

Abstract
We previously showed that γδT cells are involved in the pathogenesis of sepsis, but, the underlying mechanisms remained unclear. The present study demonstrates, for the first time, that γδT cells express the complement C5a receptor (C5aR, CD88) and that CD88 expression in γδT cells was up-regulated in mice following sepsis both at protein and mRNA levels. Complement C5a itself contributed to the regulation of C5aR expression on γδT cells, as (i) neutralization of C5a in vivo prevented the expression of C5aR on γδT cells in septic mice and (ii) incubation of mouse spleen cells or purified γδT cells with recombinant C5a in vitro increased CD88 expression by γδT cells at both protein and mRNA levels. C5a receptor on γδT cells also mediates increased interleukin-17 (IL-17) expression as incubation of mouse spleen cells or purified γδT cells with recombinant C5a promotes the IL-17 expression by γδT cells. Ligation of the C5aR on γδT cells activated the phosphoinositide 3-kinase (PI3K)/Akt signalling pathway, which enhances CD88 expression and promotes IL-17 secretion. These results demonstrate that C5a acts directly on the C5aR expressed on γδT cells, resulting in cell activation, and subsequently enhances their capacity for IL-17 production. The up-regulation of the PI3K/Akt pathway following C5a stimulation contributes to up-regulation of γδT-cell function.
Keywords: C5a, CD88, sepsis, γδT cells
Introduction
γδT cells are involved in a wide variety of disease processes and play important roles in both innate and adaptive immunity. However, the underlying mechanisms, particularly the molecules involved and the biochemical bases, are still unclear.1 Recently, data have been obtained suggesting that γδT cells act as early responders that promote inflammation through the production of interleukin-17 (IL-17).2,3γδT cells can also act as antigen-presenting cells to initiate immunity, hence acting as a bridge between innate and adaptive immunity.4 However, in most cases, the mechanisms used by γδT cells and the identity of the molecules involved and the cell surface markers with which they interact have not been fully determined.1
The complement system is part of the innate immune network and is composed of a sophisticated network of soluble and membrane-bound proteins.5 Complement C5a is an important soluble pro-inflammatory mediator and can activate innate immune cells after binding to its receptor expressed on neutrophils or dendritic cells.6,7 In addition to its role in the innate immune response, C5a has recently been shown to play an important role in the regulation of T-cell immunity.8–14 For example, Strainic et al.8 found that the locally produced complement fragments C5a and C3a provide both co-stimulatory and survival signals to naive CD4+ T cells, and Kim et al11 found that the C5a receptor (C5aR) is essential for optimal generation of anti-viral CD8+ T-cell responses. Although it is clear that C5a is actively involved in the regulation of both innate and adaptive immunity, its roles in the regulation of γδT cells, important mediators of both innate and adaptive immunity, are largely unclear.
We previously found that γδT cells play pathogenic roles in the progression of sepsis in mice and that their activity can be affected by excessive production of C5a.15 However, the underlying mechanisms remain unclear. Whether C5a is directly involved in the regulation of γδT cells in sepsis and other inflammatory diseases remains to be determined.
Sepsis is a life-threatening medical condition caused by various micro-organisms entering the bloodstream and triggering an uncontrolled inflammatory reaction. It is well known that excessive production of C5a contributes to this inflammatory response and is involved in the pathogenesis of sepsis.16,17 As excessive production of C5a is linked to worse survival in sepsis,18 the expression and regulation of the C5aR in sepsis would be expected to be important, but this has not been demonstrated.19
Our data show, for the first time, that γδT cells constitutively express the C5aR (CD88) and that C5a is directly involved in the regulation of γδT-cell activity in sepsis in a phosphoinositide 3-kinase (PI3K) -Akt signal pathway-dependent manner.
Materials and methods
Mice
Male C57BL/6 mice (7–8 weeks old) were obtained from Jackson Laboratories (Bar Harbor, ME) and bred in our facilities under specific pathogen-free conditions. All treatment of mice in this study was in strict compliance with the guidelines for the care and use of laboratory animals set out by the Institute of Basic Medical Sciences.
