

Abstract
QTL mapping in autopolyploids is complicated by the possibility of segregation for three or more alleles at a locus and by a lack of preferential pairing, however the subset of polymorphic alleles that show simplex segregation ratios can be used to locate QTLs. In autopolyploid Saccharum, 36 significant associations between variation in sugar content and unlinked loci detected by 31 different probes were found in two interspecific F1 populations. Most QTL alleles showed phenotypic effects consistent with the parental phenotypes, but occasional transgressive QTLs revealed opportunities to purge unfavorable alleles from cultivars or introgress valuable alleles from exotics. Several QTLs on homologous chromosomes appeared to correspond to one another–multiple doses of favorable ‘alleles’ at such chromosomal region(s) yielded diminishing returns–such negative epistasis may contribute to phenotypic buffering. Fewer sugar content QTLs were discovered from the highest-sugar genotype than from lower-sugar genotypes, perhaps suggesting that many favorable alleles have been fixed by prior selection, i.e. that the genes for which allelic variants (QTLs) persist in improved sugarcanes may be a biased subset of the population of genes controlling sugar content. Comparison of these data to mutations and QTLs previously mapped in maize hinted that seed and biomass crops may share a partly-overlapping basis for genetic variation in carbohydrate deposition. However, many QTLs do not correspond to known candidate genes, suggesting that other approaches will be necessary to isolate the genetic determinants of high sugar content of vegetative tissues.
Autopolyploid genomes, containing many different homologous chromosomes that can pair and recombine in most or all possible combinations, have been underexplored at the molecular level due to the special problems they pose in genetic and molecular analyses (Sreenivasan et al. 1987; Burner 1997). The importance of autopolyploidy is highlighted by its prominence among cultivated crops, including sugarcane (8–18×), sugar beet (3×), ryegrass (4×), bermuda grass (3–4×), cassava (4×), potato (4×), alfalfa (4×), red clover (4×), Grande Naine banana (3×), apple cultivars (3×), and many ornamentals (Zeven 1979).
Among the world's leading crops with an annual production at a projected record 97 million metric tons in 1999/2000 (FAS Online, 1999), sugarcane is a classical example of a complex autopolyploid. Cultivated sugarcane varieties have ∼80–140 chromosomes, comprising 8–18 copies of a basic × = 8 or × = 10 (D'Hont et al. 1995, 1998; Ha et al. 1999; Irvine 1999). Most chromosomes of cultivated sugarcane appear to be largely derived from Saccharum officinarum (Irvine 1999); however, in situ hybridization data suggest that about 10% may be derived from S. spontaneum (D'Hont et al. 1996). S. officinarum commonly has high sucrose content, low fiber content, thick stalks, little pubescence, rare flowering, and limited tillering. S. spontaneum does not accumulate sucrose, and is fibrous, thin-stalked, pubescent, profusely flowering, and abundantly tillering.
Like other vegetatively propagated plant species, cultivated sugarcane (Saccharum spp. hybrids) and its wild relatives are highly heterozygous. Pure inbred lines do not exist due to the difficulty of self-pollination and the random pairing of multiple homologous chromosomes. The segregating populations used in genetic studies are the progenies (first generation) derived from crosses between two cultivated varieties (Kang et al. 1983; Milligan et al. 1990) or cultivated varieties and wild species (Guimarães et al. 1997; Ming et al. 1998). Chromosome transmission is normal for most crosses, yielding n×+×n progeny (Burner 1997), but 2n×+×n transmission predominates in S. officinarum (2n = 80) × S. spontaneum F1 and BC1 crosses, a phenomenon known as “female restitution” (Bremer 1923; Price 1957).
The most abundant restriction fragment length polymorphisms in sugarcane are “single-dose restriction fragments” (SDRFs) showing 1:1 segregation in doubled haploid and interspecific F1 populations (Wu et al. 1992; Da Silva et al. 1995; Guimarães et al. 1997; Ming et al. 1998). SDRFs represent 70% of the detectable polymorphic loci resulting from the segregation of alleles of different dosages (Da Silva 1993). The random chromosome pairing that is characteristic of autopolyploids makes it necessary to construct linkage maps for each parent of a cross, unlike diploid species, in which allelism permits a unified map of both parents to be generated. Studies using molecular markers have begun to resolve the genetic complexity of sugarcane by analysis of SDRFs (Da Silva et al. 1995; D'Hont et al. 1995; Grivet et al. 1996; Dufour et al. 1997; Guimarães et al. 1997; Ming et al. 1998). Comparative mapping has shown striking colinearity among the genomes of grasses (Hulbert et al. 1990; Ahn and Tanksley 1993; Ahn et al. 1993; Lin et al. 1995; Moore et al. 1995; Paterson et al. 1995), and even distantly related species (Paterson et al. 1996). A comparative approach has greatly expedited sugarcane genome analysis, in particular using the small diploid genome of closely related sorghum as a guide (Dufour et al. 1997; Guimarães et al. 1997; Ming et al. 1998).
Genetic tools for sugarcane have only recently become adequate to quantify the effect of many genomic regions on a trait. Two prior studies reported the association of DNA markers with disease resistance and flowering time in sugarcane. Daugrois et al. (1996) identified a putative major gene for rust resistance linked at 10 cM with a RFLP marker CDSR0029 in sugarcane cultivar ‘R570.’ Guimarães et al. (1997) found an RFLP marker associated with short-day flowering. However, the mapping populations used in these two studies were too small (83 and 100 individuals, respectively) for comprehensive quantitative trait loci (QTL) analysis.
We report here the results of our study into the genetic basis of variation in sugar content among sugarcane genotypes using single-dose DNA markers. The fundamental complexity of autopolyploid genetics resulting from heterozygosity and lack of preferential pairing is further complicated by the fact that sugar content is a complex industrial trait influenced by variation in carbon fixation, photosynthate partitioning into sucrose, transportation and accumulation of sucrose (Berding et al. 1997; Moore et al. 1997) in harvestable biomass, and extractability of sucrose from biomass (Legendre and Henderson 1972). Our primary objectives were to determine the number and location of QTLs for sugar content in sugarcane, which is arguably the most important trait for the sugarcane industry; to investigate the molecular basis of phenotypic buffering that may contribute to the success of autopolyploid crops; and to investigate the possibility that candidate genes for QTLs affecting carbohydrate metabolism in biomass crops might be identified based on discrete mutations affecting seed development in other crops.
RESULTS
Sugar Content QTLs
GG × IND progeny values ranged from 39.4 to 249.0 lb sugar per ton of harvested biomass (lb/ton) (see Methods), a range that was ∼40.1% wider than the (albeit large) difference between the parents (IND = 53.2, GG = 202.8). A full model comprised of 14 QTLs, eight from GG and six from IND, explained 65.5% of phenotypic variation (PV). The eight GG QTLs alone explained 38.6% of PV, while the six IND QTLs alone explained 36%. Among the 14 QTLs found, only one was inconsistent with the expected parental phenotypes, a GG QTL near pSB0279d that reduced sugar content by about 20.3 lb/ton. Also, a putative (P < 0.006) IND QTL near pSB0044d increased sugar content by 14.4 lb/ton. These two loci together account for part of the transgressive variation. A total of 18 putative QTLs (0.003 < P < 0.01) were found, and five (27.8%) of these were associated with significant QTLs on homologous chromosomes, or with candidate genes (Table 1, Fig. 1).
