

Abstract
Aims
High shear stress (HSS) can have significant impact on angiogenesis and atherosclerosis in collateral arteries near the bifurcation and curvature regions. Here, we investigate the spatiotemporal pattern of HSS-induced intracellular calcium alteration.
Methods and results
Genetically encoded biosensors based on fluorescence resonance energy transfer were targeted in the cytoplasm and the endoplasmic reticulum (ER) to visualize the subcellular calcium dynamics in bovine aortic endothelial cells under HSS (65 dyn/cm2). Upon HSS application, the intracellular Ca2+ concentration ([Ca2+]i) increased immediately and maintained a sustained high level, while the ER-stored calcium had a significant decrease only after 300 s. The perturbation of calcium influx across the plasma membrane (PM) by the removal of extracellular calcium or the blockage of membrane channels inhibited the early phase of [Ca2+]i increase upon HSS application, which was further shown to be sensitive to the magnitudes of shear stress and the integrity of cytoskeletal support. In contrast, Src, phospholipase C(PLC), and the inositol 1,4,5-trisphosphate receptor (IP3R) can regulate the late phase of HSS-induced [Ca2+]i increase via the promotion of the ER calcium efflux.
Conclusion
The HSS-induced [Ca2+]i increase consists of two well-co-ordinated phases with different sources and mechanisms: (i) an early phase due to the calcium influx across the PM which is dependent on the mechanical impact and cytoskeletal support and (ii) a late phase originated from the ER-calcium efflux which is regulated by the Src, PLC, and IP3R signalling pathway. Therefore, our work presented new molecular-level insights into systematic understanding of mechanotransduction in cardiovascular systems.
Keywords: Shear stress, Laminar flow, FRET, Calcium, Live cell imaging
1. Introduction
High shear stress (HSS) can occur under various pathophysiological conditions. For example, HSS with a magnitude of 65–85 dyn/cm2 can be observed in compensatory flows inside collateral arteries where local arterial blockage occurs.1 Chronic HSS with a magnitude around 100–130 dyn/cm2 can also occur at specific locations such as the apex of a bifurcation2 or the outer side of a curvature.3 As such, HSS can have significant impact on atherosclerosis or intracranial aneurysm initiation at these local sites.2,3 It remains unclear, however, on how endothelial cells (ECs) sense HSS and modulate intracellular molecular activities to result in pathophysiological consequences.
The calcium ion is an important signalling molecule for acto-myosin contractility, which regulates endothelium permeability and atherosclerosis.4–6 Shear stress has been well documented to increase the intracellular Ca2+ concentration ([Ca2+]i). Upon flow stimulation, Ca2+ influx from the extracellular medium was shown to occur via different calcium channels on the plasma membrane (PM), such as the stretch-activated cation channel (SACC), store-operated channel (SOC), or voltage-operated channel (VOC).7 The endoplasmic reticulum (ER)-stored calcium can also be released under flow via inositol 1,4,5-trisphosphate receptor (IP3R)8 and ryanodine receptor7 on the ER membrane. Interestingly, both the extracellular Ca2+ influx across the cell membrane5,9,10 and the ER-stored Ca2+ release11–13 were reported to mediate the shear stress-induced [Ca2+]i increase. While the different types of shear stress, cell types, and experimental conditions in these studies may contribute to this discrepancy of observations in terms of the calcium source, the precise reason remains unclear.10 It is possible that the extracellular calcium influx and ER-stored calcium release dynamically co-ordinate at subcellular levels to regulate [Ca2+]i in ECs upon the shear stress stimulation.
A variety of signalling molecules have been shown to participate in the regulation of [Ca2+]i upon shear stress application. For example, the flow-induced [Ca2+]i increase could be blocked when protein kinase C (PKC) ζ was inhibited by a specific inhibitor chelerythrine.14 Other studies showed that shear stress induced a store-operated Ca2+ influx, which can be mediated by the cyclic guanosine monophosphate (cGMP) and protein kinase G (PKG) pathways.15 G-proteins can also mediate the regulation of [Ca2+]i in response to shear stress in Dicytostelium discoideum cells.8 These signalling molecules may regulate the [Ca2+]i via the manipulation of calcium channel expression and protein phosphorylation16 such as SACC,16 P2X4 purinoceptors,5 transient receptor potential vanilloid 4 (TRPV4),17 and transient receptor potential melastatin 7 (TRPM7).18 Despite this progress on the understanding of calcium signalling under flow, the exact contribution of these signalling molecules to the HSS-induced [Ca2+]i increase still needs to be elucidated and integrated to advance our systematic understanding of mechanotransduction.
In the present study, we investigated the HSS-induced [Ca2+]i alteration utilizing a calcium biosensor based on fluorescence resonance energy transfer (FRET) between an enhanced cyan fluorescent protein (ECFP) and a YPet.19 An ER-targeted version of this biosensor, which has an ER retention sequence fused to the C-terminal of YPet, was also applied to continuously visualize and quantify the levels of ER-stored calcium at subcellular regions.4,20 Our results indicate that HSS can induce a [Ca2+]i increase in two phases with different sources and mechanisms in bovine aortic endothelial cells (BAECs).
2. Methods
2.1. Flow systems
To impose a laminar flow on ECs, a parallel-plate flow chamber was applied as previously described.21,22 In brief, a glass slide seeded with a confluent BAEC monolayer forms the floor of a flow channel, created by sandwiching a silicone gasket between the cover glass slide and an acrylic plate. Cells are exposed to HSS created by flows originating from a hydrostatic pressure difference between two reservoirs positioned at different heights. The channel width is 10 mm, the channel height is 0.5 mm, and the total and entrance lengths are 45 and 15 mm, respectively. This flow chamber system has been well established to apply precisely controlled wall shear stress, which can be calculated as: 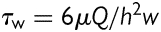 , where µ, is the fluid viscosity of solution, Q, is the flow rate, h, is the channel height, and w, is the channel width. This flow system was perfused with a medium containing polystyrene particles to confirm the laminar flow patterns. HSS was set to be 65 dyn/cm2 as previously described.1 The flow experiments were conducted at 37°C with 5% CO2 to maintain the pH at 7.4.
