

Abstract
The transporter associated with antigen processing (TAP) is essential for intracellular transport of protein fragments into the endoplasmic reticulum for loading of major histocompatibility complex (MHC) class I molecules. On the cell surface, these peptide–MHC complexes are monitored by cytotoxic T lymphocytes. To study the ATP hydrolysis of TAP, we developed an enrichment and reconstitution procedure, by which we fully restored TAP function in proteoliposomes. A TAP-specific ATPase activity was identified that could be stimulated by peptides and blocked by the herpes simplex virus protein ICP47. Strikingly, the peptide-binding motif of TAP directly correlates with the stimulation of the ATPase activity, demonstrating that the initial peptide-binding step is responsible for TAP selectivity. ATP hydrolysis follows Michaelis–Menten kinetics with a maximal velocity Vmax of 2 μmol/min per mg TAP, corresponding to a turnover number of approximately 5 ATP per second. This turnover rate is sufficient to account for the role of TAP in peptide loading of MHC molecules and the overall process of antigen presentation. Interestingly, sterically restricted peptides that bind but are not transported by TAP do not stimulate ATPase activity. These results point to coordinated dialogue between the peptide-binding site, the nucleotide-binding domain, and the translocation site via conformational changes within the TAP complex.
Keywords: membrane proteins, transport mechanism, ATPase, antigen processing
An essential step in the major histocompatibility complex (MHC) class I-mediated cellular immune defense is the transport of antigenic peptides from the cytoplasm into the endoplasmic reticulum (ER) (reviewed by ref. 1). The transporter associated with antigen processing (TAP) translocates peptides that are mainly derived from the proteasomal degradation into the ER (reviewed by ref. 2). Subsequently, peptides bind to MHC class I molecules and are presented at the cell surface, where they are recognized by cytotoxic T lymphocytes.
TAP belongs to the family of ATP-binding cassette (ABC) transporters that, under consumption of ATP, translocate a vast spectrum of substrates across membranes (3). On the basis of sequence comparison, TAP fits in the B subfamily of human ABC transporters (www.gene.ucl.ac.uk/users/hester/abc.html), including the well-characterized P-glycoprotein (ABCB1). The TAP complex forms a heterodimer of TAP1 (ABCB2) and TAP2 (ABCB3), where each subunit comprises a hydrophobic transmembrane domain and a cytoplasmic nucleotide-binding domain (NBD) containing the Walker A/B and C-loop consensus sequences. The transmembrane domains are thought to form a channel by which peptides are transported across the ER membrane driven by ATP hydrolysis. The coupling between NBDs, the substrate-binding site, and the translocation pathway is not understood and is a central topic to be studied for all ABC transporters.
TAP is part of a macromolecular peptide translocation and loading complex that comprises TAP1, TAP2, tapasin, MHC class I heavy chain, and β2-microglobulin in stoichiometrically defined ratios (4). Tapasin acts as a linker between the MHC class I heterodimer and TAP, ensuring high loading efficiency. This linker is required for stabilizing the MHC class I loading complex and up-regulates the peptide transport by increasing the steady-state level of TAP (5, 6). Viral proteins also form complexes with TAP to inhibit peptide translocation by different mechanisms and subsequently help the virus to escape immune surveillance. The herpes simplex virus protein ICP47 inhibits peptide binding to TAP (7, 8).
ATP-independent binding of peptides with a preferential length of 8–16 amino acids to TAP is a prerequisite for peptide translocation into the ER (9). However, peptides with a length of up to 40 residues are transported with decreasing efficiency (10). Also, peptides with sterically restricted side chains are transported into the ER lumen, demonstrating the plasticity of this transport protein (11, 12). To guarantee the transport of this broad spectrum of epitopes across the ER membrane, the substrate specificity is condensed onto the three amino terminal residues and the carboxyl terminal residue of the substrate (13). Peptides are fixed by TAP on both ends and seem to form only minor contacts in between. From peptide-binding studies (11, 14) and fluorescence resonance energy transfer experiments (R.A. and R.T., unpublished data), we have evidence that only one single peptide-binding site exists, which is formed by cytosolic loop regions close to the NBD domains (15). Recently, kinetic studies showed that peptide binding to TAP is a two-step process with a fast association step followed by a slow structural rearrangement possibly triggering the ATP-dependent translocation (14).
In comparison to substrate specificity and ligand binding, little is known about the ATP-dependent translocation of peptides. In early studies, it has been shown that peptide transport requires ATP hydrolysis (16–18). Both NDBs bind ATP when expressed separately, but they do not hydrolyze ATP (19, 20). Both ATP-binding sites need to be functional, because mutation of TAP1 NBD leads to a loss of transport function (21). However, the NBDs are used not only for energizing peptide translocation but also for synchronizing the entire process of peptide translocation, loading, and dissociation of MHC molecules from the TAP complex (22). Moreover, ATP and ADP also have a stabilizing effect on TAP (23). Recent studies emphasize the interaction between the NBDs because residues adjacent to the NBD core are responsible for dimerization of TAP1 and TAP2 NBDs (24). Additional evidence for the interaction of the domains comes from the observation that the lateral diffusion of green fluorescent protein-tagged TAP in the ER membrane is reduced 2-fold only in the presence of both substrates, ATP and peptide, resembling a structural rearrangement in the transmembrane domain of TAP (25). All these characteristics point to the central role of ATP in substrate translocation, stabilization of the TAP heterodimer, and release of MHC class I molecules (26). Therefore, direct analysis of ATP hydrolysis is a keystone for understanding the TAP complex. So far, attempts to study the ATPase activity of TAP have failed. Here we have developed an enrichment and reconstitution protocol. With this system in hand, an ATP-dependent peptide transport of TAP in proteoliposomes could be demonstrated for the first time. Furthermore, we could identify and characterize the ATPase activity of TAP that is strictly controlled by peptides.