Table 1.
Biometrical Parameters of QTLs Associated with Sugar Content7
| Marker(s) | LG9HG) | SLG | P (LOD)a | R2 | A |
|---|---|---|---|---|---|
| CDSB10eG1 | 6 | 0.0010(3.0) | 0.084 | 16.1 | |
| CDSB32iGb | 5(1) | 0.0065 | 0.036 | 13.3 | |
| CDSC42dG | 0.0025 | 0.044 | 14.7 | ||
| pSB103cG2 | 41(11)c | B | 0.0003(3.1) | 0.064 | 15.7 |
| CDSR17aG | 0.0024 | 0.074 | 18.1 | ||
| pshD13dG | I | 0.0011 | 0.063 | 17.8 | |
| CSU81bG | B | 0.0061 | 0.039 | 14.2 | |
| pSB279dG | 38(2) | 0.0024 | 0.074 | −20.3 | |
| CDSR125eG | 0.0016 | 0.061 | 20.0 | ||
| SG322bG | 53 | G | 0.0017 | 0.054 | 16.7 |
| CDSB10fI | 13,15(15) | 0.0009 | 0.057 | −17.3 | |
| CDSB67cI | 0.0026 | 0.044 | −14.9 | ||
| CDSR133dI | J | 0.0055 | 0.043 | −13.7 | |
| CDSR78dI | 53 | B | 0.0077 | 0.039 | −14.2 |
| CDSRiicI3 | 36(2) | A | 0.0008(3.1) | 0.086 | −19.3 |
| CDSR94aI | 0.0022 | 0.058 | −17.3 | ||
| pSB106cI | I | 0.0018 | 0.053 | −16.4 | |
| pSB44dI | A | 0.0062 | 0.042 | 14.4 | |
| UMC114hI4 | 64 | I | 0.0003(4.7) | 0.131 | −23.4 |
| CDSB32nM | 18(1) | F | 0.0006 | 0.058 | 10.3 |
| CDSB57jM | 0.0010 | 0.062 | 10.3 | ||
| CDSC42c, IMd | 20 | 0.0006 | 0.087 | Fig. 3 | |
| CDSC5b, kMd | 10 | D | 0.0001 | 0.097 | Fig. 3 |
| CDSC52gM5 | 12(2) | A | 0.0001(3.6) | 0.082 | 11.7 |
| CDSR125bM | 34 | D | 0.0010 | 0.057 | 9.6 |
| CDSR146eM | 40 | H | 0.0012 | 0.060 | 10.4 |
| CDSR15fM | 42(12) | 0.0001 | 0.075 | 11.2 | |
| CDSR35hM | 0.0001 | 0.098 | 13.4 | ||
| CDSR46dM | 0.0015 | 0.105 | 12.8 | ||
| CDSR96fM6 | 0.0001(6.6) | 0.206 | 18.8 | ||
| CSU428b, dMd | 0.0003 | 0.085 | Fig. 3 | ||
| CSU449aM | 39 | J | 0.0022 | 0.056 | 9.5 |
| pSB103cM | B | 0.0034 | 0.073 | 10.7 | |
| pSB167bM | C | 0.0001 | 0.096 | 13.2 | |
| pSB189eM | D | 0.0018 | 0.056 | 10.7 | |
| pSB289eM | A | 0.0006 | 0.060 | 10.1 | |
| pSB82eM | G | 0.0008 | 0.056 | 9.7 | |
| SHO38dM | B | 0.0057 | 0.041 | 8.1 | |
| UMC147eM | 67 | C | 0.0001 | 0.119 | 14.1 |
| CDSB32c, fPd | 4, 5(12) | A | 0.0001 | 0.129 | Fig. 3 |
| CDSB7eP | 25(4) | B | 0.0044 | 0.047 | −9.0 |
| CDSR29aP | 24 | F | 0.0017 | 0.054 | −9.4 |
| CDSR35bP | 43 | C | 0.0018 | 0.089 | −12.6 |
| pSB1368dP | A | 0.0094 | 0.056 | 9.4 | |
| RZ508bP | 13(7) | I | 0.0029 | 0.052 | −9.3 |
LT, linage group; SLG, sorghum LG; A, allele effect;
P value and LOD score.
Markers in italics associated with putative QTLs (see Methods).
The number in parenthesis is homologous group number (see Ref. 6 below).
Double dose QTLs.
-pSB1379dG interval.
-CDSR116aG.
-CDSR87aI.
-CSU395eI.
-CDSR96aM.
-CSU419fM.
Lbs sugar per ton biomass.
Figure 1.

Comparative mapping of sugar content QTLs. Bars and whiskers indicate 1 and 2 LOD likelihood intervals. Solid lines connect homologous loci on different sugarcane and sorghum linkage groups. Arrows indicate the inferred locations of markers used to align the homologous linkage groups among maize, sorghum, and sugarcane based on published (Ming et al. 1998) and unpublished data (M. McMullen and E. Coe, pers. comm.). Individual sorghum linkage groups are represented by LGs A to J. Sugarcane linkage groups from four parental varieties are indicated by the last letter of the marker name: G (Green German); M (Muntok Java); I (IND 81–146); P (PIN 84–1). Approximate map positions of double-dose (#) markers are inferred by the method of Da Silva (1995). The letters in parentheses following the marker name represent the sorghum linkage groups where the marker mapped, if different from the corresponding location shown. Only regions that contain, or are homologous to, QTLs are shown. Twenty-eight of the 36 sugar content QTLs are shown on this figure, along with nine putative QTLs. The markers associated with the remaining eight sugar content QTLs could not be mapped in sorghum.
Sugar content of PIN × MJ progeny ranged from −49.0 to 65.2 lb/ton, a range about 30.4% wider than the difference between the parents (PIN = −37.2, MJ = 50.4). Negative sugar content values reflect the lower ratio of pol to brix. If this ratio is below 35% (varies slightly at different factories), the calculated sugar content value will be negative, indicating sucrose can not be separated from other soluble solids in cane juice (Pol value is explained in Methods). A full model comprised of 22 QTLs, 18 from MJ and four from PIN, explained 68.3% of PV (Table 1). The 18 MJ QTLs alone explained 45.7% of PV, while the four PIN QTLs alone explained 33.4% of PV. Allele effects of all QTLs were consistent with expected parental phenotypes, except one putative PIN QTL near pSB1368d increased sugar content by 9.4 lb/ton, accounting for part of the progeny transgression of parental phenotypes. Three DNA probes (CDSC0005, CDSC0042, CSU0428) were each diagnostic of two MJ QTLs at unlinked loci, and one DNA probe (CDSB0032) was diagnostic of two PIN QTLs at unlinked loci. A total of 23 putative QTLs (0.003 < P < 0.01) were found, and four (17.4%) were associated with significant QTLs on homologous chromosomes, or with candidate genes (Table 1, Fig. 1).
Some sugar content QTLs showed clear patterns of association with other traits. Sugar content was positively correlated with Pol (r = 0.64 in GG × IND, r = 0.92 in PIN × MJ) and stalk weight (r = 0.50 in GG × IND, r = 0.53 in PIN × MJ), and negatively correlated with ash content (r = −0.66 in GG × IND, r = −0.55 in PIN × MJ, Table 2). Most IND sugar content QTLs were associated with increased fiber and ash, and reduced stalk weight. Most MJ sugar content QTLs were associated with increased Pol and stalk weight, and reduced ash. Most PIN QTLs were associated with reduced Pol and increased ash.