, where µ, is the fluid viscosity of solution, Q, is the flow rate, h, is the channel height, and w, is the channel width. This flow system was perfused with a medium containing polystyrene particles to confirm the laminar flow patterns. HSS was set to be 65 dyn/cm2 as previously described.1 The flow experiments were conducted at 37°C with 5% CO2 to maintain the pH at 7.4.
2.2. Cell culture and transfection
BAECs were isolated from a bovine aorta from a local slaughterhouse as described.23 Approval for handling BAECs was granted by the Institutional Review Board of University of Illinois, Urbana-Champaign. The cells were cultured in high glucose of Dulbecco's Modified Eagle Medium (DMEM, GIBCO, Invitrogen, USA) containing 10% foetal bovine serum (FBS), 2 mM l-glutamine, 100 unit/mL penicillin, and 100 µg/mL sodium pyruvate in a humidified incubator of 95% O2 and 5% CO2 at 37°C. The DNA plasmids were transfected into the cells by using Lipofectamine 2000 (Invitrogen) reagent according to the product instructions.
2.3. Gene construction and DNA plasmids
A yellow fluorescent protein (YFP) variant Citrine was replaced with YPet to enhance the dynamic range of the FRET-based Ca2+ biosensor (ECFP-CaM-M13-EYFP).20 An ER-targeted calcium biosensor was described previously.4 The membrane-targeted Src FRET biosensor (Kras-Src) was constructed by fusion of a prenylation substrate sequence (KKKKKKSKTKCVIM) from KRas to the C-terminus of the cytosolic Src biosensor.20 Both biosensor constructs for Src and Ca2+ were cloned into pcDNA3.1 (Invitrogen) using BamHI/EcoRI behind a Kozak sequence for mammalian cell expression.20
2.4. Microscopy, image acquisition, and analysis
BAECs expressing various exogenous proteins were starved with 0.5% FBS of DMEM for 24 h before imaging experiments. All images were obtained by using Zeiss Axiovert inverted microscope equipped with a cooled charge-coupled device camera (Cascade 512B, Photometrics), 440DF20 excitation filter, 455DRLP dichroic mirror, and two emission filters controlled by a filter changer [480DF30 for cyan fluorescent protein (CFP) and 535DF25 for YFP]. Time-lapse fluorescence images were acquired by MetaFluor 6.2 software (Universal Imaging) for the calcium or Src biosensor with 10 or 60 s as the time interval, respectively. The emission ratio images were computed and generated after background subtraction by the MetaFluor software to represent the FRET efficiency before being subjected to quantification and analysis by Excel (Microsoft).
2.5. Statistical analysis
All the ratio data were normalized by their basal levels before stimulation in the same cell. Statistical analysis was performed among groups by using ANOVA analysis in SPSS software first, and Student's t-test function in the Excel software (Microsoft) was applied to evaluate the statistical difference between two groups. A significant difference was determined by a P-value of <0.05.
3. Results
3.1. [Ca2+]i has a sustained increase after HSS application
Since [Ca2+]i plays a crucial role in EC physiology and pathology upon mechanical stimulation, we study the spatiotemporal characteristics of [Ca2+]i under HSS. In BAECs, [Ca2+]i measured by a cytosolic calcium FRET biosensor was observed to increase ∼two-folds within 10 s upon the HSS application and maintain a sustained high level for >900 s (Figure 1A, and Supplementary material online, Movie S1). Monitored by an ER calcium biosensor, the ER calcium concentration in BAECs only had a minor decrease (around 10%) within the first 300 s after the HSS exposure and decreased significantly afterwards to reach a level of ∼40% reduction after 1000 s (Figure 1B, and Supplementary material online, Movie S2). These results suggest that the ER-stored calcium release did not contribute significantly in the first 300 s under HSS (the early phase), but may do so in the late phase.
Figure 1.

HSS-induced cytoplasmic calcium increase and ER calcium release. The representative YPet/ECFP emission ratio images (left panels) and averaged time courses (right panels) of the (A) cytosolic and (B) ER calcium FRET biosensors to represent the (A) cytoplasmic (n= 10) and (B) ER (n = 12) Ca2+ concentration under HSS. The standard error (SEM) bars were shown at every 20 measurements on the averaged time courses. All the scar bars represent the length of 20 µm.
3.2. HSS-induced [Ca2+]i increase in the early phase is mainly a result of the extracellular calcium influx
To identify the source of the HSS-induced [Ca2+]i increase, BAECs were treated for 15 min with 5 mmol/L of ethylene glycol tetraacetic acid (EGTA), a Ca2+ chelator, to remove the extracellular calcium. [Ca2+]i did not change significantly within the first 300 s of flow application except for a minor peak and increased gradually after 500 s to reach a level similar to the control group (Figure 2A, and Supplementary material online, Figure S1A, Movie S3). Similar responses can be observed when BAECs were kept in the Ca2+-free medium under flow (Figure 2B, and Supplementary material online, Figure S1A), suggesting that the early phase of [Ca2+]i increase may be attributed to the extracellular calcium influx.
Figure 2.

The HSS-induced [Ca2+]i increase in BAECs is originated from both the extracellular calcium influx and the ER-stored Ca2+ release. (A–C): the averaged time courses represent the cytoplasmic Ca2+ concentration in BAECs pre-treated with (A) 5 mmol/L EGTA for 15 min (n= 4), (B) Ca2+-free DMEM for 15 min (n= 8), or (C) 100 µmol/L 2-APB for 1 h (n= 9) before HSS application. (D) Bar graphs (mean ± SEM) represent the max values of YPet/ECFP emission ratios measuring the cytoplasmic Ca2+ concentration in the early and the late phase in BAECs upon HSS application under different conditions as indicated. *Represents a P< 0.05 when comparing with the same phase in the control group; + represents a P< 0.05 when comparing with different phases in the same group. (E–F): the averaged time courses represent the ER Ca2+ concentration in BAECs pre-treated with (E) Ca2+-free DMEM for 15 min (n= 5) or (F) 100 µmol/L 2-APB for 1 h (n= 10) before HSS application. The standard error (SEM) bars were shown at every 300 s on the averaged time courses.