Reagents and antibodies
Recombinant human C5a was purchased from Sigma-Aldrich (St. Louis, MO). Wortmannin, a specific inhibitor of the PI3K-Akt signalling pathway, was purchased from Cell Signaling Technology, CST (Beverly, MA). Anti-mouse C5aR (CD88) monoclonal antibody was obtained from Cell Sciences (Clone 20/70, Canton, MA). Polyclonal rabbit anti-mouse C5a antibodies were produced as described previously.15 Briefly, the C-terminal of mouse C5a (amino acids 58–77) was chosen and the peptide was synthesized and coupled to keyhole limpet haemocyanin and used to immunize rabbits. The IgG was purified by protein A chromatography and its reactivity with recombinant mouse C5a (Sigma-Aldrich) was confirmed by ELISA. Rabbit IgG was used as the isotype control.
Induction of experimental sepsis by caecal ligation and puncture
Specific pathogen-free 7- to 8-week-old male C57BL/6 mice were used to establish the sepsis model. Briefly, the mice were anaesthetized by intraperitoneal injection of ketamine, then approximately two-thirds of the caecum was ligated through a 2-cm abdominal midline incision and punctured extensively with a 21-gauge needle. After the bowel was repositioned, the abdomen was closed in layers, using a 4/0 surgical suture and metallic clips. Sham-operated mice were handled in the same manner, except that the caecum was not ligated and punctured. Immediately after caecal ligation and puncture (CLP), test animals were injected intravenously with 400 μg anti-C5a antibodies in 400 μl PBS, while the control animals received the same amount of normal rabbit IgG (Jing Mei Biotechonogy, Beijing, China). This CLP model of sepsis is believed to closely simulate clinical sepsis in humans because of the polymicrobe-driven inflammatory process.
Isolation of γδ T cells
Briefly, spleens were collected and spleen cells were enriched by Ficoll–Hypaque centrifugation (Pharmacia, Uppsala, Sweden) and washed twice with PBS containing 0·5% BSA and 2 mm EDTA. The cell pellet containing 1 × 107 cells was resuspended in 50 μl PBS and 5 μl phycoerythrin (PE) -conjugated anti-γδT-cell antibody (clone GL3; eBioscience Inc. (San Diego, CA)) was added and the mixture was incubated at 4° for 10 min. Non-bound antibodies were removed by two washes with PBS and the cells were re-suspended in 50 μl PBS. Then, 10 μl anti-PE-Microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany) was added and the suspension was incubated at 4° for 15 min. After two washes, the cells were sorted using magnetic antibody cell sorting Separator columns (Miltenyi Biotec) into bound and non-bound fractions. The bound fraction was collected and its purity was measured by flow cytometry on a FACScaliber flow cytometer using cell quest software (Becton Dickinson, San Jose, CA).
Flow cytometry analysis
To label C5aR (CD88) or C5l2 on γδT cells, spleen cells were stained for 30 min at 4° with FITC-conjugated anti-γδT-cell receptor antibody (eBioscience), allophycocyanin-conjugated anti-mouse CD3 antibody (eBioscience), and then for surface staining, PE-conjugated anti-mouse CD88 antibody (C5aR; Abcam, Cambridge, MA) were washed and used for flow cytometry analysis; for intracellular (C5L2) staining, the above cells were fixed overnight at 4° with 1 ml fixation buffer (Fix and Perm cell permeabilization kit; eBioscience). After washing, purified rat anti-mouse C5L2 (R&D, Minneapolis, MN) was added, followed by incubation with PE-conjugated anti-rat IgG (Santa Cruz Biotechnology, Santa Cruz, CA). After 30 min of incubation, the cells were washed and collected for flow cytometry analysis. For intracellular cytokine (IL-17) labelling, spleen cells were incubated for 4–6 hr at 4° with 1 μg/ml brefeldin A, 50 ng/ml PMA and 1 μg/ml ionomycin (all from Sigma-Aldrich), then they were stained for 30 min at 4° with FITC-conjugated anti-γδT-cell receptor antibody (eBioscience) and allophycocyanin-conjugated anti-mouse CD3 antibody and fixed overnight at 4° with 1 ml fixation buffer (Fix and Perm cell permeabilization kit; eBioscience). After washing, PE-conjugated anti-IL-17 antibody (eBioscience) was added, then, after 30 min of incubation at 4° and washed twice with 1 × permeabilization Buffer (eBioscience), the cells were collected for flow cytometry analysis on a FACSCalibur flow cytometer using cell quest software.
