

Abstract
Glioblastoma multiforme (GBM) is the most common and aggressive malignant brain tumour. Patients afflicted with this disease unfortunately have a very poor prognosis, and fewer than 5% of patients survive for 5 years from the time of diagnosis. Therefore, improved therapies to treat this disease are sorely needed. One such class of drugs that have generated great enthusiasm for the treatment of numerous malignancies, including GBM, is histone deacetylase (HDAC) inhibitors. Pre-clinical data have demonstrated the efficacy of various HDAC inhibitors as anticancer agents, with the greatest effects shown when HDAC inhibitors are used in combination with other therapies. As a result of encouraging pre-clinical data, numerous HDAC inhibitors are under investigation in clinical trials, either as monotherapies or in conjunction with other treatments such as chemotherapy, biologic therapy or radiation therapy. In fact, two actively studied HDAC inhibitors, vorinostat and depsipeptide, were recently approved for the treatment of refractory cutaneous T cell lymphoma. In this review, we first present a patient with GBM, and then discuss the pathogenesis, epidemiology and current treatment options of GBM. Finally, we examine the translation of pre-clinical studies that have demonstrated HDAC inhibitors as potent radiosensitizers in in vitro and in vivo models, to a phase II clinical trial combining the HDAC inhibitor, valproic acid, along with temozolomide and radiation therapy for the treatment of GBM.
Keywords: glioblastoma multiforme, GBM, histone deacetylase inhibitor, HDAC inhibitor, valproic acid, VPA, radiosensitization, radiosensitizer
Case presentation
A previously healthy 41-year-old women was evaluated by her primary care physician for 6 months of light intolerance and headaches. She was initially diagnosed with migraines and started on Imitrex. However, subsequently she noticed a decrease in the peripheral vision of her right eye. A CT scan was performed and revealed a 2 cm × 2 cm × 3 cm lesion in her left parietal occipital lobe. An MRI confirmed a 3.5 cm × 3.4 cm × 3.4 cm cystic enhancing lesion, typical of a glial neoplasm (Fig. 1A). The patient underwent gross total surgical resection of the mass. The pathology of the resected tumour was reported as a malignant astrocytic neoplasm with anaplastic appearing nuclei, mitotic figures, microvascular changes and necrosis, consistent with the diagnosis of GBM [World Health Organization (WHO) Grade IV]. Figure 1B is a characteristic histological image of GBM, similar to what was reported for our patient.
Fig 1.
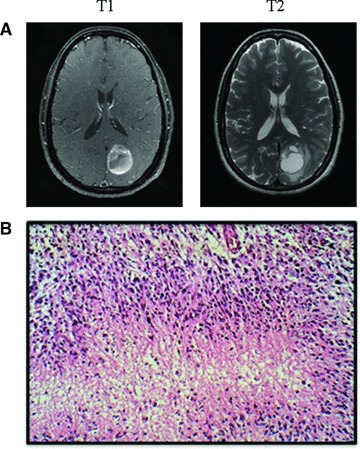
(A) T1 and T2 representative MRI images of the patient at diagnosis. These images display a ring-enhancing lesion in the left parietal occipital lobe, characteristic of GBM. (B) Representative pathological image of GBM displaying pseudopalisading formation of malignant cells surrounding areas of necrosis (Frontalcortex.com).
Following an uneventful surgical recovery, she presented to the Radiation Oncology Clinic at the National Cancer Institute (NCI) of the National Institutes of Health for consultation regarding post-operative radiation therapy and the possibility of enrolling in a clinical trial. At that time, she continued to complain of impaired right peripheral vision and a mild gait disturbance.
The patient had no significant past medical or surgical history, with no known radiation exposure. Family history was significant for a paternal grandfather with lung cancer and a maternal grandmother with colon cancer, with no family history of intracranial neoplasms. She has never smoked cigarettes. On physical exam, she was well appearing, with a neurological exam significant for a wide-based gait and a right peripheral visual field deficit.
Following a comprehensive discussion regarding the risks and benefits of either enrolling in a Phase II clinical trial (NCT00302159) combining the HDAC inhibitor, valproic acid (VPA), with low-dose temozolomide and radiation, versus the standard of care regimen of low-dose temozolomide and radiation, the patient opted to enroll in the clinical trial.
Epidemiology of GBM
The incidence of primary central nervous system malignant tumours is approximately seven cases per 100,000 person years. GBM specifically has an incidence of about three cases per 100,000 person years, and it remains the most common primary malignant brain tumour, comprising 53.8% of such tumours. Disease incidence rises with increasing age, and the majority of cases occur in individuals between 65 and 80 years of age. Unfortunately, survival rates remain poor with approximately only 34% of patients surviving at one year, 12% at 2 years, and less than 5% at 5 years following initial diagnosis [1]. Older age and incomplete surgical resection typically portend a worse prognosis [2].
Prior radiation therapy is a significant environmental risk factor associated with the development of GBM. Neglia et al. examined a cohort of 14,000 survivors from a broad range of paediatric cancers and found that radiation therapy was significantly associated with an increased risk for gliomas (OR = 6.78, 95% CI = 1.54–29.7). The risk increased linearly with increasing doses of radiation. Furthermore, radiation exposure to children under the age of 5 years carried the highest risk of subsequent glioma development, suggesting that the developing brain is more susceptible to radiation-induced carcinogenesis [3]. Interestingly, low-dose ionizing radiation is also a risk factor for the development of malignant glioma, as children treated with 1.5 Gray (Gy) for tinea capitis had an increased incidence of gliomas [4].
Pathogenesis of GBM
GBM tumours are classified by WHO grading system as grade IV astrocytic tumours. All grades of gliomas tend to occur in the white matter of the cerebral hemispheres [5]. GBM is the highest grade glioma and is typically recognized on histology by large areas of necrosis and microvascular proliferation. In addition, GBM tumours characteristically have the appearance of a pseudopalisading formation of malignant cells surrounding areas of necrosis and haemorrhage [5]. Figure 1B demonstrates the typical pathological features of GBM.