Materials and Methods
Cell Propagation and Microsome Preparation.
Human Burkitt lymphoma cells (Raji cells) were propagated in roller bottles at 37°C and 5% CO2 in RPMI medium 1640 (GIBCO/BRL) supplemented with 10% FCS, 1 mM sodium pyruvate, and 40 units/ml each of penicillin and streptomycin. Microsomes were prepared as described previously (27). Microsomal membranes were resuspended with Hepes buffer (20 mM Hepes–NaOH/145 mM NaCl/2 mM KCl/1 mM 1,4-dithio-dl-threitol/20% glycerol, pH 7.4). Protein concentration of the microsomes was determined by microbicinchoninic acid assay (Pierce) and adjusted to 6 mg/ml. Aliquots were frozen in liquid nitrogen and stored at −80°C.
Reconstitution of the TAP Complex.
Microsomal pellets were thawed on ice, washed, and resuspended with solubilization buffer (Hepes buffer supplemented with 0.5 mM MnCl2 and 2 mM MgCl2, pH 7.4). For solubilization, n-decyl-β-d-maltopyranoside (DM) (Calbiochem) were added to reach a ρ value of 2, where ρ is given by:
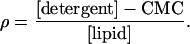 |
1 |
CMC is the critical micellar concentration of the detergent (1.6 mM for DM). In microsomes, the lipid-to-protein ratio (wt/wt) was assumed to be one. The mixture was incubated for 20 min at 4°C, and insoluble material was removed by centrifugation at 23,000 × g for 15 min. Solubilized TAP complex was purified by Superose 6 PC3.2/30 (Amersham Pharmacia Biotech) equilibrated with solubilization buffer containing 2.4 mM DM. TAP-containing fractions were pooled and subsequently used for reconstitution.
For liposome preparation, a lipid mixture containing 23% (wt/wt) cholesterol, 10% (wt/wt) phosphatidic acid, and 67% (wt/wt) phosphatidylcholine (Avanti Polar Lipids) was dissolved in chloroform. The organic solvent was removed under a stream of nitrogen, and lipids were dried in a vacuum. The lipid film was hydrated in Hepes buffer yielding a lipid concentration of 8 mg/ml. Liposomes were extruded through a 200-nm-pore-size polycarbonate filter by using a LipoFast-Extruder (Avestin, Ottawa) followed by four cycles of freeze and thaw. For reconstitution, a pellet of 200 μl unilamellar liposomes was resuspended with 180 μl of size-exclusion chromatography-purified TAP and incubated for 20 min at 4°C. The ρ value for reconstitution was optimized to 0.14. Detergent was removed on a 15-ml Sephadex G50 fine (Sigma) column with Hepes buffer containing 0.2 mM ATP and 2 mM MgCl2. In the exclusion volume, turbid fractions were collected and centrifuged for 15 min at 23,000 × g. Finally, proteoliposomes were resuspended in Hepes buffer for further analysis.
Peptide Binding and Transport.
All peptides were synthesized by solid-phase technique by applying conventional Fmoc chemistry. The branched peptides were a gift of J. Neefjes (Cancer Institute, Amsterdam) (12). Peptides were radiolabeled with 125I, as reported previously (27). The peptide-binding affinity (KD) and the amount of TAP in microsomes or proteoliposomes were determined in solubilization buffer by saturation-binding assays as described (11, 28). The data were fitted by Eq. 2:
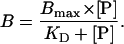 |
2 |
B is the amount of bound peptide, and [P] is the total peptide concentration. To analyze the peptide transport, 50 μl of proteoliposomes were incubated with 1 μM of 125I-labeled RRYNASTE and 3 mM ATP or ADP in 100 μl solubilization buffer for 2 minutes at 32°C. The reaction was quenched by adding 500 μl ice-cold solubilization buffer containing 9 μM of nonlabeled peptide (RRYQKSTEL). After 15 min of incubation on ice, the samples were centrifuged for 8 min (23,000 × g, 4°C). The pellet was resuspended with 600 μl of solubilization buffer and pelleted again. The supernatant was removed, and the transported substrate was determined by γ-counting.
ATPase Assay.