Real-time-PCR
Quantitative reverse transcription PCR was performed with a multicolour real-time PCR detection system (iQ5, MJ; Bio-Rad, Hercules, CA), and DyNAmo HS SYBR Green qPCR kit (MJ BioWorks Inc., South San Francisco, CA).
The real-time PCR was performed following the manufacturer's protocol and carried out in triplicate. The PCR primer sequences were: mouse CD88: sense: 5′-atgccca-accctaggccagccaag-3′; antisense: 5′-tgggccccacttcgcaggtcccga-3′; mouse C5l2: sense: 5′-GCTCCTCCGCACTCCTT-3′; anti-sense: 5′-ACCGCACTTTCCTCATCC-3′; and mouse glyceraldehyde 3-phosphate dehydrogenase (GAPDH): sense 5′-tcttgggctacactgaggacc-3′, antisense: 5′-cataccaggaaatgagcttga-3′. The GAPDH was used as an endogenous control for any sample-to-sample variation in quantity and quality of RNA and difference in efficiency of reverse transcription and PCR.
Measurement of IL-17 by ELISA
The concentration of IL-17 and interferon-γ was measured using ELISA kits (eBioscience) and an automatic plate reader.
Measurement of intracellular cAMP
The purified γδT cells were incubated for 30 min with forskolin (10 nm; Sigma Co.). In some experiments, γδT cells were further incubated in the presence or absence of recombinant human C5a (20 nm, 100 nm) for 40 min at 37°. The treated cells were then washed twice with cold PBS and lysed in 250 μl cell lysis buffer by three cycles of freeze–thaw. The cell lysates were used to measure cAMP using a Parameter cAMP assay kit according to the manufacturer's instructions (R&D Systems).
Western blot analysis
The γδT cells were stimulated with recombinant human C5a for the indicated time, then lysed. Equal amounts of protein were subjected to SDS–PAGE followed by transfer to a PVDF membrane and probing with antibodies against nuclear factor-κΒ (NF-κΒ) p65, iκΒ, Akt, or phosphor-Akt, phosphor NF-κΒ p65 (all from Cell Signaling Techniques). Bound antibody was visualized using an enhanced chemiluminescence kit (Amersham Biosciences, Piscataway, NJ).
Statistical analysis
Results were expressed as the mean ± SEM and subjected to statistical analysis. Student's t-test or analysis of variance was used where appropriate to determine significant differences between samples. Values of P < 0·05 were considered significant.
Results
γδT cells constitutively express the C5a receptor, CD88
C5a is involved in the regulation of αβT-cell immunity through the C5aR expressed on CD4+ and CD8+ T cells.8–14 Our previous data15 also suggested an interaction between C5a and γδT cells in sepsis. However, whether C5a directly regulated the function of γδT cells, a T-cell subset with important roles in both innate and adaptive immunity, remained unclear. To test this, we first examined whether γδT cells express the C5a receptor, CD88. Spleen cells and mesenteric lymph node cells were prepared from naive C57BL/6 mice and C5aR expression on γδT cells was examined by flow cytometry. As shown in Fig. 1, C5aR (CD88) was constitutively expressed on the γδT-cell population. Besides CD88, C5L2 is another C5a receptor recently identified with biological functions.20 Here we also examined the expression of C5L2 on γδT cells. There is no significant difference between the C5L2 staining group and the control group, indicating that C5L2 is not expressed on γδT cells (data not shown).
Figure 1.