Although the histologies of GBM tumours have a common set of features, it is becoming increasingly clear that GBM tumours can be more accurately characterized and distinguished by their genomic and transcriptomic profiles. As such, the NCI, in collaboration with the National Human Genome Research Institute (NHGRI), recently launched a comprehensive research network, The Cancer Genome Atlas (TCGA). TCGA was established to utilize genomic analysis technologies and large-scale sequencing, to better understand the genomic abnormalities that drive tumourigenesis, to help explain resistance to treatment and to ultimately suggest targets of susceptible pathways [6]. GBM was one of the first neoplasms studied under this program, due in large part to its extremely poor prognosis [7]. One of the initial studies using the data generated from TCGA differentiated GBM into four main subgroups, identified as neural, proneural, classical and mesenchymal. These different GBM subtypes were linked with normal neural cell types to provide insight into the possible cell of origin for each of these tumours. More importantly, these different subtypes were correlated with clinical response, which in the future could help guide therapies and inform patients about their appropriate prognoses [8]. TCGA is just one of many efforts attempting to better characterize different subgroups of GBM based on molecular features, to personalize the most effective therapeutic strategies for each individual patient’s tumour.
Standard treatment of GBM
The current front line standard of care therapy for GBM includes maximal surgical resection followed by radiotherapy with concurrent temozolomide, followed by adjuvant temozolomide. Unfortunately, surgical resection is often time compromised by the diffusely infiltrative nature of gliomas and the fact that the tumour often invades critical neurological structures [9].
Following surgical resection, radiation therapy has been shown to increase median survival from 14 to 36 weeks [10]. The initial benefits of radiation were established using whole brain radiation therapy, but improvements in technology, such as the use of involved field radiation therapy, have drastically reduced the side effects associated with radiation. For maximal survival benefit, a total dose of 60 Gy of radiation should be delivered to the tumour. Significantly lower doses of radiation are associated with a reduction in survival benefit [11], whereas doses greater than 60 Gy do not offer additional survival benefit [12].
The addition of temozolomide, an alkylating agent, to post-operative radiation is the only chemotherapeutic agent that provides significant further improvements in the survival of patients with GBM. Stupp et al. performed the landmark Phase III trial comparing post-operative concurrent temozolomide and radiation to radiation alone. The trial found significant improvement in both median survival (14.6 versus 12.1 months) and 2-year overall survival (26.5% versus 10.4%), demonstrating the efficacy of temozolomide [13]. Follow-up of this study revealed that the benefit of temozolomide lasted for at least 5 years, with overall survival rates of 10% in the combination group compared to 2% in the group treated initially with only post-operative radiation [14].
Subsequent analysis of tumour tissue samples from patients in this Phase III trial revealed that silencing of the MGMT (O6-methylguanine–DNA methyltransferase) DNA-repair gene, by promoter methylation, was associated with a greater benefit from the temozolomide therapy. Silenced MGMT expression in tumour cells impairs their ability to repair DNA damage caused by temozolomide, therefore leading to decreased tumour cell survival. Although less significant, patients with an unmethylated MGMT promoter also had modest benefit from the addition of temozolomide, thus, the combination of temozolomide with radiation remains the standard of care therapy for all patients with GBM [15].
Given that GBM tumours are particularly vascular, and overexpress numerous angiogenic factors, there has been much investigation into the utilization of anti-angiogenic therapies as potential treatment options. Recently, the antibody against vascular endothelial growth factor, bevacizumab (Avastin), was granted accelerated approval by the Food and Drug Administration (FDA) for its use as a single agent in recurrent gliomas [16]. This approval was a result of a successful Phase II trial examining the efficacy of both bevacizumab alone and the combination of bevacizumab with irinotecan (topoisomerase 1 inhibitor). When compared to historical controls, both arms had a greater radiological response, improved progression-free survival and improved median survival. However, because the trial was not designed as a comparative study, it is unclear whether bevacizumab with concurrent irinotecan is superior to bevacizumab monotherapy [17].
Bevacizumab, in combination with other therapies, is also under investigation as front line therapy for GBM. A Phase II trial combining bevacizumab with standard of care radiation and temozolomide has thus far demonstrated encouraging clinical responses [18]. Moreover, Phase III randomized clinical trials are currently underway to assess the efficacy of bevacizumab with current standard of care front line therapy for GBM (NCT00884741 and NCT00943826) [19].
Thus far, only small incremental gains have been made in the therapy of GBM, and the current standard of care therapies are limited in their ability to combat this aggressive and fatal tumour. However, there are a number of pre-clinical and clinical studies examining the utility of novel targeted therapies for the treatment of GBM [20]. HDAC inhibitors comprise one promising class of drugs under investigation for various malignancies, including GBM.
HDAC inhibitors as cancer therapy
HDAC inhibitors belong to a class of agents that target the aberrant epigenetic characteristics of tumour cells. Epigenetic changes refer to alterations that affect gene expression and cellular phenotype without modifying the DNA sequence itself. Histone modification is one such mechanism of alteration, and it plays an important role in tumour formation, progression and resistance to treatment [21]. In normal cellular biology, histone proteins help control gene expression by modulating chromatin structure and function. Post-translational modifications of histone tails including acetylation, methylation, ubiquitination and phosphorylation (the histone code), determine how these histone proteins control chromatin remodelling [22].
More specifically, histone acetylation is regulated through the opposing actions of histone acetyltransferases (HATs) and histone deacetylases (HDACs). HATs transfer acetyl moieties to lysine residues and HDACs remove the acetyl moieties. HAT activity relaxes chromatin, permitting various transcription factors to interact with DNA, thereby promoting transcription. In contrast, HDAC activity condenses chromatin, preventing access of transcription factors, which leads to transcriptional repression [23]. HAT inactivity and HDAC over activity have been associated with tumourigenesis [24]. Unlike primary genetic mutations, aberrations in an epigenome are reversible and therefore an intriguing potential target for therapy. Because it is pharmacologically much simpler to inhibit an enzyme rather than to induce one, HDAC inhibition has gained enormous clinical interest as an anticancer strategy.
However, it is not only histone modification that enables HAT and HDAC disturbances to lead to tumour development. HATs and HDACs are also able to alter the acetylation status of numerous non-histone targets, including other proteins involved in gene expression, proliferation, migration, cell death, DNA repair, angiogenesis, inflammation and the immune response [25]. These non-histone targets also likely contribute to tumour progression and resistance to treatments. Correspondingly, HDAC inhibitors have been shown, in a pre-clinical setting, to be effective anticancer agents via multiple mechanisms, including the induction of cell-cycle arrest, intrinsic and extrinsic apoptotic mechanisms, mitotic cell death, autophagic cell death, generation of reactive oxygen species, inhibition of angiogenesis [25] and improvement in NK cell mediated tumour immunity [26]. Because of the overlap of these diverse effects on cancer cells and the effects seen after irradiation (discussed later), HDAC inhibitors are attractive not only as monotherapeutic agents, but also in combination with radiation therapy.