To determine the ATPase activity of reconstituted TAP, 30 μl of solubilization buffer supplemented with 1.8 mM ouabain/9 mM NaN3 and a mixture of ATP (5 mM) and [γ-32P]ATP (73 fM; 6.7 μCi) (Amersham Pharmacia Biotech) was incubated for 1 min at 32°C. The reaction was started by addition of 20 μl of liposomes containing reconstituted TAP and incubated 4 min. Adding 1.5 ml of reagent A (2.5 M H2SO4/1% ammonium molybdate) stopped the reaction. Fifteen microliters of 20 mM H3PO4 and 3 ml of reagent B (isobutanol, cyclohexane, acetone, reagent A in a ratio of 5:5:1:0.1) were added and vigorously vortexed. After phase separation, 1 ml of the upper organic phase was mixed with scintillation fluid (Microscint, Packard Bioscience) to determine the release of inorganic phosphate by γ-counting. For calculation of Km.pep, Vmax, and KM, kinetic data were fitted by Eq. 3:
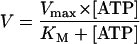 |
3 |
Results
Reconstitution of the TAP Complex.
So far, all attempts to analyze the ATPase activity of TAP have failed because of the high background activity of other ATPases present in total membrane or microsomal preparations. To circumvent this problem, we developed an enrichment and reconstitution method to analyze TAP function in proteoliposomes. To obtain large quantities of TAP for biochemical characterization, we chose human Burkitt lymphoma cells (Raji cells) that show a high level of TAP expression even without γ-interferon stimulation. Microsomes from Raji cells were solubilized with DM, which was found to be optimal for solubilization of TAP (28). To reduce the background activity from other ATPases, detergent-solubilized proteins were separated by size exclusion chromatography (Fig. 1A). Each fraction was analyzed by immunoblotting and peptide-binding assays after reconstitution into proteoliposomes. The TAP complex consisting of TAP1 (Mw,app, 71 kDa) and TAP2 (Mw,app, 75 kDa) was eluted within a narrow region, corresponding to fractions 19–21 (Fig. 1B). In contrast to tapasin (Mw,app, 48 kDa) that was coeluted with TAP, MHC class I heavy chain was detected in fractions 32. Thus DM seems to disrupt the interaction between TAP and MHC I molecules, whereas digitonin can preserve the interaction (29).
Figure 1.

Rapid and efficient reconstitution of TAP into proteoliposomes. Raji microsomes (6 mg/ml) were solubilized by DM (ρ = 2) and loaded on a Superose 6 column (A). Each fraction (60 μl) was analyzed by SDS/PAGE followed by immunoblotting by using specific monoclonal antibodies against TAP1 (mAb 148.3) (27), TAP2 (mAb 435.3) (9), MHC class I heavy chain (hc) (mAb A1.4) (38), and tapasin (mAb, R.T., unpublished work) (B). To study the efficiency of TAP reconstitution, equal aliquots of microsomal protein, after solubilization, isolation (fractions 19 to 21), and reconstitution into proteoliposomes, were analyzed by immunoblotting (C).
Partially purified TAP was reconstituted into liposomes by size-exclusion chromatography. In comparison to other established reconstitution methods such as dialysis, dilution, or hydrophobic adsorption to Bio-Beads (Bio-Rad), reconstitution by size exclusion combines two advantages: First, it is a very fast method preventing prolonged detergent exposure. Second, it can be used for detergents with low critical micellar concentration, such as DM or digitonin. Optimal conditions were observed when TAP is reconstituted into preformed unilamellar liposomes at defined phospholipid, cholesterol, and detergent ratios (for details see Materials and Methods). The optimized reconstitution procedure turned out to be very efficient. As demonstrated by immunodetection (Fig. 1C) and by the Bmax value of peptide binding, approximately 50% of microsomal TAP was reconstituted (Table 1). On the basis of these results and peptide-binding assays with semipermeabilized proteoliposomes (data not shown), the majority of TAP molecules (>95%) seems to have the same orientation with the peptide-binding site facing to the exterior of the proteoliposomes. In binding assays with radiolabeled peptides comprising various affinities for TAP, reconstituted TAP displayed the same ligand affinity as TAP in microsomes (Table 1). In conclusion, peptide affinity and TAP selectivity remain fully preserved after reconstitution into proteoliposomes.
Table 1.
TAP activity in microsomes and proteoliposomes
| Peptides | TAP in
microsomes
|
TAP in proteoliposomes
|
|||||
|---|---|---|---|---|---|---|---|
| Peptide binding
|
Peptide
binding
|
ATPase activity
|
|||||
| KD, nM | Bmax, cpm/ng | KD, nM | Bmax, cpm/ng | TAP recovery | Km.pep, nM | Vmax, μmol/min × mg TAP | |
| RRYQKSTEL | 154 ± 9 | 0.469 ± 0.004 | 164 ± 22 | 3.245 ± 0.219 | 49.7% | 161 ± 15 | 2.02 ± 0.13 |
| RRYNASTEL | 503 ± 54 | 0.465 ± 0.024 | 516 ± 52 | 3.184 ± 0.213 | 50.2% | 574 ± 134 | 1.93 ± 0.02 |
| EPGNTWDED | >106 | ND | >106 | ND | ND | >106 | ND |
ND, not determined.