Table 2.
Allele Effect of Other Associated Traits Affected by Sugar Content QTLs
| Marker(s) | Pol a (d) | Fiber a | Ash a (D) | Stk Wt a (d) |
|---|---|---|---|---|
| CDSB10fI | – | 1.27** | 0.99** | −3.03** |
| CDSB67cI | – | 1.03* | – | – |
| CDSR128bI | – | 1.31** | 0.69* | – |
| CDSR88cI1 | – | 1.8** | 0.7* | −2.77** |
| CDSR94aI | – | – | 0.85** | −3.29** |
| pSB106cI | –1.02* | – | – | |
| pSB44dI | 0.92** | – | – | – |
| UMC114hI2 | – | 2.67** | – | −4.45** |
| % of R2 explained | 100 | 16.2 | 43.1 | 25.3 |
| Corr. with SC | 0.64*** | 0.21* | −0.66** | 0.50** |
| CDSB32nM | 0.53* | – | −0.91** | 2.13** |
| CDSC42c, 1M | 0.46* | – | – | 0.85* |
| (0.08*) | (1.01*) | |||
| CDSC5b, kM | 0.47* | – | – | 1.45* |
| (0.07*) | (0.86*) | |||
| CDSC52gM3 | – | – | – | 0.22** |
| CDSR125bM | 0.85 | – | −0.62* | 1.37** |
| CDSR146eM | – | – | – | – |
| CDSR15fM | 0.55** | – | – | 1.77** |
| CDSR35hM | – | – | −0.88** | 2.09** |
| CDSR46dM | 0.71** | – | −0.80* | – |
| CDSR96fM4 | 0.98** | – | −0.78** | 2.36** |
| CSU428b, dM | 0.40* | – | – | 1.49* |
| (0.29*) | (1.30*) | |||
| CSU449aM | 0.57** | – | −0.75** | 1.81** |
| pSB167bM | 0.64** | – | −0.75** | 1.96** |
| pSB289eM | 0.51* | – | – | 1.88** |
| pSB82eM | 0.49* | – | −0.60** | 1.63** |
| UMC147eM | 0.75** | – | – | 1.04* |
| % of R2 explained | 73.7 | 55.2 | 21.7 | |
| CDSB32c, fP | −0.43* | – | 0.57* | −1.3 |
| (−0.07) | (−0.08) | (−0.08) | ||
| CDSR29aP | −0.64** | – | 0.59* | – |
| CDSR35bP | −0.78** | – | – | – |
| pSB82e | – | – | 0.55* | – |
| RZ508bP | – | – | 0.64** | – |
| % of R2 explained | 60.9 | 53.3 | ||
| Corr. with SC | 0.92*** | 0.14* | −0.55** | 0.53*** |
P <0.01.
P <0.003.
(a) Additive effect; (d) dominant effect; (Stk Wt) stalk weight.
-CDSR87aI interval.
-CSU395eI.
-CDSR96aM.
-CSU419fM.
Dosage Effects of Individual QTLs
In the four cases where single DNA probes detected sugar content QTLs at each of two or more unlinked loci, it was possible to investigate whether the dosage (zero, one, or two ‘copies’) of the chromosomal region(s) containing the favorable allele(s) had nonadditive (i.e., nonlinear) effects on phenotype (Fig. 2), as described in the Methods section. All four showed nonlinear tendencies suggesting less-than-additive effects, but in only one case (CSU0428b, dM) did the regression line have a significant nonlinear (in this case, quadratic) component. Other traits for which significant effects were linked to larger numbers of loci detected by common probes provided a test of higher dosages. For example, two DNA probes each detected three loci associated with plant height in MJ, and another two DNA probes each detected four loci associated with plant height in MJ (R. Ming et al., in prep.). In all four cases, the regression lines showed less-than-additive gene action, with significant (P < 0.05) quadratic trends in three cases, and a significant quartic trend in one case.
Figure 2.

Dosage effects of QTLs for sugar content and plant height (HT). Lowercase letters after the probe name represent the loci detected by this probe; the capital letter M or P represents parental variety MJ or PIN. Phenotypic effects of allele substitution at each locus are shown next to the locus (letter); the asterisk indicates the significant level at P < 0.01. Pl, Pq, Pc, and Pqt are the probabilities that linear, quadratic, cubic, and quartic components of the dosage curve are equal to zero.
Comparative Analysis of QTLs
Alignment with the high-density sorghum linkage map has enabled us to fill gaps in the sugarcane map (Ming et al. 1998), and also to evaluate correspondence of sugarcane QTLs to structural genes, phenotypic mutants, and QTLs affecting carbohydrate metabolism. Major genes or QTLs affecting maize seed carbohydrate status or levels of carbohydrate-metabolizing enzyme activities were placed on sorghum linkage groups (LGs) based on DNA markers mapped on both the 1998 maize linkage map (Davis et al. 1999), and our sorghum map (Ming et al. 1998). A total of 11 candidate genes or QTLs from maize in nine genomic regions were evaluated for correspondence to sugar content QTLs (Table 3). Four of these corresponded to at least six (and possibly as many as eight) sugar content QTLs in MJ, GG, and IND (none corresponded in PIN), and one corresponded to a putative MJ QTL. Among the 18 MJ QTLs, two corresponded to candidate genes, a level of correspondence that could be explained by chance in 94.6% of cases. Among the eight GG QTLs, at least three (possibly four) corresponded to candidate genes, a level of correspondence that could be explained by chance in only 4% of cases. Among the six IND QTLs, at least one (possibly two) corresponded to candidate genes, a level of correspondence that could be explained by chance in 33.5% of cases. The uncertainty in the number of GG and IND QTLs that correspond to candidate genes is related to the uncertain location of the probe CDSB0010 in the sorghum genome, where it could not be directly mapped due to lack of polymorphism. Its proximity to anchor markers that could be mapped in both sorghum and sugarcane suggest two possible locations, one near the bottom of LG D (with no candidate gene) and the other on LG G near the location corresponding to the su2 mutation of maize. CDSB0010 is being further investigated by physical mapping–if it should prove to map near su2, then the rates of correspondence of GG and IND QTLs to candidate genes would be explicable by chance in only 0.5% and 8.3% of cases, respectively.
Table 3.
Probability of Correspondence between Maize Candidate Genes and Surgarcane QTLs as Ranhdom Events
| Maize | GG | IND | MJ | PIN | |
|---|---|---|---|---|---|
| * of QTLs | 9 | 9 | 7 | 16 | 4 |
| # of matches | 4 | 4 | 4 | 1 | |
| # of interval | 56 | 56 | 43 | 43 | |
| P value | 0.026 | 0.009 | 0.261 | 0.433 |
Comparative QTL analyses were summarized in reference to sorghum linkage groups as follows:
Sorghum Linkage Group A
This chromosome contains the sps1 gene and sh2 mutant of maize (Mains 1949; Hannah and Nelson 1976) (Table 4), as well as four sugarcane QTLs and one putative QTL. Three sugar content QTLs and one putative QTL corresponded to a region between markers CDSR0067a and pSB0044. The location of sh2 inferred from the maize-sorghum alignment was 3 cM from UMC63 toward marker CSU0451b (Ming et al. 1998; Davis et al. 1999). However, direct mapping of an Sh2 clone (provided by C.L. Hannah, University of Florida) in sorghum showed a different location, near sps1. Further, direct mapping of Sh2 in sugarcane on five homologous LGs showed no association with sugar content, and mostly corresponded to the mapped Sh2 position in sorghum. We tentatively conclude that the inferred location of sh2 based on maize-sorghum alignment is doubtful and that the QTLs are not associated with sh2, although the possibility of a proximal duplication of the sh2 locus is under investigation.