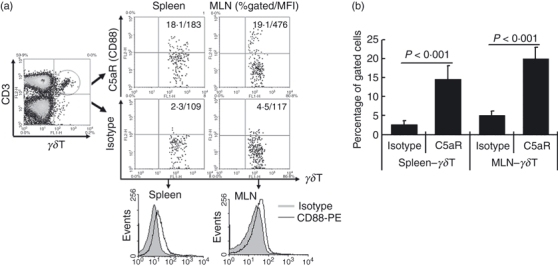
γδT cells constitutively express the C5a receptor, CD88. Spleen cells and mesenteric lymph node (MLN) cells were isolated from naive C57BL/6 mice and stained with antibodies against mouse CD3 (allophycocyanin-conjugated), the γδT T-cell receptor (FITC-conjugated), and the C5a receptor (CD88) (phycoerythrin-conjugated), then CD3+γδT cells were gated to analyse CD88 expression. (a) Representative flow cytometry dot plot and histogram picture analysing the expression of CD88 on gated CD3+γδT cells. (b) Statistical analysis of the expression of CD88 on γδT cells. The data shown are representative of the results from three independent experiments with at least four mice per group.
C5aR expression on γδT cells is up-regulated in mice following sepsis
Previous data showed that C5aR expression in different tissues is up-regulated following sepsis in mice.19 To test whether C5aR expression on γδT cells was also inducible, we used the CLP sepsis model described previously15 and measured C5aR expression on γδT cells by flow cytometry. As shown in Fig. 2(a,b), C5aR expression on γδT cells in a spleen cell suspension was significantly up-regulated in mice following sepsis. We also analysed the total numbers of CD88+γδT cells in spleen. We found that after CLP, the total number of CD88+γδT cells also increased (Fig. 2c), indicating that there was an enhanced CD88+γδT-cell response in mice following sepsis. The γδT cells in mesenteric lymph node cells and peritoneal lavage fluid also showed increased C5aR expression in mice following sepsis (data not shown). We examined whether there was enhanced CD88 expression in γδT cells collected from septic mice compared with those from sham controls by real-time-PCR. Our results showed that γδT cells from septic mice showed enhanced CD88 expression at mRNA levels compared with those from sham control mice (Fig. 2d). Here we also examined whether there was C5L2 mRNA expression in γδT cells from naive or septic mice. Consistent with the above data of flow cytometry, no C5L2 mRNA was detected in murine γδT cells (data not shown).
Figure 2.
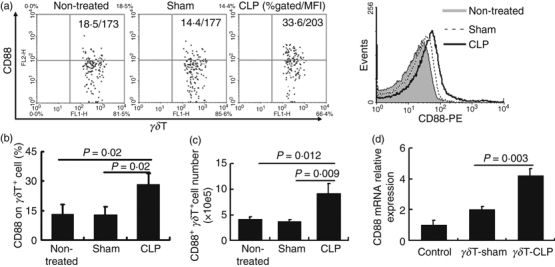
Increased expression of CD88 on γδT cells in mice following sepsis. Spleen cells were isolated from untreated, sham-operated, and septic (caecal ligation and puncture; CLP) mice and stained with antibodies against mouse CD3 (allophycocyanin-conjugated), the γδT T-cell receptor (FITC-conjugated), and the C5a receptor (CD88) (phycoerythrin-conjugated), then CD3+γδT cells were gated to analyse CD88 expression. (a) Representative flow cytometry dot plot and histogram picture analysing the expression of CD88 on γδT cells. (b) Statistical analysis of CD88 frequency on γδT cells. The data shown are representative of the results for three independent experiments with at least four mice per group. (c) The total number of CD3+ CD88+γδT in spleen cells from different group were analysed. (d) γδT cells were collected from sham control mice and septic mice, respectively. Then CD88 expression at mRNA level was examined by real-time PCR. Un-fractionated total lymphocytes were used as control.