KD and Bmax values for microsomes and proteoliposomes were determined by peptide-binding assays with radiolabeled peptides and fitted as a hyperbolic curve fit. Bmax is given per nanogram total protein. TAP recovery was calculated by comparing the total amount of binding sites before and after reconstitution. Km.pep and Vmax were determined by ATPase assays, as described in Materials and Methods.
Although substrate-binding activity is well conserved after reconstitution, ATP-dependent transport activity seems to be a very fragile property of TAP (28, 30). Here we were able to demonstrate that reconstituted TAP not only binds peptides but also displays ATP-dependent transport activity (Fig. 2). In the absence of ATP, no peptide translocation is observed. Transport is peptide specific, as it can be inhibited in the presence of an excess of unlabeled peptide. Moreover, the TAP-specific viral inhibitor ICP47 also inhibits peptide transport. The possibility that peptides are only bound to TAP or proteoliposomes can be excluded, first because peptide binding to TAP is nucleotide independent (11), and second, proteoliposomes are washed after transport with an excess of high-affinity peptide RRYQKSTEL, which removes bound but not transported peptides. In summary, we have established a fast and sparing method for reconstitution of TAP restoring peptide binding and translocation activity.
Figure 2.

ATP-dependent peptide transport into proteoliposomes. Peptide translocation into proteoliposomes was assayed at 32°C for 2 min by using 1 μM of the 125I-labeled peptide, RRYNASTEL (KD = 516 nM), in the presence or absence of ATP (3 mM). TAP-bound but not transported peptide was competed out by washing with 9 μM of the unlabeled peptide RRYQKSTEL (KD = 164 nM). To demonstrate that transport is peptide specific, the assay was performed in the presence of a 400-fold molar excess of unlabeled peptide and ATP. To show TAP-specific transport, the TAP-inhibitor ICP47 (10 μM) was added in the presence of ATP. The background results from peptide bound to but not transported by TAP. Data were obtained from duplicate measurements.
Peptide-Stimulated ATPase Activity of the TAP Complex.
The ATPase activity of reconstituted TAP was assayed by release of inorganic phosphate from [γ-32P]ATP, which increases linearly within the first 4 minutes of analysis. Therefore, initial rate constants were measured. In the absence of peptides, we observed an ouabain- and NaN3-insensitive ATPase activity of TAP-containing proteoliposomes (Fig. 3A). This basal activity was reduced to 65% by ortho-vanadate that inhibits ABC transporters and P-type ATPases, indicating that additional ATPases are still present after TAP enrichment. A similar situation (reduction of 30–40%) was found for P-glycoprotein expressed in insect cells (31). To analyze the coupling between peptide binding, transport, and ATP hydrolysis, the release of inorganic phosphate was determined in the presence or absence of peptides RRYQKSTEL and EPGNTWDED. These peptides are known to be a high- and low-affinity substrate of TAP (KD = 164 nM and KD > 1 mM, respectively) (Table 1) (13). In the presence of vanadate, no peptide stimulation of the ATPase activity was detected, indicating that TAP is in a vanadate-trapped state. A 2.8-fold stimulation of the vanadate-sensitive ATPase activity was observed in the presence of the high-affinity peptide (0.8 μM), whereas the low-affinity peptide does not stimulate ATP hydrolysis (Fig. 3A). Additional experiments were performed with the herpes simplex virus protein ICP47, which specifically inhibits peptide binding to TAP (7) and destabilizes TAP complex formation (32). ICP47 blocked the stimulation of the ATPase activity even in the excess of the high-affinity peptide. Note that ICP47 did not affect basal ATPase activity. This observation provides indirect evidence that basal ATPase activity derives from other ATPases. However, we cannot formally exclude the possibility that TAP possesses a (low) basal ATPase activity that is insensitive to ICP47. In summary, these data demonstrate that TAP-specific and peptide-stimulated ATPase activity was identified that is vanadate sensitive.
Figure 3.

Peptide-stimulated ATPase activity of reconstituted TAP. (A) For determination of the ATPase activity of TAP, the release of inorganic γ-32Pi from [γ-32P]ATP was measured for 4 min at 32°C in the presence or absence of ortho-vanadate (1 mM) and/or peptides (0.8 μM) (R9LQK = RRYQKSTEL; E9D = EPGNTWDED). To demonstrate TAP-specific ATPase activity, proteoliposomes were incubated with TAP-inhibitor ICP47 (5 μM) for 40 min on ice before addition of peptides. (B) Specificity pattern of the vanadate-sensitive ATPase activity analyzed by combinatorial peptide libraries. Combinatorial peptide libraries X8O (2.4 μM) were incubated with TAP-containing proteoliposomes.
To correlate TAP selectivity with ATP hydrolysis, we applied combinatorial peptide libraries that have been successfully used to decipher the binding motif of TAP (2, 13). In Fig. 3B, only a small set of sublibraries (X8O) is shown that point to the most drastic effects within the binding motif of human TAP. All peptides were used at concentrations of 2.4 μM, corresponding to the KD value of the library X9 (13). The XRX7 library with the favored arginine (KD = 0.56 μM) stimulated the vanadate-sensitive ATPase activity by 2.5-fold. A similar enhancement was observed in presence of the X8O library carrying favored hydrophobic or basic residues such as tyrosine at the C terminus (X8Y; KD = 0.29 μM). In contrast, libraries with disfavored residues, such as proline at position two (XPX7; KD = 46 μM) or aspartate at the C terminus (X8D; KD = 18 μM), did not stimulate ATPase activity. From these experiments, one can generalize that TAP selectivity is mirrored in the stimulation of the ATPase activity.