HDAC inhibitors as radiosensitizers
The full therapeutic potential of HDAC inhibitors is likely to be achieved when they are combined with other cancer therapies such as radiation. Particularly, there is accumulating evidence from pre-clinical studies, which shows that HDAC inhibitors from diverse chemical classes are effective radiosensitizers in a variety of malignancies [27]. Pre-clinical examples of malignancies for which HDAC inhibitors have exhibited a radiosensitizing effect include GBM [28–30], head and neck squamous cell cancer [31], non–small-cell lung cancer [32], colorectal cancer [33, 34], prostate cancer [29, 35], melanoma [36] and metastatic breast cancer [37].
The exact mechanism by which HDAC inhibitor induced radiosensitization occurs is currently unknown, but it may be due at least in part to the prevention of the DNA damage repair process. HDAC inhibitors prevent DNA double-strand break (DSB) repair, as demonstrated by prolonged expression of phosphorylated H2AX (γH2AX), a marker for DNA DSBs, following radiation [38]. A variety of HDAC inhibitors have been shown to delay the dispersal of radiation-induced γH2AX foci [28–29, 32, 36, 39–40]. Thus, HDAC inhibitors appear to prevent DNA DSB repair leading to enhanced tumour cell death.
The mechanism for this impaired DNA DSB repair process is not entirely clear. Possible explanations include the down-regulation of certain DNA repair molecules, including Ku70, Ku86, Rad50, Ku80 and Rad51 [39, 41]. In addition, the interaction of HDACs with DNA damage response proteins, such as 53BP1 [42], may play an important role in HDAC inhibitor radiosensitization. Furthermore, HDAC inhibitors may affect a tumour cell’s response to radiation by altering chromatin condensation patterns. Chromatin exists in a condensed form, known as heterochromatin in which there is decreased gene expression, and a more open form, known as euchromatin in which there is increased gene expression [22]. A recent study reported that tumour cells grown in a three-dimensional matrix exhibit an elevated amount of heterochromatin, similar to tumour xenografts, and they are more resistant to ionizing radiation when compared to cells grown in two-dimensional monolayer cultures. Treatment with the HDAC inhibitor, LBH589, induced chromatin decondensation in cells grown in a three-dimensional culture, which correlated with an increase in DNA DSBs and radiosensitivity. Therefore, the relaxation of chromatin induced by HDAC inhibition may increase radiation-induced DNA DSBs, and thereby increase killing of tumour cells [43].
A better understanding of the mechanism of HDAC inhibitor induced radiosensitization will help guide the effective and safe use of HDAC inhibitors in combination with radiation. At present, it appears that the pre-clinical protocol for maximal radiosensitization induced by a chemically diverse set of HDAC inhibitors requires drug exposure both pre- and post-irradiation [28, 35, 44]; cells exposed to drug only pre-irradiation typically display a very modest radiosensitization [35]. Interestingly, the HDAC inhibitors MS-275 and VPA have also been studied when administered only post-irradiation, with differing results. MS-275 displays only minimal radiosensitization when given exclusively post-irradiation [35]. However, VPA effectively radiosensitizes cells when administered up to 24 hrs following radiation treatment, although the degree of enhancement is not as robust compared to cells treated both pre- and post-irradiation [45]. Taken together, these studies suggest the importance of maintaining sufficiently high HDAC inhibitor levels both before the tumour cells are irradiated and after irradiation, as the cells attempt to repair the radiation-induced DNA damage. A further understanding of this mechanism of radiosensitization should aid in determining the optimal timing for drug administration relative to the radiation treatment, to maximize the therapeutic effect of this combined modality.
HDAC inhibitors and normal tissue radioresponse
To maximize the therapeutic ratio of a radiosensitizing agent, it is important that the drug not enhance the radiation-induced toxicity of normal cells or tissue. It is hypothesized that the aberrant HDAC activity exhibited in tumour cells makes them more susceptible to HDAC inhibitors, and thus, normal tissue toxicity should be minimized. However, there is conflicting pre-clinical data regarding the specificity of HDAC inhibitors for cancer cells compared to normal cells, which should be further clarified given the important clinical safety implications.
Several in vitro studies have reported that exposure to HDAC inhibitors does not increase the radiosensitivity of various normal tissue cell lines, including fibroblasts and osteoblasts [36, 41, 45]. In addition, when examined in normal breast and intestinal epithelial cell lines, HDAC inhibitors did not enhance the cellular toxicity induced by a variety of chemotherapeutic DNA damaging agents [46]. Moreover, in vitro data suggest that HDAC inhibitors may actually protect normal tissue from radiation-induced toxicities. For example, the HDAC inhibitor phenylbutyrate has been shown to improve both DNA repair and cell survival in normal fibroblast cell lines [47].
In contrast, it has also been reported that some HDAC inhibitors do inhibit the repair of radiation-induced DNA DSBs and increase cellular radiosensitivity of normal cells, specifically lymphocytes [48] and primary skin fibroblasts in vitro [49]. In the study examining lymphocytes, the HDAC inhibitor sodium butyrate inhibited radiation-induced DNA DSBs. However, there was no mention of cell survival, which makes it difficult to assess for a change in radiation sensitivity as measured by cell death [48]. In the study using fibroblasts, the radiosensitizing effect was demonstrated to vary as a function of the type of HDAC inhibitor used. Of note, fibroblasts treated with VPA did not have an increase in DNA DSBs compared to radiation alone and displayed only a very modest increase in radiation sensitivity [49]. These results indicate that HDAC inhibitors are a complex class of molecules with varied responses to similar assays requiring in-depth study of each drug for its effects on tumour and normal cells.