Table 4.
List of Maize Candidate Genes and QTLs for Sugar Content
| Symbol | Bin(maize) | Name and/or gene product |
|---|---|---|
| su1 | 4.05 | sugary1, isoamylase |
| su2 | 6.04–6.05 | sugary2, starch branching enzyme? |
| sh1 | 9.01 | shrunken1, sucrose synthase |
| sh2 | 3.09 | Shrunken2, ADP glucose Pyrophosphorylase |
| se1 | 2.10 | sugary-enhancer1 |
| bt2 | 4.05 | brittle endosperm2, ADP glucose pyrophosphorylase |
| sps1 | 8.06 | sucrose-phosphate synthase1 |
| sus1 | 9.04 | sucrose synthase |
| Qagp3 | 10.05–10.07 | QTL ADP glucose pyrophosphorylase activity 3 |
| Qsps1 | 1.08–1.10 | SPS QTL1 |
| Qsps2 | 2.06–2.07 | SPS QTL2 |
| Qsps3 | 4.07–4.08 | SPS QTL3 |
Sorghum Linkage Group B
Four putative QTLs, one from each sugarcane genotype, corresponded to a genomic region that contains the maize sugar mutant se1 (Ferguson et al. 1978) and also Qsps2, a QTL that modifies the activity of sucrose phosphate synthase in maize (Causse et al. 1995b). One QTL from GG and a putative QTL from MJ corresponded to a common region between CSU0460 and pSB0500 but not to any candidate genes.
Sorghum Linkage Group C
One MJ QTL corresponded to a genomic region that contains Qsps1, another QTL that modifies sucrose phosphate synthase activity in maize (Causse et al. 1995a). One QTL each from MJ and PIN corresponded to a common region near CDSR0035 but not to any candidate genes.
Sorghum Linkage Group D
One QTL each from GG and MJ corresponded to a genomic region that contains Qagp3, a QTL modifying ADP glucose pyrophosphorylase activity in maize (Causse et al. 1995a). Another two QTLs, one each from GG and MJ, corresponded to a region between markers CDSR0046 and CSU0423 but not to any candidate genes.
Sorghum Linkage Group F
Three QTLs, one each from GG and PIN, and a double-dose QTL from MJ, corresponded to a common region between markers CDSC0042 and CDSR0029 that did not contain any candidate genes.
Sorghum Linkage Group G
One QTL each from GG and IND corresponded to a genomic region that contains the maize su2 mutant, thought to encode a starch branching enzyme (Eyster 1934; Baba et al. 1991). Two QTLs, one from MJ and the other double-dose QTL from PIN, and a putative QTL from GG, corresponded to a common region between markers CDO0202 and CDSB0032 that did not contain any candidate genes.
Sorghum Linkage Group H
One putative MJ QTL associated with increased sugar content corresponded to a genomic region that contains Qsps3, the third of three known maize QTL(s) that modifies sucrose phosphate synthase activity (Causse et al. 1995a).
Sorghum Linkage Group I
One QTL from GG and two from IND corresponded to a genomic region that contains the maize sus1 mutant, encoding sucrose synthase (McCarty et al. 1986). The GG QTL was detected by the maize sucrose synthase clone pshD13.
Sorghum Linkage Group J
One putative QTL each from MJ and IND corresponded to a common region near CDSR0133. The maize su1 (encoding isoamylase) (McCarty et al. 1986) and bt2 (encoding ADP glucose pyrophosphorylase) (Emerson et al. 1935; Baba et al. 1991; Miller and Chourey 1995) mutants are located nearby, but appear to be too far away to correspond to the QTLs. The inferred (left of sorghum LG J) and mapped (right) locations of bt2 correspond very well.
DISCUSSION
The autopolyploidy of sugarcane was reflected in a high level of apparent duplication of QTLs, reflected by both correspondence of QTLs from different genotypes and by segregation for QTLs at multiple, apparently homologous locations in individual genotypes. Across the 36 genomic regions that showed significant association with variation in sugar content in the four genotypes, single homologous genomic regions accounted for three QTLs in three cases (CDSR0067a–pSB0044 on LG A, CDSC0042–CDSR0029 on LG F, and pSB0106–UMC0114a on LG I), and two QTLs in five cases (CDSR0035 on LG C, UMC0044–CDSR0125 and CDSR0046–CSU0423 on LG D, CDO0202–CDSB0032 and SG0322–CSU0063b on LG G). In one of these cases (CDSR0067a–pSB0044), plus at least three additional cases (CSU0460–pSB0101 and SHO0038–CDSB0007 on LG B, CDSR0133 on LG J) putative QTLs that fell slightly below our stringent significance threshold also corresponded to significant QTLs or to each other.
The 36 sugar content QTLs (Table 1) correspond to only eight nonoverlapping regions of the sorghum genome (on LGs A, B, C, D, F, G, and I). This suggests that the observed QTLs may be accounted for by a much smaller number of ancestral genes that have been multiplied by the rapid duplication of chromosomes that has characterized sugarcane genome evolution since its divergence from a common ancestor shared with sorghum (Ming et al. 1998).
In six (75%) of these eight nonoverlapping regions, we find both QTLs from high sugar content parents (GG or MJ) that increase sugar content, and also QTLs from low sugar content parents (IND or PIN) that decrease sugar content. While such a result would simply reflect allelism in a diploid, this explanation is not adequate for an autopolyploid F1 population. The mapping of an interspecific F1 population means that the S. officinarum and S. spontaneum alleles have little chance to pair, as the map is based on heterozygosity and recombination that occurs in the S. officinarum and S. spontaneum parents, respectively (and therefore we made maps of each parent; see Ming et al. 1998). Therefore, the effect of each allele is estimated based on the average phenotype(s) of individuals that differ by the presence/absence of the allele from one parent, which is completely independent of the presence/absence of the allele from the other parent. The discovery of QTLs in the same genomic region(s) from S. officinarum (GG or MJ) that increase sugar content, and from S. spontaneum (IND or PIN) that decrease sugar content, provides independent confirmation of the importance of these genomic regions in the control of this trait. Confirmation from different varieties and/or species increases the level of confidence that a QTL exists in the region, and also suggests that DNA markers linked to the QTL may be useful in other germplasm.
One QTL (pSB0279dG) and two putative QTLs (pSB0044dI, pSB1368dP) showed phenotypic effects that were the opposite of what would be predicted based on the phenotypes of the parents contributing these alleles. The discovery of exceptional QTLs from low-sugar wild genotypes that increase sugar content, and QTLs from high-sugar cultivars that reduce sugar content, confer added incentive for incorporating marker-assisted selection into sugarcane breeding programs. Deleterious QTLs from the high-sugar parent could be purged, and favorable QTLs from exotic sources could be introgressed. The phenotypic effects of these unexpected alleles explain part of the observed transgressive segregation for sugar content.