3.3. HSS-induced ER calcium release in the late phase is dependent on IP3R
When BAECs were pre-treated for 1 h with 100 µmol/L 2-aminoethoxydiphenyl borate (2-APB), an IP3R inhibitor to prevent the ER calcium release, [Ca2+]i had an initial peak within 200 s and declined to the baseline after ∼400 s upon HSS application (Figure 2C, and Supplementary material online, Figure S1B, Movie S4). Hence, the inhibition of IP3R can block the late phase of the HSS-induced [Ca2+]i increase, which may originate from the ER-stored Ca2+ release. We further separated the profile of HSS-induced [Ca2+]i increase into an early phase (0–200 s), a transition phase (200–500 s), and a late phase (500–1200 s) to perform statistical analysis. The maximal values of the early and late phases were calculated from each individual cell and averaged to examine the effect of different treatments. The statistical results confirmed that the removal of extracellular calcium inhibited the early phase, while the inhibition of IP3R on the ER membrane had more effect on the late phase (Figure 2D). Indeed, a significant inhibition of the HSS-induced ER calcium release could be observed after 2-APB pre-treatment (Figure 2F, and Supplementary material online, Figure S1C, Movie S5). In contrast, the Ca2+-free medium did not have any obvious effect on this HSS-induced ER calcium release (Figure 2E, and Supplementary material online, Figure S1C). These results suggest that the inhibition of the early phase of the HSS-induced [Ca2+]i increase by the removal of extracellular calcium is independent of the ER calcium release. We have hence hypothesized that the [Ca2+]i increase upon HSS application may consist of the following (i) an early phase mainly engendered from the calcium influx across the PM and (ii) a late phase mainly from the ER calcium release.
3.4. The involvement of SACC and VOC in the HSS-induced [Ca2+]i increase
Different calcium channels allow the calcium influx across the PM.7 To further identify which channels contribute to the HSS-induced [Ca2+]i increase, BAECs transfected with calcium biosensor were pre-treated for 1 h with streptomycin (STM), inhibitor of SACC, or nifedipine (Nif), inhibitor of VOC. The pre-treatment with 200 µmol/L of STM significantly inhibited the early phase with no effect on the late phase (Figure 3A and D, and Supplementary material online, Figure S2, Movie S6). Gadolinium trichloride (GdCl3; 10 µmol/L), which can inhibit both SACC and ER Ca2+ channels,24 significantly inhibited both early and late phases (Figure 3B and D, and Supplementary material online, Figure S2). Interestingly, 10 µmol/L of Nif pre-treatment did not have any significant effect on the [Ca2+]i increase upon HSS, suggesting that VOC may play a minor role herein (Figure 3C and D). BAECs expressing the ER calcium biosensor were then pre-treated with STM or GdCl3. STM has no significant effect on the HSS-induced calcium decrease inside ER (Supplementary material online, Figure S3). In contrast, GdCl3 appeared to affect the calcium traffic across both the PM (Figure 3A and D) and ER membrane (Supplementary material online, Figure S3), which is consistent with previous reports.24 Therefore, these results suggest that SACCs on the PM may constitute the main contributors to the early phase of the [Ca2+]i increase in response to HSS application.
Figure 3.

The early phase of HSS-induced [Ca2+]i increase in BAEC is mainly regulated by Ca2+ influx across the PM. (A–C): the time course represents the cytoplasmic Ca2+ concentration in cells pre-treated for 1 h with (A) 200 µmol/L STM, a SACC blocker (n= 6); (B) 5 µmol/L GdCl3, an inhibitor affecting both SACC and ER Ca2+ channels (n= 12); or (C) 10 µmol/L nifedipine, a l-type voltage-operated Ca2+ channel blocker (n= 4) before the application of HSS. (D) Bar graphs (mean ± SEM) represent the max values of YPet/ECFP emission ratios measuring the cytoplasmic Ca2+ concentration in the early and the late phase in BAECs upon HSS application under different conditions as indicated. *Represents a P < 0.05 when comparing with the same phase in the control group; + represents a P< 0.05 when comparing with different phases in the same group. The standard error (SEM) bars were shown at every 20 measurements on the averaged time courses.
3.5. The early phase of [Ca2+]i increase upon HSS is sensitive to the mechanical impact and dependent on the cytoskeletal integrity
We further examined the mechanism regulating the HSS-induced [Ca2+]i increase. The magnitude of shear stress was altered to examine its effect on the HSS-induced [Ca2+]i increase. Shear stress with a lower magnitude of 35 dyn/cm2 caused a significantly weaker [Ca2+]i response in the early phase but a similar response in the late phase, when compared with the HSS of 65 dyn/cm2. Shear stress at 15 dyn/cm2 caused lower responses in both early and late phases (Figure 4A and B), with the response having decreased more in the early phase (38.7%) than in the late phase (27.9%; Figure 4B, and Supplementary material online, Figure S4A). These results suggest that the early phase of [Ca2+]i increase and hence the calcium influx via the PM may be more sensitive to the direct mechanical impact caused by shear stress.
Figure 4.