Neutralization of C5a in vivo down-regulates CD88 expression on γδT cells
We previously found that blocking C5a in vivo increases the survival rate and decreases γδT-cell activity in septic mice.15 To test whether excessive production of C5a directly affected the phenotype of the γδT cell, CD88 expression on γδT cells was examined in septic mice with or without intraperitoneal anti-C5a antibody administration. As shown in Fig. 3, C5a neutralization significantly down-regulated CD88 expression on γδT cells, indicating that excessive production of C5a contributed to the up-regulation of CD88 on γδT cells in sepsis. Considering our previous findings that neutralization of C5a in vivo significantly increases survival rate in sepsis and down-regulates IL-17 expression by γδT cells,15 these data further indicate that excessive production of C5a is directly involved in the regulation of γδT-cell activity in sepsis by up-regulating C5aR expression and enhancing cytokine production by γδT cells.
Figure 3.
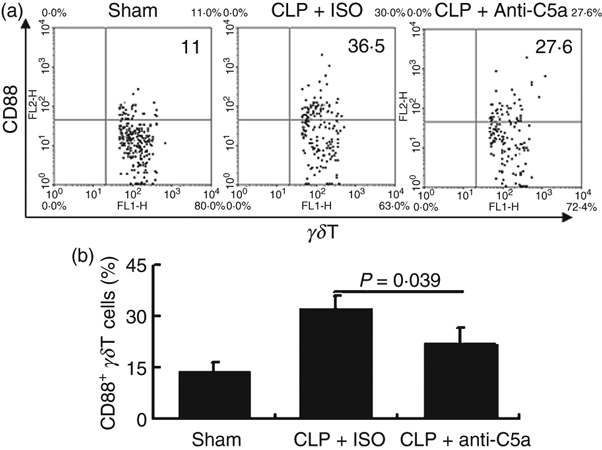
Neutralization of C5a partially prevents the up-regulation of CD88 expression on γδT cells in septic mice. Spleen cells from sham-operated mice and mice that have undergone caecal ligation and puncture (CLP) treated with anti-C5a antibody or control IgG were stained with antibodies against mouse CD3 (allophycocyanin-conjugated), the γδ T-cell receptor (FITC-conjugated), and the C5areceptor (CD88) (phycoerythrin-conjugated), then CD3+γδT cells were gated to analyse CD88 expression. (a) Representative flow cytometry dot plot. (b) Statistical analysis of the expression of CD88 on γδT cells. The data shown are representative of the results from three independent experiments with at least four mice per group.
Recombinant C5a promotes up-regulation of CD88 expression by γδT cells at both protein and mRNA level
To directly investigate the effects of C5a on CD88 expression by γδT cells in vitro, we incubated mouse spleen cells with recombinant human C5a for 12 hr, and then measured CD88 expression by γδT cells. As shown in Fig. 4(a,b), stimulation with C5a significantly up-regulated CD88 expression by γδT cells.
Figure 4.
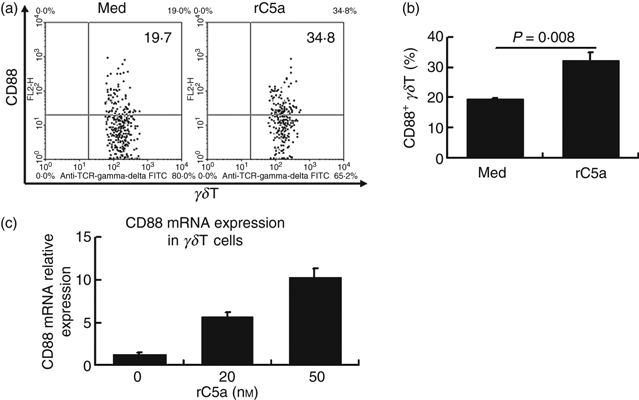
Recombinant C5a promotes up-regulation of CD88 expression by γδT cells at both protein and mRNA level. Spleen cells from untreated C57BL/6 mice were incubated with recombinant human C5a (rC5a) (20 nm) or medium (Med) for 12 hr, then were stained with antibodies against mouse CD3 (allophycocyanin-conjugated), the γδT T-cell receptor (FITC-conjugated), and the C5a receptor (CD88) (phycoerythrin-conjugated) then CD3+γδT cells were gated to analyse CD88 expression. (a, b) Representative flow cytometry dot plots analysing expression of CD88 (a) by γδT cells. (b) Statistical analysis of the expression of CD88 by γδT cells. (c) γδT cells were purified from septic mice as described in the Materials and Methods, and incubated with recombinant human C5a at the indicated dosage. Twelve hours later, cells were collected and used for real-time PCR analysis. The results shown are representative of those from four independent experiments. We also examined the effects of C5a receptor ligation on the activation of γδT cells using different markers. Although CD69, CD44 and CD62 ligand expression on γδT cells was significantly up-regulated in mice following sepsis, incubation of spleen cells with C5a did not affect the expression of these molecules on γδT cells (data not shown).