Dosage Effects of Individual QTLs
Multiplex segregation at QT loci may be partly responsible for the phenotypic buffering that is one factor in the success of many autopolyploid crops, as reported in alfalfa for physiological measurements such as net CO2 exchange, acetylene reduction, activities of ribulose-1,5-bisphosphate carboxylase, and leaf tissue concentrations of buffer-soluble protein, chlorophyll, and DNA (Leps et al. 1980; Pfeiffer et al. 1980; Meyers et al. 1982a,b; Molin et al. 1982).
In four cases, two or more loci detected by the same DNA probe were each associated with variation in sugar content, enabling us to investigate the possibility of such phenotypic buffering in sugarcane. Three DNA probes (CDSC0005, CDSC0042, CSU0428) were each diagnostic of two MJ QTLs at unlinked loci, and one DNA probe (CDSB0032) was diagnostic of two PIN QTLs at unlinked loci. Such associations may reflect multiplex segregation at orthologous genetic loci, or perhaps just coincidence of different QTL alleles at nearby loci, but in either case permit us to evaluate the net consequences of stacking multiple copies of a genomic region each associated with common phenotypic effects. “Stacking” of multiple doses of chromosomal segments containing favorable QTLs generally yielded diminishing effects on phenotype, especially in cases where high-order duplication could be tested (Fig. 2). This is similar to the results reported from stacking unlinked QTLs in a diploid, tomato, which were attributed to epistasis (Eshed and Zamir 1996). Epistasis in sugarcane is complicated by the possibility of nonlinear interactions between loci at homologous sites (such as we report), in addition to nonlinear interactions between unrelated loci (Eshed and Zamir 1996).
Detecting this type of phenotypic buffering provides strategic information for marker-assisted selection in autopolyploid crops. Although diagnostic DNA markers enable us to pyramid multiple QTLs in a polyploid, incorporating any one copy of the multiple alleles may obtain most of the desired effect in the breeding population.
Nonadditive gene action in multiple-dose QTLs may also confer evolutionary opportunities. If a single copy of a gene/QTL is physiologically sufficient, the extra copies are free to collect mutations, often becoming nonfunctional, but perhaps occasionally resulting in a distinctive new function which improves fitness.
An important future investigation regards the contribution of multilocus QTL genotypes to stability of performance across different environments. Sugar content is a trait of relatively high heritability (Kang et al. 1983); however, a role of multiple-dose QTLs in enhancing environmental stability would be of potentially great importance for less heritable traits.
It was curious that the highest-sugar genotype, Green German, showed only eight sugar content QTLs—far fewer than the 18 found in Muntok Java, which had much lower sugar content. The high ploidy of Green German (2n = 97–117) would make it possible, at least in principle, that additional favorable QTLs may be present in Green German but in so many doses that most progeny have several copies—and consequently phenotypic variation cannot be associated with marker segregation. This may suggest that our experiment has only detected a subset of the QTLs that are responsible for sugar content—specifically, overlooking those QTLs that have large additive effects and have been driven to high frequencies by selection. This notion could be tested by crossing GG × IND progeny back to IND, and doing further QTL mapping. If this notion is true, then the tendency of multiple-dose QTLs for showing nonlinear dosage effects may be representative only of the subset of alleles that have not yet reached high frequency in improved sugarcane populations.
Candidate Genes for Sugar Content
The complexity of measuring sugar content, and large number of genes influencing the trait, suggest that it will be very difficult to identify the underlying genes using positional approaches alone. An overlapping genetic basis for variation in seed carbohydrate metabolism of grain crops and variation in stem carbohydrate accumulation of biomass crops would be a useful aid in the identification of candidate genes for QTLs affecting sugar content, a complex industrial trait. Perturbations in seed carbohydrate metabolism often result in discrete visible phenotypes, and several underlying genes have been cloned. A growing collection of maize mutants promises to provide additional candidate genes.
Several of the sugarcane QTLs we mapped correspond approximately to the genomic locations of maize mutants: two to sus1, and at least one (perhaps as many as three) to su2. Four additional sugarcane QTLs corresponded approximately to previously mapped maize QTLs that modify the activities of key sugar-metabolizing enzymes: two to ADP glucose pyrophosphorylase (Qagp3), one to sucrose phosphate synthase (Qsps1), and one putative QTL to Qsps3. The rice sps1 gene was also mapped on rice chromosome 1 and corresponded to maize chromosome bin 3.09 (Fig. 1; Sakamoto et al. 1995; Davis et al. 1999). A rice cDNA clone, R1966, which is homologous to the barley sus gene, was mapped on rice chromosome 6 and corresponded to maize chromosome bin 9.03 and sugarcane sus QTLs (Kurata et al. 1994). Because three copies of rice Sucrose Synthase (Wang et al. 1995) and two copies each of maize Sucrose Synthase (Causse et al. 1995b; Miller and Chourey 1995) and Sucrose Phosphate Synthase (McCarty et al. 1986; Binh et al. 1995) have been reported, it is possible that some additional sugar content QTLs in sugarcane might encode or modify the same enzyme but locate on different genomic regions. Even if the candidate genes identified to date prove helpful, other approaches will clearly also be necessary to reveal most (or even much) of the molecular basis for variation in the sugar content of sugarcane. Candidate genes were found for only 17.5% of the sugar content QTLs, and half of the candidates were themselves QTLs. Over all loci examined, the extent of association between sugar content QTLs and candidate genes was strong for Green German, suggestive but equivocal for IND, weak for MJ, and nonexistent for PIN. While sus1 and su2 are strongly implicated as candidates for a direct role in the genetic determination of sugar content, no candidates were found in several genomic regions that showed much stronger evidence of a role in sugar content based on numerous QTLs corresponding to these regions. For example, a sugarcane genomic region corresponding to the central region of sorghum LG A was associated with three QTLs, and one additional putative QTL, but no candidate genes have been found. Similarly, a region near CDSC0042 on sorghum LG F corresponds to four sugarcane QTLs but not to any candidate genes. The recent completion of a database of nearly 300,000 ESTs for sugarcane (http://sucest.lbi.dcc.unicamp.br/en/) and identification of genes involved in most steps of carbohydrate metabolism provide a valuable starting point.
METHODS
Mapping Populations
Two interspecific segregating populations were studied, each made by P. Tai, USDA-ARS, Canal Point, Florida. Due to the heterozygous nature of the sugarcane varieties and species, the progenies of these interspecific crosses were segregating like F2 intercross populations in diploid species based on two populations investigated in this experiment. (1) 264 plants from S. officinarum ‘Green German’ (GG, 2n = 97–117) × S. spontaneum ‘IND 81–146’ (IND, 2n = 52–56) (GG × IND). (2) 239 F1 plants from S. spontaneum ‘PIN 84–1’ (PIN, 2n = 96) × S. officinarum ‘Muntok Java’ (MJ, 2n = 140) (PIN × MJ). The chromosome numbers of a sampling of the progenies from these two crosses were 2n = 73–85 for GG × IND and 2n = 99–121 for PIN × MJ, indicating n + n transmission (Burner 1997).
Each individual plant of the progenies was vegetatively propagated through cuttings for three replications. Both populations were grown from November 1994 to February 1996, as randomized complete block designs with rows 1.5 m apart and plants 0.6 m apart in the row, at the Texas A&M Agricultural Research and Extension Center. As significant replication effects were found at frequencies lower than the Type I error rate, phenotypes of the three replications were averaged for analysis.