In addition, there is growing in vivo evidence that HDAC inhibitors can actually protect normal tissue from radiation-induced side effects. When applied topically to mice, HDAC inhibitors such as phenylbutyrate, trichostatin A and VPA provide protection against numerous well-defined toxicities of radiation therapy, including cutaneous radiation syndrome, long-term skin fibrosis and radiation-induced malignancies [50]. These observations are correlated at a molecular level with inhibition of pro-inflammatory cytokines such as tumour necrosis factor (TNF)-α and transforming growth factor (TGF)-β, which may be one of the mechanisms by which HDAC inhibitors protect normal tissue [50]. Similarly, an additional study in mice demonstrates that topical phenylbutyrate effectively reduces the severity of radiation-induced oral mucositis, a common and morbid side effect of radiotherapy treatment for head and neck cancers. These findings are similarly associated with a decrease in the expression of TNF-α[47]. Therapies with the potential to mitigate the incidence and severity of radiation-induced normal tissue toxicity would have tremendous clinical benefit, as often times it is the normal tissue toxicity that limits the delivery of the radiation dose necessary for tumour control.
Clinical safety of HDAC inhibitors
With HDAC inhibitors now entering the clinical arena as radiosensitizing agents, there is growing clinical evidence to support the safety of these compounds when delivered in combination with radiotherapy. Initial data from our trial involving VPA, temozolomide and radiation for the treatment of GBM have not shown significantly increased toxicity compared to what is reported in the standard of care treatment regimen of radiation and temozolomide alone. The most significant side effects observed have been drug-related neurological toxicities, which are reversible within 72 hrs after the cessation of VPA [51].
In addition, a retrospective study of paediatric patients with high-grade gliomas who were treated with VPA as an anti-seizure medication, along with standard radiochemotherapy, did not experience an increase in toxicity. Although this was a retrospective study, it offers encouraging results, which suggest that VPA can be safely combined with radiation and chemotherapy [52].
Furthermore, Ree et al. recently published the first Phase I trial combining an HDAC inhibitor with radiation. Specifically, they assessed the use of vorinostat combined with pelvic palliative radiation for the treatment of various gastrointestinal carcinomas. Although the study was not designed to determine the additive toxicity of vorinostat to the radiation regimen, the authors determined a safe maximum tolerated dose (MTD) of vorinostat in conjunction with pelvic radiation. The results from this study now pave the way to use the MTD of vorinostat in conjunction with more long-term curative radiation treatment of certain pelvic malignancies [53]. Our understanding of the tolerability and toxicity profiles of HDAC inhibitors combined with radiation will continue to improve as more HDAC inhibitors are introduced into the clinical setting as potential radiosensitizing agents.
Biomarkers and HDAC inhibitors
When investigating a pharmaceutical agent, it is useful to identify a safe and accurate method to measure drug delivery and activity. Because histone acetylation is a direct downstream consequence of HDAC inhibition, one of the most common methods of measuring the activity of HDAC inhibitors is to test for the hyperacetylation of histone H3 and histone H4. Although the most direct method of testing for tumour histone hyperacetylation is via tumour biopsy, such a procedure is too invasive to consistently use for determining drug activity in the clinical setting. Therefore, there is interest in utilizing peripheral lymphocytes as surrogate biomarkers for global acetylation after HDAC inhibitor treatment. Although testing peripheral lymphocyte histone hyperacetylation is a potential strategy for optimizing protocols that involve HDAC inhibition and radiation therapies [29], this method does not account for inherent tumour resistance mechanisms, such as drug efflux pumps or the inability of the drug to penetrate bulky tumour masses.
If histone acetylation status proves to be an effective method for measuring drug delivery, it will be additionally important to find susceptibility biomarkers to identify patients who will most likely benefit from HDAC inhibitor therapy. As such, predictive biomarkers are vitally important when testing drugs and tracking patients in clinical trials. For example, if only a small subset of patients benefit from therapy, the success of these responders will likely be lost in the statistics of the larger trial if the subset cannot be adequately identified and analysed.
Interestingly, HR23B, a protein that transports ubiquitinated cargo to proteosomes, was identified as a marker for the sensitivity of HDAC inhibitor induced apoptosis. The proposed mechanism involves disruption of HDACs’ normal inhibition of HR23B, and thus stabilization of proteosome activity. Without inhibition of HR23B, the proteosome becomes overloaded, leading to ER stress and apoptosis [54–55]. In patients with cutaneus T cell lymphoma (CTCL), a disease that typically responds to HDAC inhibitors, there is an association of HR23B expression with the clinical response to vorinostat. Specifically, up-regulated HR23B levels were shown to have a positive predictive value of 71.7% when measuring patient response to vorinostat [55]. Therefore, measuring a tumour’s HR23B status could provide an important predictive value for determining which patients with CTCL are likely to respond to HDAC inhibitor based therapy. This study also opens the door for assessing HR23B expression in other neoplasms and measuring how the levels correlate with tumour response to HDAC inhibitors.
Unlike HR23B in CTCL, to date there are no known biomarkers that characterize patients who are more likely to respond to HDAC inhibitors in combination with radiation. However, as we better understand how HDAC inhibitors enhance tumour cell radiosensitivity, we can utilize this knowledge to search for molecular biomarkers to predict this response.
Clinical protocols and HDAC inhibitors
Thus far, vorinostat and depsipeptide are the only HDAC inhibitors that have achieved FDA approval for cancer therapy, specifically for the treatment of refractory CTCL [56–58]. However, there are many ongoing clinical trials assessing the efficacy and safety of other HDAC inhibitors, used alone or in combination therapies, in both solid and haematologic malignancies. Recently, a number of clinical trials have begun to specifically examine the combination of HDAC inhibitors with radiation for various malignancies (Table 1) [19].
Table 1.