ATPase Activity and TAP Selectivity.
To understand the ATP hydrolysis by TAP in quantitative detail, we analyzed the ATPase activity in dependence of the peptide concentration (Fig. 4). We chose three peptides resembling substrates with high (RRYQKSTEL; KD = 164 nM), intermediate (RRYNASTEL; KD = 516 nM), and very low affinity (EPGNTWDED; KD > 1 mM) (Table 1). The ATPase activity of TAP showed a hyperbolic dependence on the peptide concentration for the high- and intermediate-affinity substrates. The concentrations of half-maximal ATPase stimulation (Km.pep) amount to 161 ± 15 nM and 574 ± 132 nM for RRYQKSTEL and RRYNASTEL, respectively. For the “nonbinding” peptide, only a lower limit of the dissociation constant (KD) and of the half-maximal ATPase stimulation (Km.pep) could be given. It must be stressed that Km.pep does not reflect the classical Michaelis–Menten constant Km for ATP hydrolysis. These half-maximal ATPase values (Km.pep) correlate directly with the affinity constants (KD) for all three substrates (Table 1). Note that the maximal ATPase stimulation (Vmax) for various peptides is the same (Fig. 4, Table 1). Saturation of all substrate-binding pockets leads to a characteristic maximal stimulation of ATPase activity that is identical for different peptides. Accordingly, 50% occupation of the peptide-binding sites results in half-maximal stimulation of the ATPase activity. From this impressive correlation between TAP specificity and ATPase activity, we deduce that peptide binding is the key step in stimulating ATP hydrolysis.
Figure 4.

Coupling between peptide binding and ATP hydrolysis. Release of γ-32Pi from [γ-32P]ATP was analyzed after 4 min at 32°C in the presence of various peptides. RRYQKSTEL (circles), RRYNASTEL (squares), and EPGNTWDED (triangles) have high, intermediate, and low binding affinity for TAP, respectively. Half-maximal stimulation (Km,pep) was obtained at 161 ± 15 nM and 574 ± 132 nM for RRYQKSTEL and RRYNASTEL, respectively. For the peptide with low affinity, only a lower limit of the half-maximal ATPase stimulation could be given. An identical maximal peptide-stimulated ATPase activity (Vmax) of 2 μmol/min per milligram TAP was found for RRYQKSTEL and RRYNASTEL. Data were obtained from duplicate measurements.
By comparing the amount of released inorganic phosphate in the presence and absence of the peptide RRYQKSTEL (saturating concentration of 1 μM), the TAP-specific ATPase activity was analyzed, even in the presence of other ATPases, which are not peptide sensitive (Fig. 5). The difference of released inorganic phosphate was fitted to Michaelis–Menten kinetics with a Km of 0.3 ± 0.06 mM. Based on the TAP concentration determined by saturation peptide-binding experiments, the maximal rate of ATP hydrolysis Vmax is approximately 2 μmol/min per mg TAP. This leads to a turnover number of 5 ATP per TAP complex per second and a catalytic activity of 20,000 M−1 s−1.
Figure 5.

Michaelis–Menten kinetics of TAP-specific ATPase activity. The stimulated ATPase kinetics of TAP were determined by subtracting the amount of released γ-32Pi in the presence and absence of 1 μM RRYQKSTEL after 4 min at 32°C. Data were fitted by using the Michaelis–Menten equation. Based on this curve fit, a Km of 0.3 ± 0.06 mM ATP was calculated (see Table 1). Data were obtained from duplicate measurements.
Crosstalk Between Peptide-Binding, Transport, and ATP Hydrolysis.
To investigate the puzzling question whether substrate binding alone or subsequent downstream events might also affect ATP hydrolysis, we used sterically restricted peptides. These peptides are based on the nonapeptide ARDNATKDY carrying a polylysine chain of up to 20 residues linked to the ɛ-amino group of lysine. Interestingly, all of these peptides bind to TAP with similar affinity (IC50 values of 0.5 to 1.2 μM) with no correlation between length of the polylysine side chain and affinity. In contrast to binding affinity, transport efficiency decreases drastically for peptides containing more than eight lysine units (12). Therefore, these peptides are an ideal tool to distinguish between peptide binding and subsequent translocation. As shown in Fig. 6, peptide-stimulated ATPase activity depends on the length of the polylysine branch. For up to five lysine residues, no effect on peptide-induced ATPase activity is observed. However, with increasing length of the polylysine branch (eight residues or more), a drastic decrease of peptide-stimulated ATPase activity was observed. A peptide consisting of the 20-mer polylysine side chain did not stimulate ATPase activity. This reduced stimulation of the ATPase activity with increasing length of the polylysine side chain directly correlates with the peptide transport efficiency (Fig. 6), indicating that ATP hydrolysis reflects peptide translocation.