The parents of these crosses were chosen for their differences in sugar content, and also based on the inference that two were S. officinarum, and the other two were S. spontaneum (in order to maximize DNA polymorphism). The levels and patterns of DNA polymorphisms among the four genotypes generally supported this inference. However, determining the taxonomic affinity of sugarcane genotypes is often complex. For example, Green German and Muntok Java are listed as S. officinarum in the world catalog of sugarcane genetic stocks (Rao and Vijayalakshmi 1962). The chromosome numbers of original Green German and Muntok Java were 2n = 80 (Bremer 1923; Rao and Vijayalakshmi 1962). A recent investigation revealed the chromosome numbers of modern accessions of GG as 2n = 97–117 and MJ as 2n = 140 (Burner 1997). This raises the possibility that the clones of these two varieties used in the mapping project may be hybrids of unknown ancestry, although morphologically they resemble S. officinarum.
Phenotyping
Sugar content was expressed in the industry standard units of pounds of sugar per ton of cane, equivalent to the content of sucrose at 96% purity, calculated based on Brix and Pol values as described (Legendre and Henderson 1972). Brix is the percentage of all soluble solids, mostly sugars, minerals, and organic acids, in the sugarcane stalk. Pol is the level of sucrose in stalk juice determined by polarimetry; a “clarified” juice sample from which optically active nonsugar compounds have been removed (Birkett and Seip 1975) is placed in a standard optical cylinder and polarized light is passed through the cylinder. The degree of rotation of the plane of light exiting the tube is recorded. Sucrose and glucose are dextro-rotatory, while fructose is levo-rotatory. In sugarcane juice, glucose and fructose levels are usually similar and small, so cancel each other out. Percentages of phenotypic value were calculated from the range of phenotypic value in the segregating population divided by the difference of parental phenotypic value.
Genotyping
RFLP analysis used laboratory methods as previously described (Chittenden et al. 1994). DNA probes used for QTL mapping were selected based on preliminary analysis of 1255 single-dose RFLP markers (i.e., alleles segregating in simplex segregation ratios) for association with phenotypes in 85 plants (Ming et al. 1998); additional probes were picked at 20 cM or smaller intervals for a more comprehensive search of particular regions of the genome showing even tenuous associations with sugar content in the subpopulations. A total of 186 probes were mapped in both populations using methods described (Ming et al. 1998), and generated 243, 232, 122, and 138 single-dose markers for GG, IND, MJ, and PIN, respectively.
Data Analyses
Single-factor ANOVA was conducted (SAS/GLM, SAS Institute 1989) to determine the associations between RFLP markers and sugar content in sugarcane. Correlations among traits were calculated using SAS/CORR. When flanking markers were available, MAPMAKER/QTL version 1.1 was used to calculate LOD scores by interval mapping. Because single-dose markers of sugarcane were segregating in a 1:1 ratio, the same as a backcross population in diploid species, MAPMAKER/QTL for backcross populations was chosen for map construction and QTL analysis (Da Silva et al. 1995; Grivet et al. 1996; Guimarães et al. 1997; Ming et al. 1998). Significance thresholds of LOD gt; 2.5 (interval mapping) or P < 0.003 (analysis of variance) were used to declare QTLs, based on the genome size and marker density in our sugarcane maps (Lander and Botstein 1989). Markers associated with sugar content at P < 0.01 (LOD > 2.0) were deemed “putative QTLs,” but shown on maps only when they corresponded to a genomic region that contained a significant QT locus or loci for sugar content, or a candidate gene in maize. The coefficient of determination R2 was calculated using SAS/GLM for each marker or QTL, as the percentage of phenotypic variation explained by each marker or QTL. The total phenotypic variance explained was estimated by including all significant single- and multiple-dose QTLs in a full model for multiple regression analysis. The allele effect of each single-dose QTL was the average difference in phenotype of individuals differing by one copy of the indicated allele (single-dose versus zero-dose).
When two or more loci detected by the same probe were each associated with a trait at P < 0.01 by single-factor ANOVA, these loci were combined to investigate the effects of double-dose (0–2 copies), triple-dose (0–3) or quadruple-dose (0–4) genotypes for the genomic region(s) containing the favorable alleles using trend analysis (Gomez and Gomez 1984). The average phenotypic value of each class (0 copy, 1 copy, 2 copies, etc.) was used for testing the dosage effect. Linear, quadratic, cubic, and quartic trends for the effect of marker dosage on phenotype were tested using the CONTRAST statement of the SAS/GLM procedure. Strictly additive dosage effects would result in a significant (nonzero) linear trend, but nonsignificant higher-order trends. Significant higher-order trends reflect nonadditive QTL dosage effects. To declare correspondence between candidate genes and sugar content QTLs, the probability of these corresponding QTLs occurring by chance was calculated using the hypergeometric probability distribution as previously described (Lin et al. 1995; Paterson et al. 1995). Specifically, this used the following equation:
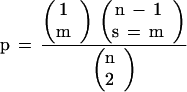 |
where n = the number of intervals which can be compared (defined as 30 cM, approximating a QTL likelihood interval); m = the number of ‘matches’ declared; l = the total number of genes/QTLs found in the larger sample, and s = the number of genes/QTLs found in the smaller sample. The average genomic interval that correspondence between QTLs could be assessed was ∼30 cM, and our sugarcane mapping data afforded comparisons across 1700 cM between maize, GG, and IND, and 1300 cM between maize, MJ, and PIN linkage maps. The number of intervals that could be compared were therefore estimated at 56 (= 1700/30) for GG and IND, and 43 (= 1300/30) for MJ and PIN. A match was declared between maize and sugarcane when corresponding intervals harbor candidate genes/QTLs in each. In the case of unlinked markers associated with sugar content QTLs, a match was declared when a marker associated with sugar content corresponded to the maximum-likelihood location of a maize sugar mutant/QTL within ∼5 cM (since most QTLs spanned ∼30 cM genomic region on average).
Acknowledgments
We thank P. Tai and J.D. Miller for making the crosses; M. McMullen and E. Coe for unpublished data and maize probes; C. Hannah for Sh2 and Bt2 probes; K. K. Wu, J. Betran, and P. Morrell for helpful comments; X. Draye and T. DelMonte for technical help; and the American Sugar Cane League, Australian Sugar Research and Development Corp., Cenicana, Centro de Tecnologia Copersucar, Florida Sugar Cane League, Hawaiian Sugar Planters' Association, Mauritius Sugar Industry Research Institute, USDA Plant Genome Program, Texas Higher Education Coordinating Board, and Texas Agricultural Experiment Station for funding.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL paterson@dogwood.botany.uga.edu; FAX (706) 583-0160.
Article published on-line before print: Genome Res., 10.1101/gr.198801.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.198801.