Current clinical trials involving radiation and HDAC inhibitors
| HDAC inhibitor | Chemotherapeutic or biological agent(s) | Type of malignancy | Phase | Trial identifier |
|---|---|---|---|---|
| Panobinostat | None | Prostate, oesophageal and head and neck cancer | I | NCT00670553 |
| Vorinostat | None | Brain metastases | I | NCT00838929 |
| Vorinostat | None | Brain metastases from non-small cell lung cancer | I | NCT00946673 |
| Vorinostat | None | Non-small cell lung cancer | I | NCT00821951 |
| Vorinostat | None | Pelvic cancer | I | NCT00455351 |
| Vorinostat* | None | Resistant/relapsed neuroblastoma | I | NCT01019850 |
| Vorinostat | Capecitabine | Non-metastatic pancreatic cancer | I | NCT00983268 |
| Vorinostat | Cisplatin | Squamous cell carcinoma of the oropharynx | I | NCT01064921 |
| Vorinostat | None | Locally advanced pancreatic cancer | I/II | NCT00831493 |
| Vorinostat | Temozolomide | Glioblastoma multiforme | I/II | NCT00731731 |
| Vorinostat | Paclitaxel | Inoperable stage III non-small cell lung cancer | I/II | NCT00662311 |
| Vorinostat | 5-FU | Pancreatic cancer | I/II | NCT00948688 |
| Valproic acid | Hydralazine, Cisplatin | Cervical cancer | II | NCT00404326 |
| Valproic acid | Temozolomide | Glioblastoma multiforme | II | NCT00302159 |
| Valproic acid | Bevacizumab | Paediatric high-grade gliomas | II | NCT00879437 |
Radiation form is the radioactive drug, iobenguane I 131.
Reports from Phase II data on HDAC inhibitors as monotherapies for various solid malignancies demonstrate that they are well tolerated with good toxicity profiles compared to current standard cancer therapies. In general, the side effects of HDAC inhibitors are reversible with drug cessation and primarily include fatigue, nausea, dehydration, diarrhoea, prolonged QT, thrombocytopenia, lymphopenia and neutropenia [59].
Despite the favourable toxicity profile, HDAC inhibitors used as monotherapies in solid cancers do not tend to significantly improve outcomes compared to current standard therapies [60–64]. However, a Phase II study of vorinostat for refractory GBM offers some promise. Although displaying only modest efficacy, this study met its primary endpoint with 9/52 patients progression-free at 6 months. Encouragingly, the patients who met this primary endpoint had a long duration of stable disease, indicating that there may be a subset of patients who benefit from HDAC inhibitor therapy [63].
More encouraging examples of the potential for HDAC inhibitors in cancer therapy are evident from trials combining HDAC inhibitors with chemotherapy. A Phase II randomized, double-blind clinical trial showed that vorinostat enhances the efficacy of carboplatin and paclitaxel for advanced stage non–small-cell lung cancer (NSCLC). Specifically, upon comparison of chemotherapy plus vorinostat to chemotherapy alone, there was an increase in confirmed response rates (34% versus 12.5%, P = 0.02). There was also an increase in overall and progression-free survival, but these endpoints did not reach statistical significance. However, these gains were associated with increased toxicity in the vorinostat group, particularly with a significant increase in grade 4 platelet toxicity [65].
In addition, another Phase II clinical trial examined the utility of VPA, hydralazine (a demethylating agent), and different chemotherapies for various solid malignancies refractory to chemotherapy [66]. In this study, the investigators noted an 80% clinical response, defined as either a partial response or stable disease. Again, the toxicity profile was not negligible, consisting of a significant amount of grade 4 haematologic toxicity. Because this study enrolled patients with multiple types of neoplasms (ovarian, cervical, breast, testicular and lung), the results specific to each type of neoplasm are difficult to analyse. Yet, the apparent efficacy in multiple malignancies provides some evidence that combining chemotherapy and HDAC inhibitors may have utility in a large spectrum of cancers [66].
Ongoing NIH/NCI HDAC inhibitor (VPA) trial
VPA is an ideal agent to study as a GBM radiosensitizer for multiple reasons. First, it shows impressive pre-clinical efficacy as a radiosensitizer in glioma cell lines in vitro and in vivo, at a dose comparable to one that can be achieved in the clinic (Fig. 2) [28]. Furthermore, VPA is FDA approved, and it is safely and commonly used for the treatment of epilepsy, bipolar disorder and migraine prophylaxis [67]. Finally, VPA effectively crosses the blood–brain barrier, as demonstrated by its anti-seizure, anti-migraine and mood modulatory activity.
Fig 2.
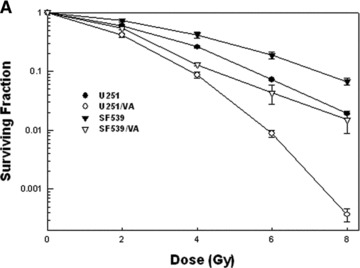
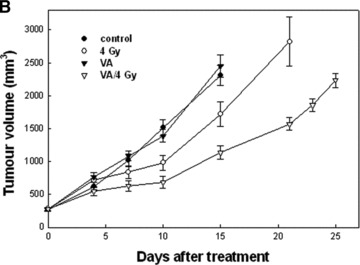
Excerpt graphs from Camphausen et al. showing in vitro and in vivo radiosensitization of glioma cells by valproic acid treatment (28). (A) U251 and SF539 glioma cells were treated with valproic acid both before and following radiation. Clonogenic survival curves reveal an increase in radiosensitivity in valproic acid treated cells. (B) The effects of valproic acid and radiation on tumour growth delay. U251 glioma cells were implanted into the hind leg of mice and divided into four treatment groups: (1) control, (2) 4 Gy radiation, (3) valproic acid, (4) valproic acid and 4 Gy radiation. The combination treatment regimen showed the most significant tumour growth delay.
Therefore, starting in 2006, we opened a multi-institutional Phase II clinical trial of VPA in combination with temozolomide and radiation therapy for patients with high-grade gliomas. The primary endpoints of this trial are both progression-free survival and overall survival. The secondary endpoint is regimen toxicity. As a surrogate marker for drug activity, peripheral lymphocytes are assessed for histone acetylation. The treatment regimen includes (1) fractionated radiotherapy for 6.5 weeks to a total dose of 60 Gy, (2) oral VPA (25mg/kg/day) twice daily on days 7–49 and (3) oral temozolomide daily throughout the course of radiation. Four weeks following the completion of the radiation regimen, patients receive adjuvant temozolomide. Patients are followed with MRI scans, including 1 month after the completion of the radiation treatment, every 3 months for the first 2 years, and then every 6 months. The total accrual goal for this study is 41 patients and 21 patients have been accrual as of January 2011.
