences. 2009;1153:256–263. doi: 10.1111/j.1749-6632.2008.03959.x. [DOI] [PubMed] [Google Scholar]
- WHO. Severe falciparum malaria: World Health Organization, Communicable Diseases Cluster. Transactions of the Royal Society of Tropical Medicine and Hygiene. 2000;94:S1–S90. [PubMed] [Google Scholar]