REFERENCES
- Ahn S, Anderson JA, Sorrells ME, Tanksley SD. Homoeologous relationships of rice, wheat, and maize chromosomes. Mol Gen Genet. 1993;241:483–490. doi: 10.1007/BF00279889. [DOI] [PubMed] [Google Scholar]
- Ahn S, Tanksley SD. Comparative linkage maps of rice and maize genomes. Proc Natl Acad Sci. 1993;90:7980–7984. doi: 10.1073/pnas.90.17.7980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T, Kimura K, Mizuno K, Etoh H, Ishida Y, Shida O, Arai Y. Sequence conservation of the catalytic regions of amylolytic enzymes in maize branching enzyme I. Biochem Biophys Res Commun. 1991;181:87–94. doi: 10.1016/s0006-291x(05)81385-3. [DOI] [PubMed] [Google Scholar]
- Berding N, Moore PH, Smith GR. Advancing in breeding technology for sugarcane. In: Keating BA, Wilson JR, editors. Intensive sugarcane production: Meeting the challenges beyond 2000. Wallingford, UK: CAB International; 1997. pp. 189–220. [Google Scholar]
- Binh NQ, Michaud D, Yelle S, Krivitzky M, Lecharny A. Purification of maize sucrose synthase 1-specific polyclonal antibodies by affinity chromatography. Plant Physiol Biochem. 1995;33:419–422. [Google Scholar]
- Birkett HS, Seip JJ. Core sampling studies. Proc Am Soc Sugar Cane Technol. 1975;4 (New Series):163–177. [Google Scholar]
- Bremer G. A cytological investigation of some species and species-hybrids of the genus Saccharum. Genetica. 1923;5:273–326. [Google Scholar]
- Burner DM. Chromosome transmission and meiotic behavior in various sugarcane crosses. J Am Soc of Sugar Cane Technologists. 1997;17:38–50. [Google Scholar]
- Causse M, Rocher JP, Henry AM, Charcosset A, Prioul JL, de Vienne D. Genetic dissection of the relationship between carbon metabolism and early growth in maize, with emphasis on key-enzyme loci. Mol Breeding. 1995a;1:259–272. [Google Scholar]
- Causse M, Rocher JP, Pelleschi S, Barriere Y, de Vienne D, Prioul JL. Sucrose phosphate synthase: An enzyme with heterotic activity correlated with maize growth. Crop Sci. 1995b;35:995–1001. [Google Scholar]
- Chittenden, L., Schertz, M., K.F., Lin, Y.-R., Wing, R.A., and Paterson, A.H. 1994. A detailed RFLP map of Sorghum bicolor × S. propinquum, suitable for high-density mapping, suggests ancestral duplication of Sorghum chromosomes or chromosomal segments. Theor. Appl. Genet. 87: 925–933. [DOI] [PubMed]
- Da Silva J. “A methodology for genome mapping of autopolyploids and its application to sugarcane (Saccharum spp.).” Ph.D thesis. Ithaca, NY: Cornell University; 1993. [Google Scholar]
- Da Silva J, Honeycutt RJ, Burnquist W, Al-Janabi SM, Sorrells ME, Tanksley SD, Sobral BWS. Saccharum spontaneum L. ‘SES 208’ genetic linkage map combining RFLP- and PCR-based markers. Mol Breeding. 1995;1:165–179. [Google Scholar]
- Daugrois JH, Grivet L, Roques D, Hoarau JY, Lombard H, Glaszmann JC, D'Hont A. A putative major gene for rust resistance linked with a RFLP markers in sugarcane cultivar ‘R570’. Theor Appl Genet. 1996;92:1059–1064. doi: 10.1007/BF00224049. [DOI] [PubMed] [Google Scholar]
- Davis G, McMullen M, Baysdorfer C, Musket T, Grant D, Staebell M, Xu G, Polacco M, Koster L, Melia-Hancock S, et al. A maize map standard with sequenced core markers, grass genome reference points and 932 expressed sequence tagged sites (ESTs) in a 1736-locus map. Genetics. 1999;152:1137–1172. doi: 10.1093/genetics/152.3.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Hont A, Grivet L, Feldmann P, Rao PS, Berding N, Glaszmann JC. Characterisation of the double genome structure of modern sugarcane cultivars (Saccharun spp.) by molecular cytogenetics. Mol Gen Genet. 1996;250:405–413. doi: 10.1007/BF02174028. [DOI] [PubMed] [Google Scholar]
- D'Hont A, Ison D, Alix K, Roux C, Glaszmann JC. Determination of basic chromosome numbers in the genus Saccharum by physical mapping of ribosomal RNA genes. Genome. 1998;41:221–225. [Google Scholar]
- D'Hont A, Rao PS, Feldmann P, Grivet L, Islam-Faridi N, Taylor P, Glaszmann JC. Identification and characterization of sugarcane intergeneric hybrids, Saccharum officinarum × Erianthus arundinaceus, with molecular markers and DNA in situ hybridization. Theor Appl Genet. 1995;91:320–326. doi: 10.1007/BF00220894. [DOI] [PubMed] [Google Scholar]
- Dufour P, Deu M, Grivet L, D'Hont A, Paulet F, Bouet A, Lanaud C, Glaszmann JC, Hamon P. Construction of a composite sorghum genome map and comparison with sugarcane, a related complex polyploid. Theor Appl Genet. 1997;94:409–418. [Google Scholar]
- Emerson RA, Beadle GW, Fraser AC. A summary of linkage studies in maize. Cornell Univ Agric Exp Stn Memor. 1935;180:1–83. [Google Scholar]
- Eshed Y, Zamir D. Less-than-additive epistatic interactions of quantitative trait loci in tomato. Genetics. 1996;143:1807–1817. doi: 10.1093/genetics/143.4.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyster WH. Genetics of Zea mays. Bibliographia Genetica. 1934;11:187–392. [Google Scholar]
- FAS Online. World Sugar Situation. U.S. Department of Agriculture, Foreign Agricultural Service.; 1999. http://www.fas.usda.gov/htp2/sugar/1999/november/world.html http://www.fas.usda.gov/htp2/sugar/1999/november/world.html. Accessed: Dec. 1999. . Accessed: Dec. 1999. [Google Scholar]
- Ferguson JE, Rhodes CA, Dickinson DB. The genetics of sugary enhancer (se), and independent modifier of sweet corn (su) J Hered. 1978;69:377–380. [Google Scholar]
- Gomez KA, Gomez AA. In: Statistical procedures for agricultural research. Gomez KA, Gomez AA, editors. New York: John Wiley & Sons; 1984. pp. 225–233. [Google Scholar]
- Grivet L, D'Hont A, Roques D, Feldmann P, Lanaud C, Glaszmann JC. RFLP mapping in cultivated sugarcane (Saccharum spp.): Genome organization in a highly polyploid and aneuploid interspecific hybrid. Genetics. 1996;142:987–1000. doi: 10.1093/genetics/142.3.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimarães CT, Sills GR, Sobral BWS. Comparative mapping of Andropogoneae: Saccharum L. (sugarcane) and its relation to sorghum and maize. Proc Natl Acad Sci. 1997;94:14261–14266. doi: 10.1073/pnas.94.26.14261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha S, Moore PH, Heinz D, Kato S, Ohmido N, Fukui K. Quantitative chromosome map of the polyploid Saccharum spontaneum by multicolor fluorescence in situ hybridization and imaging methods. Plant Molecular Biology. 1999;39:1165–1173. doi: 10.1023/a:1006133804170. [DOI] [PubMed] [Google Scholar]
- Hannah LC, Nelson OE., Jr Characterization of ADP-glucose pyrophosphorylase from shrunken-2 and brittle-2 mutants of maize. Biochem Genet. 1976;14:547–560. doi: 10.1007/BF00485834. [DOI] [PubMed] [Google Scholar]