Patient follow-up
The patient presented earlier completed the experimental protocol of 6 weeks of concurrent VPA, temozolomide and radiation to a total dose of 60 Gy, with minimal toxicity. She continued with a total of 20 cycles of adjuvant temozolomide. At the time that this patient was presented at the Grand Rounds lecture, it had been approximately 18 months since her last dose of temozolomide and 3.5 years since her initial diagnosis of GBM. The patient remains clinically and radiologically stable with no evidence of disease progression. Figure 3 shows the patient’s most recent MRI image compared to her initial scans at diagnosis and the scans from post-surgical resection. She continues to be followed with regular follow-up visits and MRI scans. Aside from the presenting visual field defect, she remains symptom-free.
Fig 3.
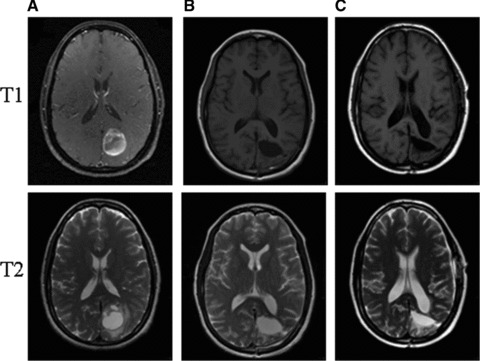
A series of T1 and T2 weighted Brain MRIs from the presented patient with GBM. (A) Represents images from the initial diagnosis. (B) Represents images shortly after gross surgical resection, showing typical post-surgical changes. (C) Represents the most recent MRI images with no evidence of disease recurrence 3 years after the initial diagnosis of GBM.
Conclusion
Given the number of active clinical trials combining HDAC inhibitors and ionizing radiation, in addition to the trials involving HDAC inhibitors and chemotherapy or biologic agents, the next few years will be revealing regarding the efficacy and safety of HDAC inhibitors as anticancer agents.
Acknowledgments
This research was supported in part by the Intramural Research Program of the National Institutes of Health, National Cancer Institute.
Conflict of interest
The authors confirm that there are no conflicts of interest.
References
- 1.Central Brain Tumor Registry of the United States. 2010. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2004–2006. Source: Central Brain Tumor Registry of the United States, Hinsdale, IL. Website: http://www.cbtrus.org/2010-NPCR-SEER/CBTRUS-WEBREPORT-Final-3-2-10.pdf.
- 2.Lamborn KR, Chang SM, Prados MD. Prognostic factors for survival of patients with glioblastoma: recursive partitioning analysis. Neuro-oncol. 2004;6:227–35. doi: 10.1215/S1152851703000620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neglia JP, Robison LL, Stovall M, et al. New primary neoplasms of the central nervous system in survivors of childhood cancer: a report from the Childhood Cancer Survivor Study. J Natl Cancer Inst. 2006;98:1528–37. doi: 10.1093/jnci/djj411. [DOI] [PubMed] [Google Scholar]
- 4.Ron E, Modan B, Boice JD, Jr, et al. Tumors of the brain and nervous system after radiotherapy in childhood. N Engl J Med. 1988;319:1033–9. doi: 10.1056/NEJM198810203191601. [DOI] [PubMed] [Google Scholar]
- 5.Ironside JW, Weller RO, Moss TH. Astrocytic tumours. In: Ironside JW, Moss TH, Louis DN, Lowe JS, Weller RO, et al., editors. Diagnostic pathology of nervous system tumours. London: Churchill Livingstone; 2002. pp. 88–96. [Google Scholar]
- 6.The Cancer Genome Atlas. http://cancergenome.nih.gov.
- 7.McLendon R, Friedman A, Bigner D, et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008 doi: 10.1038/nature07385. 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kanu OO, Mehta A, Di C, et al. Glioblastoma multiforme: a review of therapeutic targets. Expert Opin Ther Targets. 2009;13:701–18. doi: 10.1517/14728220902942348. [DOI] [PubMed] [Google Scholar]
- 10.Walker MD, Alexander E, Jr, Hunt WE, et al. Evaluation of BCNU and/or radiotherapy in the treatment of anaplastic gliomas. A cooperative clinical trial. J Neurosurg. 1978;49:333–43. doi: 10.3171/jns.1978.49.3.0333. [DOI] [PubMed] [Google Scholar]
- 11.Coffey RJ, Lunsford LD, Taylor FH. Survival after stereotactic biopsy of malignant gliomas. Neurosurgery. 1988;22:465–73. doi: 10.1227/00006123-198803000-00003. [DOI] [PubMed] [Google Scholar]
- 12.Chang CH, Horton J, Schoenfeld D, et al. Comparison of postoperative radiotherapy and combined postoperative radiotherapy and chemotherapy in the multidisciplinary management of malignant gliomas. A joint Radiation Therapy Oncology Group and Eastern Cooperative Oncology Group study. Cancer. 1983;52:997–1007. doi: 10.1002/1097-0142(19830915)52:6<997::aid-cncr2820520612>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 13.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 14.Stupp R, Hegi ME, Mason WP, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–66. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 15.Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 16.US Food and Drug Administration. Website: http://www.fda.gov.
- 17.Friedman HS, Prados MD, Wen PY, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27:4733–40. doi: 10.1200/JCO.2008.19.8721. [DOI] [PubMed] [Google Scholar]
- 18.Lai A, Nghiemphu P, Green R, et al. Phase II trial of bevacizumab in combination with temozolomide and regional radiation therapy for up-front treatment of patients with newly diagnosed glioblastoma multiforme (abstract) J Clin Oncol. 2009;27:87s. doi: 10.1016/j.ijrobp.2007.11.068. [DOI] [PubMed] [Google Scholar]
- 19.Clinicaltrials.gov. A service of the US National Institutes of Health. Website: http://www.clinicaltrials.gov.