The roles of shear stress magnitude and cytoskeleton on the HSS-induced [Ca2+]i increase in BAECs. (A): the time courses of the YPet/ECFP emission ratio of the calcium biosensor measuring the cytoplasmic Ca2+ concentration in BAECs exposed to the shear stress with magnitude of 65 (n= 10), 35 (n= 5), or 15 (n= 6) dyn/cm2. (B): bar graphs (mean ± SEM) represent the max values of YPet/ECFP emission ratios measuring the cytoplasmic Ca2+ concentration in the early and the late phase in BAECs upon HSS application under different conditions as indicated. (C–E): the time course represents the cytoplasmic Ca2+ concentration in BAECs pre-treated for 1 h with (C) Cyto D (2 µmol/L, n= 7), (D) ML-7 (5 µmol/L, n= 6), or (E) Noco (1 µmol/L, n= 4) before HSS application. (F) Bar graphs (mean ± SEM) represent the max values of YPet/ECFP emission ratios measuring the cytoplasmic Ca2+ concentration in the early and the late phase in BAECs upon HSS application under different conditions as indicated. *Represents a P< 0.05 when comparing with the same phase in the control group; + represents P< 0.05 when comparing with different phases in the same group. The standard error (SEM) bars were shown at every 20 measurements on the averaged time courses.
Since the cytoskeleton provides the mechanical support for the cell, we hypothesized that the cytoskeleton may mediate the HSS-induced [Ca2+]i increase. BAECs expressing the calcium biosensor were pre-treated with 2 µmol/L Cytochalasin D (Cyto D) for 1 h to destroy the F-actin stress fibres before flow application. Cyto D had more significant inhibition of the early phase [Ca2+]i response after flow (Figure 4C and F, and Supplementary material online, Figure S4B). The inhibition of myosin light chain kinase (MLCK) by 5 µmol/L ML-7, which can prevent the active force transmission through F-actin while maintaining the structure of the filament system, significantly inhibited the early, but not the late phase of the [Ca2+]i increase (Figure 4D and F, and Supplementary material online, Figure S4B). Similarly, the disruption of the microtubule by 1 µmol/L of nocodazole (Noco) also inhibited the early phase, but not the late phase (Figure 4E and F, and Supplementary material online, Figure S4B). Therefore, the cytoskeleton and acto-myosin apparatus play crucial roles in mediating the early phase of HSS-induced [Ca2+]i increase.
To confirm whether the simultaneous perturbation of both mechanical properties and ER calcium release can affect both early and late phases, cells were pre-treated with 2-APB and Cyto D simultaneously. The [Ca2+]i response in both phases was inhibited (Figure 5A and D, and Supplementary material online, Figure S4C). 2-APB and the removal of extracellular calcium can also eliminate the HSS-induced [Ca2+]i response (Figure 5B and D, and Supplementary material online, Figure S4C). In contrast, Cyto D and the removal of extracellular calcium together only eliminate the early, but not the late, phase (Figure 5C and E, and Supplementary material online, Figure S4C). These results confirmed that the HSS-induced [Ca2+]i increase consists of a late phase from the ER-stored calcium release, and an early phase from the calcium influx through the PM, which is dependent on the cytoskeleton.
Figure 5.

The combined effect of inhibition of both calcium influx and ER calcium release on the HSS-induced [Ca2+]i increase in BAECs. (A–C): the YPet/ECFP emission ratio time courses of the calcium biosensor representing the cytoplasmic Ca2+ concentration in BAECs pre-treated with (A) 2-APB and Cyto D together (n= 6), or (B) 2-APB and Ca2+-free DMEM together (n= 8), or (C) Cyto D and Ca2+-free DMEM together (n= 5) before HSS application. (D and E): bar graphs (mean ± SEM) represent the max values of YPet/ECFP emission ratios measuring the cytoplasmic Ca2+ concentration in the early and the late phase in BAECs upon HSS application under different conditions as indicated. *Represents a P< 0.05 when comparing with the same phase in the control group; + represents a P< 0.05 when comparing with different phases in the same group. The standard error (SEM) bars were shown at every 20 measurements on the averaged time courses.
3.6. Src and phospholipase C mediate the late phase of HSS-induced [Ca2+]i increase
We then explored the molecular mechanisms by which the late phase of HSS-induced [Ca2+]i increase is regulated. Since Src can phosphorylate phospholipase C (PLC)-γ and induce IP3 production to affect the ER calcium release,25 the role of Src in mediating the HSS-induced [Ca2+]i increase was examined. BAECs were transfected with a membrane-bound Src FRET biosensor, Kras-Src biosensor, which can visualize the Src activity with high specificity and sensitivity at the PM where Src activation occurs.26 Src activity changed gradually upon the HSS application, which can be blocked by a specific Src inhibitor 4-amino-5-(4-methylphenyl)-7-(t-butyl)pyrazolo[3,4-d]- pyrimidine (PP1) (Figure 6A). This HSS-induced FRET response can be eliminated if the biosensor SH2 domain is mutated to disable the FRET function but leave the fluorescent proteins intact (data not shown).26 This result confirmed that the FRET responses observed with the wild-type biosensor are not due to non-specific signals originated from the morphological changes of the cells upon mechanical loading. When Src was inhibited by PP1, the late phase of the HSS-induced [Ca2+]i increase was inhibited while the early phase was only moderately affected (Figure 6B and C, and Supplementary material online, Figure S5A, Movie S7). The inhibition of PLC with 10 µmol/L U73122 also inhibited the late phase (Figure 6B and C, and Supplementary material online, Figure S5A, Movie S8). Further results revealed that the inhibition of either PP1 or U73122 can block the HSS-induced ER calcium release (Figure 6D and E, and Supplementary material online, Figure S5B). These results indicate that the late phase of the HSS-induced [Ca2+]i increase is mediated by Src and PLC, which regulate the ER calcium release.
Figure 6.

The HSS-induced [Ca2+]i increase in BAECs was affected by the Src and PLC signalling pathway. (A) The representative image and time course represent the Src activation before and after HSS stimulation in BAECs pre-treated with (n= 6) or without (n= 9) 50 µmol/L PP1 for 1 h. (B and D) The time courses represent the (B) cytoplasmic or (D) ER Ca2+ concentration in BAECs pre-treated with 50 µmol/L PP1 (n= 9) for 1 h, or 10 µmol/L U73122 (n= 17) for 15 min before HSS application. (C and E) Bar graphs (mean ± SEM) represent the max values of YPet/ECFP emission ratios measuring the (C) cytoplasmic Ca2+ concentration in the early and the late phase or (E) ER Ca2+ concentration after HSS application for 20 min in BAECs pre-treated with PP1 or U73122. *Represents a P< 0.05 when comparing with the control group, and + represents a P< 0.05 when comparing with different phases in the same group. The standard error (SEM) bars were shown at every (B) 5 or (A and D) 10 measurements on the averaged time courses. All the scar bars represent the length of 20 µm.