- Hulbert SH, Todd ER, Axtell JD, Bennetzen JL. Genetic mapping and characterization of sorghum and related crops by means of maize DNA probes. Proc Natl Acad Sci. 1990;87:4251–4255. doi: 10.1073/pnas.87.11.4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine JE. Saccharum species as horticultural classes. Theor Appl Genet. 1999;98:186–194. [Google Scholar]
- Kang MS, Miller JD, Tai PY P. Genetic and phenotypic path analyses and heritability in sugarcane. Crop Sci. 1983;23:643–647. [Google Scholar]
- Kurata N, Nagamura Y, Yamamoto K, Harushima Y, Sue N, Wu J, Antonio BA, Shomura A, Shimizu T, Lin SY. A 300 kilobase interval genetic map of rice including 883 expressed sequences. Nat Genet. 1994;8:365–372. doi: 10.1038/ng1294-365. [DOI] [PubMed] [Google Scholar]
- Lander ES, Botstein D. Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics. 1989;121:185–199. doi: 10.1093/genetics/121.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendre BL, Henderson MT. The history and development of sugar yield calculations. J Am Soc Sugar Cane Technol. 1972;2:10–18. [Google Scholar]
- Leps WT, Brill WJ, Bingham ET. Effect of alfalfa ploidy on nitrogen fixation. Crop Sci. 1980;20:427–430. [Google Scholar]
- Lin Y-R, Schertz KF, Paterson AH. Comparative analysis of QTLs affecting plant height and maturity across the Poaceae, in reference to an interspecific sorghum population. Genetics. 1995;141:391–411. doi: 10.1093/genetics/141.1.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mains EB. Heritable characters in maize. Linkage of a factor for shrunken endosperm with the a1 factor for aleurone color. J Hered. 1949;40:21–24. doi: 10.1093/oxfordjournals.jhered.a105946. [DOI] [PubMed] [Google Scholar]
- McCarty DR, Shaw JR, Hannah LC. The cloning, genetic mapping, and expression of the constitutive sucrose synthase locus of maize. Proc Natl Acad Sci. 1986;83:9099–9103. doi: 10.1073/pnas.83.23.9099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers SP, Molin WT, Selman BR, Schrader LE. Ploidy effects in isogenic populations of alfalfa III. Chloroplast thylakoid-bound coupling factor 1 in protoplasts and leaves. Plant Physiol. 1982a;70:1715–1717. doi: 10.1104/pp.70.6.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers SP, Nichols SL, Baer GR, Molin WT, Schrader LE. Ploidy effects in isogenic populations of alfalfa I. Ribulose-1,5-bisphosphate carboxylase, soluble protein, chlorophyll, and DNA in leaves. Plant Physiol. 1982b;70:1704–1709. doi: 10.1104/pp.70.6.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M, Chourey P. Intracellular immunolocalization of adenosine 5′-diphosphoglucose pyrophosphorylase in developing endosperm cells of maize (Zea mays L) Planta. 1995;197:522–527. [Google Scholar]
- Milligan SB, Gravois KA, Bischoff KP, Martin FA. Crop effects on broad-sense heritabilities and genetic variances of sugarcane yield components. Crop Sci. 1990;30:344–349. [Google Scholar]
- Ming R, Liu S-C, Lin Y-R, da Silva J, Wilson W, Braga D, Van Deynze A, Wenslaff TF, Wu KK, Moore PH, et al. Detailed alignment of Saccharum and Sorghum chromosomes: Comparative organization of closely-related diploid and polyploid genomes. Genetics. 1998;150:1663–1682. doi: 10.1093/genetics/150.4.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molin GR, Meyers SP, Baer WT, Schrader LE. Ploidy effects in isogenic populations of alfalfa II. Photosynthesis, ghloroplast number, ribulose-1,5-bisphosphate carboxylase, chlorophyll, and DNA in protoplasts. Plant Physiol. 1982;70:1710–1714. doi: 10.1104/pp.70.6.1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore G, Foote T, Helentjaris T, Devos KM, Kurata N, Gall MD. Was there a single ancestral cereal chromosome? Trends Genet. 1995;11:81–82. doi: 10.1016/S0168-9525(00)89005-8. [DOI] [PubMed] [Google Scholar]
- Moore PH, Botha FC, Furbank RT, Grof CRL. Potential for overcoming physio-biochemical limits to sucrose accumulation. In: Keating BA, Wilson JR, editors. Intensive sugarcane production: Meeting the challenges beyond 2000. Wallingford, UK: CAB International; 1997. pp. 141–156. [Google Scholar]
- Paterson AH, Lan T-H, Reischmann KP, Chang C, Lin Y-R, Liu S-C, Burrow MD, Kowalski SP, Katsar CS, DelMonte TA, Feldmann KA, et al. Toward a unified genetic map of higher plants, transcending the monocot-dicot divergence. Nat Genet. 1996;14:380–382. doi: 10.1038/ng1296-380. [DOI] [PubMed] [Google Scholar]
- Paterson AH, Lin Y-R, Li ZK, Schertz KF, Doebley J, Pinson SRMm, Liu S-C, Stansel JW, Irvine JE. Convergent domestication of cereal crops by independent mutations at corresponding genetic loci. Science. 1995;269:1714–1718. doi: 10.1126/science.269.5231.1714. [DOI] [PubMed] [Google Scholar]
- Pfeiffer T, Schrader LE, Bingham ET. Physiological comparison of isogenic diploid-tetraploid, tetraploid-octoploid alfalfa populations. Crop Sci. 1980;20:299–303. [Google Scholar]
- Price S. Cytological studies in Saccharum and allied genera II. Chromosome numbers in interspecific hybrids. Bot Gaz. 1957;118:146–159. [Google Scholar]
- Rao JT, Vijayalakshmi . World catalog of sugarcane genetic stocks. Coimbatore, India: Sugarcane Breeding Institute; 1962. [Google Scholar]
- Sakamoto M, Satozawa T, Kishimoto N, Higo K, Shimada H, Fujimura T. Structure and RFLP mapping of a rice sucrose phosphate synthase (SPS) gene that is specifically expressed in the source organ. Plant Sci. 1995;112:207–217. [Google Scholar]
- SAS Institute. SAS/STAT user's guide, version 6, 4th ed. Gary, NC: SAS Institute; 1989. [Google Scholar]
- Sreenivasan TV, Ahloowalia BS, Heinz DJ. Cytogenetics. In: Heinz DJ, editor. Sugarcane improvement through breeding. Amsterdam: Elsevier Press; 1987. pp. 211–253. [Google Scholar]
- Wang A-Y, Yu W-P, Juang R-H, Huang J-W, Sung H-Y, Su J-C. Presence of three rice sucrose synthase genes as revealed by cloning and sequencing of cDNA. Plant Mol Biol. 1992;18:1191–1194. doi: 10.1007/BF00047725. [DOI] [PubMed] [Google Scholar]
- Wu KK, Burnquist W, Sorrells ME, Tew TL, Moore PH, Tanksley SD. The detection and estimation of linkage in polyploids using single-dose restriction fragments. Theor Appl Genet. 1992;83:294–300. doi: 10.1007/BF00224274. [DOI] [PubMed] [Google Scholar]
- Zeven AC. Polyploidy and domestication: The origin and survival of polyploids in cytotype mixtures. In: Lewis WH, editor. Polyploidy, biological relevance. New York: Plenum Press; 1979. pp. 385–408. [DOI] [PubMed] [Google Scholar]