Figure 6.

ATPase stimulation and peptide transport by sterically restricted peptides. To correlate vanadate-sensitive ATPase activity and peptide transport, the release of inorganic γ-32Pi after 4 min at 32°C and the peptide transport were analyzed in presence of 2.5 μM ARDNATK(-ɛKn)DY carrying different polylysine extensions (n). IC50 were taken from (12) (a) or determined by peptide competition assays as described (11) (b). Details of the ATPase, peptide-binding, and transport assay are described in Materials and Methods. Data are background-subtracted and obtained from duplicate measurements.
Discussion
In previous studies, the peptide-binding mechanism and substrate specificity of TAP have been analyzed in detail (reviewed by ref. 2). However, little is known about the pivotal role of ATP hydrolysis in TAP function. ATP is required for peptide translocation into the ER lumen (16–18). In addition, experimental data support the model that ATP synchronizes the release of peptide-loaded MHC class I molecules from TAP for subsequent journey to the cell surface (22). Furthermore, ATP stabilizes the structure of TAP probably as a consequence of structural rearrangement (23). To analyze the essential role of ATP hydrolysis, we developed a fast and minimally invasive method to solubilize, enrich, and reconstitute TAP. Peptide binding and transport activity of TAP were demonstrated in a reconstituted in vitro system. The ATPase activity of TAP is vanadate-sensitive and stimulated by peptides. By comparing the ATPase stimulation in more detail by analyzing peptides with different affinities for TAP, we observed a direct correlation between the binding constant KD and the half-maximal stimulation of the ATPase activity (Km.pep). These results were generalized by using combinatorial peptide libraries, demonstrating that stimulation of the ATPase activity correlates with the peptide-binding motif of TAP. The peptide-stimulated ATPase becomes decoupled from substrate binding only in the case of sterically restricted peptides.
Substrate-stimulated ATPase activity has been analyzed in detail for P-glycoprotein (31, 33). However, many puzzling questions remain unanswered: First, different values of the ATPase activity have been reported. The ATPase activity reaches different Vmax values in the presence of different substrates. Moreover, some substrates bind to the drug transporter but do not stimulate ATP hydrolysis. Second, at high substrate concentrations, ATPase activity is inhibited by some substrates. Third, a direct correlation between substrate-binding affinity and substrate-stimulated ATPase activity has not been observed. It should be noted that P-glycoprotein and other drug pumps transport hydrophobic substrates. Thus, membrane partition of these compounds as well as accessibility to the transporter may explain these discrepancies. In contrast, TAP, as demonstrated in this study, shows a strict correlation between peptide binding (KD) and stimulation of ATP hydrolysis (Km.pep). In addition, the maximal ATPase activity (Vmax) is independent of substrate affinity because peptides with different KD values for TAP displayed the same Vmax value. Assuming that ATP-hydrolysis reflects transport, one must conclude that the maximal translocation rate is independent of substrate affinity.
The allosteric interaction between peptide-binding, ATP hydrolysis, and peptide translocation implies a dialogue between the NBDs and substrate-binding pocket. On the basis of the process of peptide-binding and substrate-stimulated ATP hydrolysis, we propose a four-step model of TAP function: First, peptide and ATP bind independently to TAP (11). Peptide association to TAP is a fast process, probably diffusion controlled. Second, loading of the peptide-binding pocket is transmitted to the NBDs via a slow conformational change (14). Most likely binding of the peptide shifts the equilibrium toward a conformation that forms a tight interface of both NBDs, which is required for allosteric activation of ATP hydrolysis. A decrease in lateral diffusion of the TAP complex caused by structural reorganization is observed only if both ATP and peptide are bound (25). Third, the NBDs are activated, and ATP hydrolysis causes transport of peptide into the lumen of the ER. The structural change in the peptide-binding pocket is inhibited by sterically restricted peptides preventing ATP hydrolysis and consequently peptide translocation. Fourth, TAP relaxes into its initial state to complete the transport cycle. On the basis of the presented data, we cannot distinguish whether the hydrolysis of one or two ATP molecules is required for the transport cycle. However, it seems very likely that the two NBDs get in close contact in the presence of peptide and ATP and might disassemble after ATP hydrolysis.
As highlighted by sterically restricted peptides, the crosstalk within TAP is more complex than mentioned above. Peptides with polylysine extensions longer than five lysines stimulate the ATPase less than the original peptide, consistent with a decreased transport efficiency (12). However, all peptides have the same affinity independent of the length of the side-chain extension. Therefore, the peptide transporter TAP must sense the quality of the bound substrate. This quality control must be beyond the binding step and helps the cell to save energy. How this downstream control mechanism functions on molecular level remains unknown.