4. Discussion
Our results indicate that the HSS-induced [Ca2+]i increase clearly consists of two sources, the calcium influx from the extracellular space and the calcium release from ER. More importantly, the calcium from these two sources contributes to the [Ca2+]i increase at different phases. The early phase mainly originates from the calcium influx through the PM channels such as SACC, dependent on direct mechanical impact and cytoskeletal integrity, and contributing to the acute response of the HSS application. In contrast, the late phase is mainly from the ER calcium release, which is dependent on the biochemical signalling transduction through Src kinase, PLC, and ER membrane channel IP3R (Supplementary material online, Figure S6). This late phase may play roles in regulating the continuous cellular adjustments upon HSS application. Therefore, the calcium signalling in response to HSS involves multiple mechanisms well co-ordinated in space and time, which may contribute to the angiogenesis and atherosclerosis. Our findings can thus shed new light on our systematic understanding of the molecular mechanisms regulating cardiovascular diseases.
4.1. The roles of membrane channels
When an HSS with higher magnitude was applied onto the surface of ECs, a stronger Ca2+ response was observed to transmit the mechanical signals (Figure. 4A and B). Consistently, faster and longer durations of [Ca2+]i increase were observed upon HSS application, while slower and shorter durations occurred when lower shear stress was applied.7,10 It is hence clear that shear stress had a direct mechanical impact to cause a rapid calcium influx and [Ca2+]i increase, possibly via the PM channels. This direct mechanical impact may mechanically alter some of the PM channels to allow the calcium influx. Indeed, the inhibition of SACC, but not VOC, significantly inhibited the early phase of the HSS-induced [Ca2+]i increase. While the detailed mechanism by which mechanical impact alters the SACC functions remains unclear, it is believed that mechanical shear stress can cause a mechanical alteration on the PM and actin cytoskeleton, which can transmit this mechanical force to the membrane channel SACC physically coupled to the membrane and the cytoskeleton. This transmitted mechanical perturbation on SACC can cause it to undergo a conformational change or be stretched open passively to activate SACC and trigger the calcium influx (Figure 4).27
4.2. The roles of the cytoskeleton
Calcium channels on the PM have been reported to be associated with the cytoskeleton.28 In fact, the disruption of the actin cytoskeleton by Cyto D inhibited the effect of hyperosmotic stress on [Ca2+]i29 and the capacitative Ca2+ entry induced by the depletion of Ca2+ stores with thapsigargin.30 Disruption of actin filaments or microtubules also prevented the IP3R-mediated Ca2+ waves stimulated by ATP or bradykinin.31 This is consistent with our finding that the early phase of the HSS-induced [Ca2+]i increase can be significantly inhibited when actin filaments were disrupted (Figure 4). ML-7, which inhibits MLCK and acto-myosin contractility to interrupt force transmission, had shown similar inhibitory effects. Together with the results of shear stress with varying magnitudes, it is clear that the cytoskeleton and the acto-myosin apparatus play crucial roles in the early phase of the HSS-induced [Ca2+]i increase, most likely by mediating the mechanical tension transmission to open the SACC on the cell membrane.
It is interesting to note that the disruption of F-actin could inhibit not only the early but also the late phase. It is possible that actin disruption also affects the ER-stored calcium release via the coupling between the PM channels and IP3R. Indeed, the modulation of cortical F-actin by calyculin A can decouple ER from PM Ca2+ channels to prevent the activation of SOC and TRP3 channels,28 as well as the IP3R-activated [Ca2+]i increase.32 The blockage of IP3R with 2-APB can also rapidly deactivate SOC,28 suggesting that membrane channels and IP3R may be tightly coupled together, possibly via the actin cytoskeleton.
Recent evidence suggests that microtubules and actin filaments do not function in complete isolation. Instead, microtubules and actin filaments are physically coupled with each other, mediated by possible static interactions via molecular complexes or individual molecules that can bind both filaments simultaneously.33 Furthermore, guanine exchange factors (GEFs) of small GTPase RhoA can be coupled to microtubules. Hence, the disruption of microtubules may affect the subcellular functions of RhoA by releasing GEFs and subsequently acto-myosin contractility and actin stress fibres to impact on the membrane-bound channels. Therefore, the inhibition of actin filaments, MLCK, or microtubules by Cyto D, ML-7, or Noco may alter the mechanical transmission supported by the acto-myosin network to regulate the intracellular calcium under flow stimulation.
4.3. Src and PLC mediate the late phase of the HSS-induced [Ca2+]i increase
Shear stress has been shown to activate Src.34 Src can also phosphorylate PLC-γ and induce IP3 production to regulate the ER calcium release.25 So Src is a natural candidate to link the mechanical impact on the PM and the calcium release from ER under HSS. Indeed, we showed that HSS can activate Src and the inhibition of Src can significantly suppress the late phase of the HSS-induced [Ca2+]i increase. This clearly indicates that the HSS-induced Src activation is essential for the late phase of calcium response. This role of Src under HSS may be mediated by the PLC activation.35 Since the linker region connecting the catalytic domain of PLC-γ36 contains two Src homology SH2 domains and one SH3 domain,37 Src may bind to this exposed docking region to phosphorylate and activate PLC-γ, which further leads to the production of IP3 and the ER-stored calcium release.25 Consistently, our results confirmed that the PLC inhibitor U73122 can block the Src-mediated late phase of the HSS-induced [Ca2+]i increase.