Given that CD69, CD44 and CD62 ligand (CD62L) have been implicated as activation markers of γδT cells,21,22 we also investigated whether ligation of the C5aR also affected their expression. Although CD69, CD44 and CD62L expression on γδT cells was significantly up-regulated in mice following sepsis, incubation of spleen cells with C5a did not affect the expression of these molecules on γδT cells (data not shown).
To test whether C5a induced CD88 expression because of increased gene transcription, we purified γδT cells and incubated them with recombinant complement C5a for 12 hr. Then cells were collected and analysed for CD88 expression. As shown in Fig. 4(c), C5a incubation dose-dependently increased CD88 expression at the mRNA level.
C5a receptor mediates increased IL-17 expression from γδT cells
To directly examine the effects of C5aR ligation on the activity of γδT cells, we incubated mouse spleen cells or purified γδT cells with recombinant human C5a for 12 hr, then measured IL-17 expression by γδT cells by flow cytometry. To further confirm that CD88, but not C5L2, was involved in C5a-enhanced activity of γδT cells, neutralizing antibodies active against mouse CD88 were added in combination with recombinant human C5a, and then IL-17 expression by γδT cells was measured by flow cytometry. As shown in Fig. 5(a,b), incubation of mouse spleen cells with recombinant human C5a significantly increased IL-17 expression within γδT cells. Neutralization of CD88 blocked the activity of C5a-enhanced IL-17 expression, again indicating that CD88 but not C5L2 is involved in the activation of γδT cells after the addition of C5a.
Figure 5.
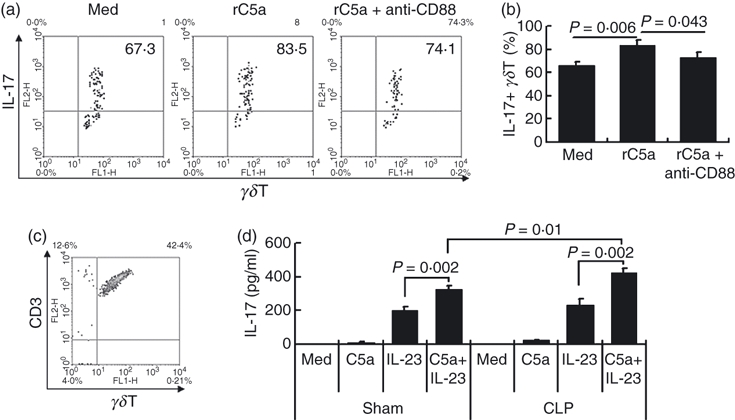
C5a receptor CD88 mediates increased interleukin-17 (IL-17) expression by γδT cells. Spleen cells from untreated C57BL/6 mice were incubated with recombinant human C5a (rC5a; 20 nm) or medium (Med) or rC5a plus anti-mouse CD88 monoclonal antibody (2 μg/ml) for 12 hr, then were stained with antibodies against mouse CD3 (allphycocyanin-conjugated), the γδT-cell receptor (FITC-conjugated), and IL-17 (intracellular staining; phycoerythrin-conjugated), then CD3+γδT cells were gated to analyse IL-17 expression (a, b). (c) γδT cells were purified from septic or sham control mice and (d) were incubated with recombinant human C5a (20 nm) and/or recombinant mouse IL-23 (10 ng/ml) for 24 hr, then the culture supernatants were assayed for IL-17 by ELISA. The results shown are representative of four independent experiments.