The Michaelis–Menten constant Km (0.3 mM), determined by the release of inorganic Pi, corresponds well with the Km measured by the amount of glycosylated peptides transported into the lumen of microsomes with increasing ATP concentration (34). The agreement between the Km values of these two different methods shows the direct coupling between peptide-binding, transport, and ATP hydrolysis, supporting the idea that each bound peptide is transported by TAP under consumption of energy. The Michaelis–Menten constant of the related P-glycoprotein ranges from 0.3 to 1.4 mM depending on species, expression system, and purification procedure (35, 36). In the presence of different drugs, the Km value of P-glycoprotein changes slightly (0.4 and 1.4 mM) and shows only drug dependence in some cases. Interestingly, the turnover numbers of the related ABC-transporter P-glycoprotein and TAP (5 ATPs) are almost identical and approximately 5-fold higher than the kcat of CFTR. In contrast to P-glycoprotein, TAP obviously has no or a very low basal ATPase activity, as deduced from the ICP47 inactivation experiments.
Assuming that one or two ATP molecules are needed to complete the translocation cycle, the transport turnover number derived from peptide-stimulated ATPase activity is much higher than anticipated from transport assays trapping peptides in the ER by glycosylation. This approach relies on various steps possibly leading to an underestimation of the transport rate. A maximum transport velocity of 2.9 fmol/min⋅micrograms microsomal protein was reported (37), corresponding to a turnover rate of 0.4 peptides per minute, taking a microsomal TAP concentration of 0.1% (wt/wt) into account. In contrast, the turnover number measured directly by ATP hydrolysis is 400-fold faster. Assuming 105 TAP complexes per cell, we can estimate the maximal intracellular flux into the ER to be 1.5 × 107 peptides/minute⋅cell. This turnover rate is sufficient to account for the role of TAP in peptide loading of MHC molecules and the overall process of antigen presentation, making evident that the intracellular transport by TAP is not the rate-limiting step in antigen processing.
Acknowledgments
We are indebted to Eckhard Linker and Karlheinz Burk for excellent technical assistance. We thank Stefan Uebel for helpful discussion and assistance and Jacque Neefjes for the generous gift of sterically restricted peptides. We are grateful to Lutz Schmitt and Jacob Piehler for critically reading the manuscript. The Deutsche Forschungsgemeinschaft supported this work.
Abbreviations
- ABC
ATP-binding cassette
- DM
n-decyl-β-d-maltopyranoside
- ER
endoplasmic reticulum
- MHC
major histocompatibility complex
- NBD
nucleotide-binding domain
- TAP
transporter associated with antigen processing
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Pamer E, Cresswell P. Annu Rev Immunol. 1998;16:323–358. doi: 10.1146/annurev.immunol.16.1.323. [DOI] [PubMed] [Google Scholar]
- 2.Uebel S, Tampé R. Curr Opin Immunol. 1999;11:203–208. doi: 10.1016/s0952-7915(99)80034-x. [DOI] [PubMed] [Google Scholar]
- 3.Higgins C F. Annu Rev Cell Biol. 1992;8:67–113. doi: 10.1146/annurev.cb.08.110192.000435. [DOI] [PubMed] [Google Scholar]
- 4.Ortmann B, Copeman J, Lehner P J, Sadasivan B, Herberg J A, Grandea A G, Riddell S R, Tampé R, Spies T, Trowsdale J. Science. 1997;277:1306–1309. doi: 10.1126/science.277.5330.1306. [DOI] [PubMed] [Google Scholar]
- 5.Lehner P J, Surman M J, Cresswell P. Immunity. 1998;8:221–231. doi: 10.1016/s1074-7613(00)80474-4. [DOI] [PubMed] [Google Scholar]
- 6.Li S, Paulsson K M, Chen S, Sjogren H O, Wang P. J Biol Chem. 2000;275:1581–1586. doi: 10.1074/jbc.275.3.1581. [DOI] [PubMed] [Google Scholar]
- 7.Ahn K, Meyer T H, Uebel S, Sempé P, Djaballah H, Yang Y, Peterson P A, Früh K, Tampé R. EMBO J. 1996;15:3247–3255. [PMC free article] [PubMed] [Google Scholar]
- 8.Tomazin R, Hill A B, Jugovic P, York I, van Endert P, Ploegh H L, Andrews D W, Johnson D C. EMBO J. 1996;15:3256–3266. [PMC free article] [PubMed] [Google Scholar]
- 9.van Endert P M, Tampé R, Meyer T H, Tisch R, Bach J F, McDevitt H O. Immunity. 1994;1:491–500. doi: 10.1016/1074-7613(94)90091-4. [DOI] [PubMed] [Google Scholar]