- 20.Clarke J, Butowski N, Chang S. Recent advances in therapy for glioblastoma. Arch Neurol. 2010;67:279–83. doi: 10.1001/archneurol.2010.5. [DOI] [PubMed] [Google Scholar]
- 21.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–92. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 23.Thiagalingam S, Cheng KH, Lee HJ, et al. Histone deacetylases: unique players in shaping the epigenetic histone code. Ann N Y Acad Sci. 2003;983:84–100. doi: 10.1111/j.1749-6632.2003.tb05964.x. [DOI] [PubMed] [Google Scholar]
- 24.Marks PA, Richon VM, Miller T, et al. Histone deacetylase inhibitors. Adv Cancer Res. 2004;91:137–68. doi: 10.1016/S0065-230X(04)91004-4. [DOI] [PubMed] [Google Scholar]
- 25.Marks PA, Xu WS. Histone deacetylase inhibitors: potential in cancer therapy. J Cell Biochem. 2009;107:600–8. doi: 10.1002/jcb.22185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.López-Soto A, Folgueras AR, Seto E, et al. HDAC3 represses the expression of NKG2D ligands ULBPs in epithelial tumour cells: potential implications for the immunosurveillance of cancer. Oncogene. 2009;28:2370–82. doi: 10.1038/onc.2009.117. [DOI] [PubMed] [Google Scholar]
- 27.Camphausen K, Tofilon PJ. Inhibition of histone deacetylation: a strategy for tumor radiosensitization. J Clin Oncol. 2007;25:4051–6. doi: 10.1200/JCO.2007.11.6202. [DOI] [PubMed] [Google Scholar]
- 28.Camphausen K, Cerna D, Scott T, et al. Enhancement of in vitro and in vivo tumor cell radiosensitivity by valproic acid. Int J Cancer. 2005;114:380–6. doi: 10.1002/ijc.20774. [DOI] [PubMed] [Google Scholar]
- 29.Camphausen K, Burgan W, Cerra M, et al. Enhanced radiation-induced cell killing and prolongation of gamma H2AX foci expression by the histone deacetylase inhibitor MS-275. Cancer Res. 2004;64:316–21. doi: 10.1158/0008-5472.can-03-2630. [DOI] [PubMed] [Google Scholar]
- 30.Kim JH, Shin JH, Kim IH. Susceptibility and radiosensitization of human glioblastoma cells to trichostatin A, a histone deacetylase inhibitor. Int J Radiat Oncol Biol Phys. 2004;59:1174–80. doi: 10.1016/j.ijrobp.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Y, Jung M, Dritschilo A, et al. Enhancement of radiation sensitivity of human squamous carcinoma cells by histone deacetylase inhibitors. Radiat Res. 2004;1:667–74. doi: 10.1667/rr3192. [DOI] [PubMed] [Google Scholar]
- 32.Geng L, Cuneo KC, Fu A, et al. Histone deacetylase (HDAC) inhibitor LBH589 increases duration of gamma-H2AX foci and confines HDAC4 to the cytoplasm in irradiated non-small cell lung cancer. Cancer Res. 2006;66:11298–304. doi: 10.1158/0008-5472.CAN-06-0049. [DOI] [PubMed] [Google Scholar]
- 33.Folkvord S, Ree AH, Furre T, et al. Radiosensitization by SAHA in experimental colorectal carcinoma models-in vivo effects and relevance of histone acetylation status. Int J Radiat Oncol Biol Phys. 2009;74:546–52. doi: 10.1016/j.ijrobp.2009.01.068. [DOI] [PubMed] [Google Scholar]
- 34.Chen X, Wong P, Radany E, et al. HDAC inhibitor, valproic acid, induces p53-dependent radiosensitization of colon cancer cells. Cancer Biother Radiopharm. 2009;24:689–99. doi: 10.1089/cbr.2009.0629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Camphausen K, Scott T, Sproull M, et al. Enhancement of xenograft tumor radiosensitivity by the histone deacetylase inhibitor MS-275 and correlation with histone hyperacetylation. Clin Cancer Res. 2004;10:6066–71. doi: 10.1158/1078-0432.CCR-04-0537. [DOI] [PubMed] [Google Scholar]
- 36.Munshi A, Kurland JF, Nishikawa T, et al. Histone deacetylase inhibitors radiosensitize human melanoma cells by suppressing DNA repair activity. Clin Cancer Res. 2005;11:4912–22. doi: 10.1158/1078-0432.CCR-04-2088. [DOI] [PubMed] [Google Scholar]
- 37.Baschnagel A, Russo A, Burgan WE, et al. Vorinostat enhances the radiosensitivity of a breast cancer brain metastatic cell line grown in vitro and as intracranial xenografts. Mol Cancer Ther. 2009;8:1589–95. doi: 10.1158/1535-7163.MCT-09-0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rogakou EP, Pilch DR, Orr AH, et al. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–68. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 39.Munshi A, Tanaka T, Hobbs ML, et al. Vorinostat, a histone deacetylase inhibitor, enhances the response of human tumor cells to ionizing radiation through prolongation of gamma-H2AX foci. Mol Cancer Ther. 2006;5:1967–74. doi: 10.1158/1535-7163.MCT-06-0022. [DOI] [PubMed] [Google Scholar]
- 40.Zhang Y, Adachi M, Zhao X, et al. Histone deacetylase inhibitors FK228, N-(2-aminophenyl)-4-[N-(pyridin-3-yl-methoxycarbonyl) amino-methyl]benzamide and m-carboxycinnamic acid bis-hydroxamide augment radiation-induced cell death in gastrointestinal adenocarcinoma cells. Int J Cancer. 2004;110:301–8. doi: 10.1002/ijc.20117. [DOI] [PubMed] [Google Scholar]
- 41.Blattmann C, Oertel S, Ehemann V, et al. Enhancement of radiation response in osteosarcoma and rhabomyosarcoma cell lines by histone deacetylase Inhibition. Int J Radiat Oncol Biol Phys. 2010;78:237–45. doi: 10.1016/j.ijrobp.2010.03.010. [DOI] [PubMed] [Google Scholar]
- 42.Kao GD, McKenna WG, Guenther MG, et al. Histone deacetylase 4 interacts with 53BP1 to mediate the DNA damage response. J Cell Biol. 2003;160:1017–27. doi: 10.1083/jcb.200209065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Storch K, Eke I, Borgmann K, et al. Three-dimensional cell growth confers radioresistance by chromatin density modification. Cancer Res. 2010;70:3925–34. doi: 10.1158/0008-5472.CAN-09-3848. [DOI] [PubMed] [Google Scholar]
- 44.Cerna D, Camphausen K, Tofilon PJ. Histone deacetylation as a target for radiosensitization. Curr Top Dev Biol. 2006;73:173–204. doi: 10.1016/S0070-2153(05)73006-4. [DOI] [PubMed] [Google Scholar]