It is possible that shear stress may induce the engagement of integrins38 to subsequently activate Src.39,40 Several putative mechanisms have been proposed for the Src activation induced by shear stress and integrin engagement. (i) The shear-modulated cytoplasmic tail of integrin β3 may directly recruit Src kinase through its SH3 domain. This action disrupts the intra-molecular interactions between different domains of Src and activates Src41. (ii) The shear-activated integrins can cause the myristoylation-mediated translocation of Src to focal adhesion sites.40 FAK Y397, in a high-affinity pYAEI context, competes with the Src C-terminal pY527 for its intra-molecular interaction and thus activates Src.40,42 (iii) Recent evidence has also shown that the integrin engagement leads to the association of integrin αvβ3 with receptor-linked protein-tyrosine phosphatases (RPTP) α, a well-characterized activator of Src family kinases.43 It is possible that the shear-regulated integrins recruit RPTPα to de-phosphorylate the Y527 on the C-terminal tail of Src and release it from the kinase domain, thus activating Src.43 Our results indicate that the inhibition of FAK activity does not block the Src activation upon HSS application (data not shown), suggesting that the shear-induced Src activation is not dependent on FAK activity. Therefore, molecular/structural interactions other than the FAK signalling pathway may couple the integrin and Src activation under flow, possibly via the integrin/Src binding or integrin/RPTPα pathway.
4.4. The role of G proteins
G proteins were found to mediate the regulation of [Ca2+]i in response to shear stress in D. discoideum cells.8 The inhibition of either Gs or Gi in BAECs by cholera toxin or pertussis toxin,44 however, did not affect the shear-induced [Ca2+]i increase (data not shown). This is consistent with a previous report that the manipulation of G proteins did not affect the stretch-induced calcium responses in myocytes.45 Other reports indicate that G proteins may modulate the voltage-operated calcium channels (VOC) in various types of cells.45–49 Since VOC channels play a minor role in our system, these reports support the note that G proteins may not have a significant contribution to the shear-induced [Ca2+]i increase in BAECs.
4.5. Non-specific oscillation of intracellular calcium
Non-specific periodic intracellular calcium oscillation was reported as a common calcium alternation in various cell types such as mesenchymal stem cells,4,50 astrocyte,51 and myofibroblasts.52 These calcium oscillations were shown to correlate with micro-contractile events of dorsal stress fibres,52 which are dependent on RhoA-activated kinase (ROCK), a downstream effector molecule of RhoA.4,44 However, these non-specific oscillations of intracellular calcium levels were not observed in BAECs (data not shown). This discrepancy of calcium oscillation in different cell types may reflect the different regulation mechanisms of calcium signals, which can be governed by different membrane potentials, physical structures, or structural supports for the membrane channels in different cell types.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Conflict of interest: none declared.
Funding
This work was supported in part by grants from National Institute of Health (NIH) HL098472, CA139272, and NS063405; National Science Foundation (NSF) CBET0846429 and CMMI0800870; the Wallace H. Coulter Foundation; and National Natural Science Foundation of China (NSFC) 10972139.
Supplementary Material
References
- 1.Sho E, Sho M, Singh TM, Nanjo H, Komatsu M, Xu C, et al. Arterial enlargement in response to high flow requires early expression of matrix metalloproteinases to degrade extracellular matrix. Exp Mol Pathol. 2002;73:142–153. doi: 10.1006/exmp.2002.2457. [DOI] [PubMed] [Google Scholar]
- 2.Meng H, Wang Z, Hoi Y, Gao L, Metaxa E, Swartz DD, et al. Complex hemodynamics at the apex of an arterial bifurcation induces vascular remodeling resembling cerebral aneurysm initiation. Stroke. 2007;38:1924–1931. doi: 10.1161/STROKEAHA.106.481234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Metaxa E, Meng H, Kaluvala SR, Szymanski MP, Paluch RA, Kolega J. Nitric oxide-dependent stimulation of endothelial cell proliferation by sustained high flow. Am J Physiol Heart Circ Physiol. 2008;295:H736–H742. doi: 10.1152/ajpheart.01156.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim TJ, Seong J, Ouyang M, Sun J, Lu S, Hong JP, et al. Substrate rigidity regulates Ca2+ oscillation via RhoA pathway in stem cells. J Cell Physiol. 2009;218:285–293. doi: 10.1002/jcp.21598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sato H, Takino T, Okada Y, Cao J, Shinagawa A, Yamamoto E, et al. A matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature. 1994;370:61–65. doi: 10.1038/370061a0. [DOI] [PubMed] [Google Scholar]
- 6.Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86:279–367. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- 7.Sharma R, Yellowley CE, Civelek M, Ainslie K, Hodgson L, Tarbell JM, et al. Intracellular calcium changes in rat aortic smooth muscle cells in response to fluid flow. Ann Biomed Eng. 2002;30:371–378. doi: 10.1114/1.1470179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fache S, Dalous J, Engelund M, Hansen C, Chamaraux F, Fourcade B, et al. Calcium mobilization stimulates Dictyostelium discoideum shear-flow-induced cell motility. J Cell Sci. 2005;118:3445–3457. doi: 10.1242/jcs.02461. [DOI] [PubMed] [Google Scholar]