- 10.Koopmann J O, Post M, Neefjes J J, Hämmerling G J, Momburg F. Eur J Immunol. 1996;26:1720–1728. doi: 10.1002/eji.1830260809. [DOI] [PubMed] [Google Scholar]
- 11.Uebel S, Meyer T H, Kraas W, Kienle S, Jung G, Wiesmüller K H, Tampé R. J Biol Chem. 1995;270:18512–18516. doi: 10.1074/jbc.270.31.18512. [DOI] [PubMed] [Google Scholar]
- 12.Grommé M, van der Valk R, Sliedregt K, Vernie L, Liskamp R, Hämmerling G J, Koopmann J-O, Momburg F, Neefjes J J. Eur J Immunol. 1997;27:898–904. doi: 10.1002/eji.1830270415. [DOI] [PubMed] [Google Scholar]
- 13.Uebel S, Kraas W, Kienle S, Wiesmüller K-H, Jung G, Tampé R. Proc Natl Acad Sci USA. 1997;94:8976–8981. doi: 10.1073/pnas.94.17.8976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neumann L, Tampé R. J Mol Biol. 1999;294:1203–1213. doi: 10.1006/jmbi.1999.3329. [DOI] [PubMed] [Google Scholar]
- 15.Nijenhuis M, Hämmerling G J. J Immunol. 1996;157:5467–5477. [PubMed] [Google Scholar]
- 16.Neefjes J J, Momburg F, Hämmerling G J. Science. 1993;261:769–771. doi: 10.1126/science.8342042. [DOI] [PubMed] [Google Scholar]
- 17.Androlewicz M J, Anderson K S, Cresswell P. Proc Natl Acad Sci USA. 1993;90:9130–9134. doi: 10.1073/pnas.90.19.9130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shepherd J C, Schumacher T N, Ashton-Rickardt P G, Imaeda S, Ploegh H L, Janeway C A J, Tonegawa S. Cell. 1993;74:577–584. doi: 10.1016/0092-8674(93)80058-m. [DOI] [PubMed] [Google Scholar]
- 19.Müller K M, Ebensperger C, Tampé R. J Biol Chem. 1994;269:14032–14037. [PubMed] [Google Scholar]
- 20.Wang K N, Früh K, Peterson P A, Yang Y. FEBS Lett. 1994;350:337–341. doi: 10.1016/0014-5793(94)00806-x. [DOI] [PubMed] [Google Scholar]
- 21.Chen H L, Gabrilovich D, Tampé R, Girgis K R, Nadaf S, Carbone D P. Nat Genet. 1996;13:210–213. doi: 10.1038/ng0696-210. [DOI] [PubMed] [Google Scholar]
- 22.Knittler M R, Alberts P, Deverson E V, Howard J C. Curr Biol. 1999;9:999–1008. doi: 10.1016/s0960-9822(99)80448-5. [DOI] [PubMed] [Google Scholar]
- 23.van Endert P M. J Biol Chem. 1999;274:14632–14638. doi: 10.1074/jbc.274.21.14632. [DOI] [PubMed] [Google Scholar]
- 24.Lapinski P E, Miller G G, Tampe R, Raghavan M. J Biol Chem. 2000;275:6831–6840. doi: 10.1074/jbc.275.10.6831. [DOI] [PubMed] [Google Scholar]
- 25.Reits E A J, Vos J C, Grommé M, Neefjes J. Nature (London) 2000;404:774–778. doi: 10.1038/35008103. [DOI] [PubMed] [Google Scholar]
- 26.Karttunen J T, Trowsdale J, Lehner P J. Curr Biol. 1999;9:R820–R824. doi: 10.1016/s0960-9822(99)80499-0. [DOI] [PubMed] [Google Scholar]
- 27.Meyer T H, van Endert P M, Uebel S, Ehring B, Tampé R. FEBS Lett. 1994;351:443–447. doi: 10.1016/0014-5793(94)00908-2. [DOI] [PubMed] [Google Scholar]
- 28.Uebel S, Plantinga T, Weber P J, Beck-Sickinger A G, Tampé R. FEBS Lett. 1997;416:359–363. doi: 10.1016/s0014-5793(97)01222-2. [DOI] [PubMed] [Google Scholar]
- 29.Li Y, Salter-Cid L, Vitiello A, Preckel T, Lee J D, Angulo A, Cai Z, Peterson P A, Yang Y. J Biol Chem. 2000;275:24130–24135. doi: 10.1074/jbc.M003617200. [DOI] [PubMed] [Google Scholar]
- 30.Stephens D B, Androlewicz M J. FEBS Lett. 1997;416:353–358. doi: 10.1016/s0014-5793(97)01221-0. [DOI] [PubMed] [Google Scholar]
- 31.Sarkadi B, Price E M, Boucher R C, Germann U A, Scarborough G A. J Biol Chem. 1992;267:4854–4858. [PubMed] [Google Scholar]
- 32.Lacaille V G, Androlewicz M J. J Biol Chem. 1998;273:17386–17390. doi: 10.1074/jbc.273.28.17386. [DOI] [PubMed] [Google Scholar]
- 33.Senior A E, Alshawi M K, Urbatsch I L. Methods Enzymol. 1998;292:514–523. doi: 10.1016/s0076-6879(98)92040-7. [DOI] [PubMed] [Google Scholar]
- 34.Meyer T H. Ph.D. thesis. Munich: Technical University Munich; 1996. [Google Scholar]
- 35.Shapiro A B, Ling V. J Biol Chem. 1994;269:3745–3754. [PubMed] [Google Scholar]
- 36.Sharom F J, Yu X H, Chu J W K, Doige C A. Biochem J. 1995;308:381–390. doi: 10.1042/bj3080381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang B, Braciale T J. J Immunol. 1995;155:3889–3896. [PubMed] [Google Scholar]
- 38.Bushkin Y, Posnett D N, Pernis B, Wang C Y. J Exp Med. 1986;164:458–473. doi: 10.1084/jem.164.2.458. [DOI] [PMC free article] [PubMed] [Google Scholar]