- 45.Chinnaiyan P, Cerna D, Burgan WE, et al. Postradiation sensitization of the histone deacetylase inhibitor valproic acid. Clin Cancer Res. 2008;14:5410–5. doi: 10.1158/1078-0432.CCR-08-0643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim MS, Blake M, Baek JH, et al. Inhibition of histone deacetylase increases cytotoxicity to anticancer drugs targeting DNA. Cancer Res. 2003;63:7291–300. [PubMed] [Google Scholar]
- 47.Chung YL, Lee MY, Pui NN. Epigenetic therapy using the histone deacetylase inhibitor for increasing therapeutic gain in oral cancer: prevention of radiation-induced oral mucositis and inhibition of chemical-induced oral carcinogenesis. Carcinogenesis. 2009;30:1387–97. doi: 10.1093/carcin/bgp079. [DOI] [PubMed] [Google Scholar]
- 48.Stoilov L, Darroudi F, Meschini R, et al. Inhibition of repair of X-ray-induced DNA double-strand breaks in human lymphocytes exposed to sodium butyrate. Int J Radiat Biol. 2000;76:1485–91. doi: 10.1080/09553000050176243. [DOI] [PubMed] [Google Scholar]
- 49.Purrucker JC, Fricke A, Ong MF, et al. HDAC inhibition radiosensitizes human normal tissue cells and reduces DNA Double-Strand Break repair capacity. Oncol Rep. 2010;23:263–9. [PubMed] [Google Scholar]
- 50.Chung YL, Wang AJ, Yao LF. Antitumor histone deacetylase inhibitors suppress cutaneous radiation syndrome: implications for increasing therapeutic gain in cancer radiotherapy. Mol Cancer Ther. 2004;3:317–25. [PubMed] [Google Scholar]
- 51.Kamrava M, Citrin D, Sproull M, et al. Acute toxicity in a phase II clinical trial of valproic acid in combination with temodar and radiation therapy in patients with glioblastoma multiforme (abstract 2094) Int J Radiat Oncol Biol Phys. 2008;72(Suppl):S211. [Google Scholar]
- 52.Masoudi A, Elopre M, Amini E, et al. Influence of valproic acid on outcome of high-grade gliomas in children. Anticancer Res. 2008;28:2437–42. [PubMed] [Google Scholar]
- 53.Ree AH, Dueland S, Folkvord S, et al. Vorinostat, a histone deacetylase inhibitor, combined with pelvic palliative radiotherapy for gastrointestinal carcinoma: the Pelvic Radiation and Vorinostat (PRAVO) phase 1 study. Lancet Oncol. 2010;11:459–64. doi: 10.1016/S1470-2045(10)70058-9. [DOI] [PubMed] [Google Scholar]
- 54.Fotheringham S, Epping MT, Stimson L, et al. Genome-wide loss-of-function screen reveals an important role for the proteasome in HDAC inhibitor-induced apoptosis. Cancer Cell. 2009;15:57–66. doi: 10.1016/j.ccr.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 55.Khan O, Fotheringham S, Wood V, et al. HR23B is a biomarker for tumor sensitivity to HDAC inhibitor-based therapy. Proc Natl Acad Sci U S A. 2010;107:6532–7. doi: 10.1073/pnas.0913912107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Olsen EA, Kim YH, Kuzel TM, et al. Phase IIB multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007;25:3109–15. doi: 10.1200/JCO.2006.10.2434. [DOI] [PubMed] [Google Scholar]
- 57.Duvic M, Talpur R, Ni X, et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL) Blood. 2007;109:31–9. doi: 10.1182/blood-2006-06-025999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Piekarz RL, Frye R, Turner M, et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol. 2009;27:5410–17. doi: 10.1200/JCO.2008.21.6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rasheed W, Bishton M, Johnstone RW, et al. Histone deacetylase inhibitors in lymphoma and solid malignancies. Expert Rev Anticancer Ther. 2008;8:413–32. doi: 10.1586/14737140.8.3.413. [DOI] [PubMed] [Google Scholar]
- 60.Luu TH, Morgan RJ, Leong L, et al. A phase II trial of vorinostat (suberoylanilide hydroxamic acid) in metastatic breast cancer: a California Cancer Consortium study. Clin Cancer Res. 2008;14:7138–42. doi: 10.1158/1078-0432.CCR-08-0122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Modesitt SC, Sill M, Hoffman JS, et al. A phase II study of vorinostat in the treatment of persistent or recurrent epithelial ovarian or primary peritoneal carcinoma: a Gynecologic Oncology Group study. Gynecol Oncol. 2008;109:182–6. doi: 10.1016/j.ygyno.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 62.Blumenschein GR, Jr, Kies MS, Papadimitrakopoulou VA, et al. Phase II trial of the histone deacetylase inhibitor vorinostat (Zolinza, suberoylanilide hydroxamic acid, SAHA) in patients with recurrent and/or metastatic head and neck cancer. Invest New Drugs. 2008;26:81–7. doi: 10.1007/s10637-007-9075-2. [DOI] [PubMed] [Google Scholar]
- 63.Galanis E, Jaeckle KA, Maurer MJ, et al. Phase II trial of vorinostat in recurrent glioblastoma multiforme: a North Central Cancer Treatment Group study. J Clin Oncol. 2009;27:2052–8. doi: 10.1200/JCO.2008.19.0694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Traynor AM, Dubey S, Eickhoff JC, et al. Vorinostat (NSC# 701852) in patients with relapsed non-small cell lung cancer: a Wisconsin Oncology Network phase II study. J Thorac Oncol. 2009;4:522–6. doi: 10.1097/jto.0b013e3181952478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ramalingam SS, Maitland ML, Frankel P, et al. Carboplatin and Paclitaxel in combination with either vorinostat or placebo for first-line therapy of advanced non-small-cell lung cancer. J Clin Oncol. 2010;28:56–62. doi: 10.1200/JCO.2009.24.9094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Candelaria M, Gallardo-Rincón D, Arce C, et al. A phase II study of epigenetic therapy with hydralazine and magnesium valproate to overcome chemotherapy resistance in refractory solid tumors. Ann Oncol. 2007;18:1529–38. doi: 10.1093/annonc/mdm204. [DOI] [PubMed] [Google Scholar]
- 67.Bialer M, Yagen B. Valproic acid: second generation. Neurotherapeutics. 2007;4:130–7. doi: 10.1016/j.nurt.2006.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]