- 9.Schwarz G, Callewaert G, Droogmans G, Nilius B. Shear stress-induced calcium transients in endothelial cells from human umbilical cord veins. J Physiol. 1992;458:527–538. doi: 10.1113/jphysiol.1992.sp019432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kwan HY, Leung PC, Huang Y, Yao X. Depletion of intracellular Ca2+ stores sensitizes the flow-induced Ca2+ influx in rat endothelial cells. Circ Res. 2003;92:286–292. doi: 10.1161/01.res.0000054625.24468.08. [DOI] [PubMed] [Google Scholar]
- 11.Hoyer J, Kohler R, Distler A. Mechanosensitive Ca2+ oscillations and STOC activation in endothelial cells. FASEB J. 1998;12:359–366. doi: 10.1096/fasebj.12.3.359. [DOI] [PubMed] [Google Scholar]
- 12.Nilius B, Droogmans G. Ion channels and their functional role in vascular endothelium. Physiol Rev. 2001;81:1415–1459. doi: 10.1152/physrev.2001.81.4.1415. [DOI] [PubMed] [Google Scholar]
- 13.Oike M, Droogmans G, Nilius B. Mechanosensitive Ca2+ transients in endothelial cells from human umbilical vein. Proc Natl Acad Sci USA. 1994;91:2940–2944. doi: 10.1073/pnas.91.8.2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Woo K, Dutta AK, Patel V, Kresge C, Feranchak AP. Fluid flow induces mechanosensitive ATP release, calcium signalling and Cl− transport in biliary epithelial cells through a PKCzeta-dependent pathway. J Physiol. 2008;586:2779–2798. doi: 10.1113/jphysiol.2008.153015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kwan HY, Huang Y, Yao X. Cyclic nucleotides and Ca2+ influx pathways in vascular endothelial cells. Clin Hemorheol Microcirc. 2007;37:63–70. [PubMed] [Google Scholar]
- 16.Brakemeier S, Eichler I, Hopp H, Kohler R, Hoyer J. Up-regulation of endothelial stretch-activated cation channels by fluid shear stress. Cardiovasc Res. 2002;53:209–218. doi: 10.1016/s0008-6363(01)00476-x. [DOI] [PubMed] [Google Scholar]
- 17.Kohler R, Heyken WT, Heinau P, Schubert R, Si H, Kacik M, et al. Evidence for a functional role of endothelial transient receptor potential V4 in shear stress-induced vasodilatation. Arterioscler Thromb Vasc Biol. 2006;26:1495–1502. doi: 10.1161/01.ATV.0000225698.36212.6a. [DOI] [PubMed] [Google Scholar]
- 18.Oancea E, Wolfe JT, Clapham DE. Functional TRPM7 channels accumulate at the plasma membrane in response to fluid flow. Circ Res. 2006;98:245–253. doi: 10.1161/01.RES.0000200179.29375.cc. [DOI] [PubMed] [Google Scholar]
- 19.Nguyen AW, Daugherty PS. Evolutionary optimization of fluorescent proteins for intracellular FRET. Nat Biotechnol. 2005;23:355–360. doi: 10.1038/nbt1066. [DOI] [PubMed] [Google Scholar]
- 20.Ouyang M, Sun J, Chien S, Wang Y. Determination of hierarchical relationship of Src and Rac at subcellular locations with FRET biosensors. Proc Natl Acad Sci USA. 2008;105:14353–14358. doi: 10.1073/pnas.0807537105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fillinger MF, Sampson LN, Cronenwett JL, Powell RJ, Wagner RJ. Coculture of endothelial cells and smooth muscle cells in bilayer and conditioned media models. J Surg Res. 1997;67:169–178. doi: 10.1006/jsre.1996.4978. [DOI] [PubMed] [Google Scholar]
- 22.Nackman GB, Fillinger MF, Shafritz R, Wei T, Graham AM. Flow modulates endothelial regulation of smooth muscle cell proliferation: a new model. Surgery. 1998;124:353–360. discussion 360–351. [PubMed] [Google Scholar]
- 23.Wang Y, Miao H, Li S, Chen KD, Li YS, Yuan S, et al. Interplay between integrins and FLK-1 in shear stress-induced signaling. Am J Physiol Cell Physiol. 2002;283:C1540–C1547. doi: 10.1152/ajpcell.00222.2002. [DOI] [PubMed] [Google Scholar]
- 24.Klusener B, Boheim G, Liss H, Engelberth J, Weiler EW. Gadolinium-sensitive, voltage-dependent calcium release channels in the endoplasmic reticulum of a higher plant mechanoreceptor organ. EMBO J. 1995;14:2708–2714. doi: 10.1002/j.1460-2075.1995.tb07271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tokmakov AA, Sato KI, Iwasaki T, Fukami Y. Src kinase induces calcium release in Xenopus egg extracts via PLCgamma and IP3-dependent mechanism. Cell Calcium. 2002;32:11–20. doi: 10.1016/s0143-4160(02)00078-7. [DOI] [PubMed] [Google Scholar]
- 26.Wang Y, Botvinick EL, Zhao Y, Berns MW, Usami S, Tsien RY, et al. Visualizing the mechanical activation of Src. Nature. 2005;434:1040–1045. doi: 10.1038/nature03469. [DOI] [PubMed] [Google Scholar]
- 27.Formigli L, Meacci E, Sassoli C, Squecco R, Nosi D, Chellini F, et al. Cytoskeleton/stretch-activated ion channel interaction regulates myogenic differentiation of skeletal myoblasts. J Cell Physiol. 2007;211:296–306. doi: 10.1002/jcp.20936. [DOI] [PubMed] [Google Scholar]
- 28.Ma HT, Patterson RL, van Rossum DB, Birnbaumer L, Mikoshiba K, Gill DL. Requirement of the inositol trisphosphate receptor for activation of store-operated Ca2+ channels. Science. 2000;287:1647–1651. doi: 10.1126/science.287.5458.1647. [DOI] [PubMed] [Google Scholar]
- 29.Rozanov DV, Deryugina EI, Monosov EZ, Marchenko ND, Strongin AY. Aberrant, persistent inclusion into lipid rafts limits the tumorigenic function of membrane type-1 matrix metalloproteinase in malignant cells. Exp Cell Res. 2004;293:81–95. doi: 10.1016/j.yexcr.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 30.Holda JR, Blatter LA. Capacitative calcium entry is inhibited in vascular endothelial cells by disruption of cytoskeletal microfilaments. FEBS Lett. 1997;403:191–196. doi: 10.1016/s0014-5793(97)00051-3. [DOI] [PubMed] [Google Scholar]
- 31.Beliveau E, Guillemette G. Microfilament and microtubule assembly is required for the propagation of inositol trisphosphate receptor-induced Ca2+ waves in bovine aortic endothelial cells. J Cell Biochem. 2009;106:344–352. doi: 10.1002/jcb.22011. [DOI] [PubMed] [Google Scholar]
- 32.Bose DD, Thomas DW. The actin cytoskeleton differentially regulates NG115–401L cell ryanodine receptor and inositol 1,4,5-trisphosphate receptor induced calcium signaling pathways. Biochem Biophys Res Commun. 2009;379:594–599. doi: 10.1016/j.bbrc.2008.12.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rodriguez OC, Schaefer AW, Mandato CA, Forscher P, Bement WM, Waterman-Storer CM. Conserved microtubule-actin interactions in cell movement and morphogenesis. Nat Cell Biol. 2003;5:599–609. doi: 10.1038/ncb0703-599. [DOI] [PubMed] [Google Scholar]
- 34.Jalali S, Li YS, Sotoudeh M, Yuan S, Li S, Chien S, et al. Shear stress activates p60src-Ras-MAPK signaling pathways in vascular endothelial cells. Arterioscler Thromb Vasc Biol. 1998;18:227–234. doi: 10.1161/01.atv.18.2.227. [DOI] [PubMed] [Google Scholar]
- 35.Schaloske R, Schlatterer C, Malchow D. A Xestospongin C-sensitive Ca(2+) store is required for cAMP-induced Ca(2+) influx and cAMP oscillations in Dictyostelium. J Biol Chem. 2000;275:8404–8408. doi: 10.1074/jbc.275.12.8404. [DOI] [PubMed] [Google Scholar]
- 36.Rhee SG, Bae YS. Regulation of phosphoinositide-specific phospholipase C isozymes. J Biol Chem. 1997;272:15045–15048. doi: 10.1074/jbc.272.24.15045. [DOI] [PubMed] [Google Scholar]
- 37.Carpenter G, Ji Q. Phospholipase C-gamma as a signal-transducing element. Exp Cell Res. 1999;253:15–24. doi: 10.1006/excr.1999.4671. [DOI] [PubMed] [Google Scholar]
- 38.Tzima E, del Pozo MA, Shattil SJ, Chien S, Schwartz MA. Activation of integrins in endothelial cells by fluid shear stress mediates Rho-dependent cytoskeletal alignment. EMBO J. 2001;20:4639–4647. doi: 10.1093/emboj/20.17.4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaplan KB, Swedlow JR, Morgan DO, Varmus HE. c-Src enhances the spreading of src-/- fibroblasts on fibronectin by a kinase-independent mechanism. Genes Dev. 1995;9:1505–1517. doi: 10.1101/gad.9.12.1505. [DOI] [PubMed] [Google Scholar]
- 40.Thomas SM, Brugge JS. Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol. 1997;13:513–609. doi: 10.1146/annurev.cellbio.13.1.513. [DOI] [PubMed] [Google Scholar]
- 41.Arias-Salgado EG, Lizano S, Sarkar S, Brugge JS, Ginsberg MH, Shattil SJ. Src kinase activation by direct interaction with the integrin beta cytoplasmic domain. Proc Natl Acad Sci USA. 2003;100:13298–13302. doi: 10.1073/pnas.2336149100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eide BL, Turck CW, Escobedo JA. Identification of Tyr-397 as the primary site of tyrosine phosphorylation and pp60src association in the focal adhesion kinase, pp125FAK. Mol Cell Biol. 1995;15:2819–2827. doi: 10.1128/mcb.15.5.2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.von Wichert G, Jiang G, Kostic A, De Vos K, Sap J, Sheetz MP. RPTP-alpha acts as a transducer of mechanical force on alphav/beta3-integrin-cytoskeleton linkages. J Cell Biol. 2003;161:143–153. doi: 10.1083/jcb.200211061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gil-Longo J, Dufour MN, Guillon G, Lugnier C. G proteins in aortic endothelial cells and bradykinin-induced formation of nitric oxide. Eur J Pharmacol. 1993;247:119–125. doi: 10.1016/0922-4106(93)90068-k. [DOI] [PubMed] [Google Scholar]
- 45.Ikeda SR, Dunlap K. Voltage-dependent modulation of N-type calcium channels: role of G protein subunits. Adv Second Messenger Phosphoprotein Res. 1999;33:131–151. doi: 10.1016/s1040-7952(99)80008-1. [DOI] [PubMed] [Google Scholar]
- 46.Dolphin AC. G protein modulation of voltage-gated calcium channels. Pharmacol Rev. 2003;55:607–627. doi: 10.1124/pr.55.4.3. [DOI] [PubMed] [Google Scholar]
- 47.Dolphin AC. Mechanisms of modulation of voltage-dependent calcium channels by G proteins. J Physiol. 1998;506:3–11. doi: 10.1111/j.1469-7793.1998.003bx.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bean BP. Neurotransmitter inhibition of neuronal calcium currents by changes in channel voltage dependence. Nature. 1989;340:153–156. doi: 10.1038/340153a0. [DOI] [PubMed] [Google Scholar]
- 49.Mirotznik RR, Zheng X, Stanley EF. G-Protein types involved in calcium channel inhibition at a presynaptic nerve terminal. J Neurosci. 2000;20:7614–7621. doi: 10.1523/JNEUROSCI.20-20-07614.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun S, Liu Y, Lipsky S, Cho M. Physical manipulation of calcium oscillations facilitates osteodifferentiation of human mesenchymal stem cells. FASEB J. 2007;21:1472–1480. doi: 10.1096/fj.06-7153com. [DOI] [PubMed] [Google Scholar]
- 51.Morita M, Higuchi C, Moto T, Kozuka N, Susuki J, Itofusa R, et al. Dual regulation of calcium oscillation in astrocytes by growth factors and pro-inflammatory cytokines via the mitogen-activated protein kinase cascade. J Neurosci. 2003;23:10944–10952. doi: 10.1523/JNEUROSCI.23-34-10944.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Castella LF, Buscemi L, Godbout C, Meister JJ, Hinz B. A new lock-step mechanism of matrix remodelling based on subcellular contractile events. J Cell Sci. 2010;123:1751–1760. doi: 10.1242/jcs.066795. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.