Subsequently, we purified γδT cells from septic mice or sham controls (Fig. 5c) and then co-cultured them with recombinant human C5a with or without recombinant mouse IL-23 for 24 hr, then measured IL-17 levels in the culture supernatants. The ELISA data showed that recombinant C5a alone did not induce IL-17 secretion, whereas IL-23 caused some release and the combination was more effective (Fig. 5c). We previously reported that γδT cells from septic mice showed enhanced CD88 expression. As expected, compared with γδT cells from sham control mice, γδT cells from septic mice showed enhanced IL-17 secretion after C5a stimulation (Fig. 5d). These data directly show that a C5a–C5aR–γδT–IL-17 axis exists in the inflammatory response in diseases such as sepsis.
Ligation of C5a activates the PI3K/Akt signalling pathway in γδT cells
C5aR signalling, in which the C5aR is coupled to G protein αi and βγ subunits and activates several downstream signalling pathways (e.g. PI3K, NF-κB, cAMP), has been studied in dendritic cells, macrophages, neutrophils and several other cell types.23,24 However, the mechanism of C5aR signalling in γδT cells is not known. It has been shown that C5a stimulation activates the Akt pathway in dendritic cells,7 raising the possibility that it could have a similar effect on this pathway in γδT cells. To identify the signalling pathway(s) responsible for the C5a-mediated activation of γδT cells, we first examined whether C5a could affect the activation of the PI3K/Akt and NF-κB pathways. As shown in Fig. 6(a–c), C5a stimulation enhanced Akt but inhibited NF-κB-p65 phosphorylation. To test whether and how C5a stimulation affected other signalling pathways in γδT cells, we measured cAMP levels in γδT cells after C5a stimulation, as the cAMP pathway is a key signalling pathway for G protein-coupled receptors25 and cAMP functions as an intracellular second messenger,26 which regulates a wide range of important cellular processes. In contrast to those found in dendritic cells, in which C5aR, a G protein-coupled receptor, decreases cAMP production, we found that ligation of C5aR increased cAMP levels in γδT cells (data not shown). The underlying mechanisms remain to be determined.
Figure 6.
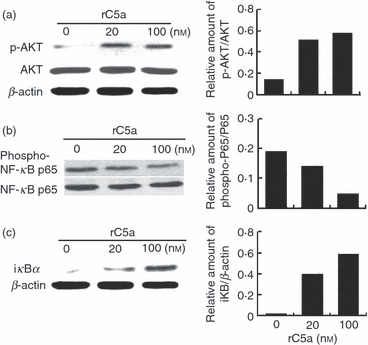
C5a stimulation activates the phosphoinositide 3-kinase (PI3K)/Akt signalling pathway while inhibit nuclear factor-κB (NF-κB) signalling in γδT cells. Purified γδT cells were stimulated with recombinant human C5a at the indicated dose for 15 min, then cell lysates were tested for phosphorylated Akt (a) and NF-κB p65 (b), as well as iκB (c) by Western blotting using antibodies against phospho-Akt, phosphor- NF-κB p65, Akt, NF-κB p65, iκB or β-actin respectively and the bands were analysed by densitometry.
Finally, to test the role of the PI3K-Akt pathway in C5a/C5aR-mediated γδT-cell activation, we pre-treated spleen cells for 1 hr with the PI3K/Akt inhibitor wortmannin, then incubated them with recombinant human C5a in the continued presence of wortmannin for 12 hr. As shown in Fig. 7(a,b), wortmannin prevented the C5a-induced up-regulation of CD88 on γδT cells. We also pre-treated γδT cells with wortmannin before stimulating them with C5a and IL-23 and measuring IL-17 secretion and found that wortmannin also inhibited C5a/IL-23-induced IL-17 secretion by γδT cells (Fig. 7c). As we use a combination of IL-23 and C5a here, to test whether wortmannin affects pathways downstream of the IL-23R or the C5aR, we examined whether wortmannin affects the signalling of IL-23 in γδT cells. Our data showed that wortmannin did not significantly affect IL-23-induced IL-17 production by γδT cells (data not shown), indicating that in the above combination, wortmannin mainly affects the signalling of C5aR.
Figure 7.
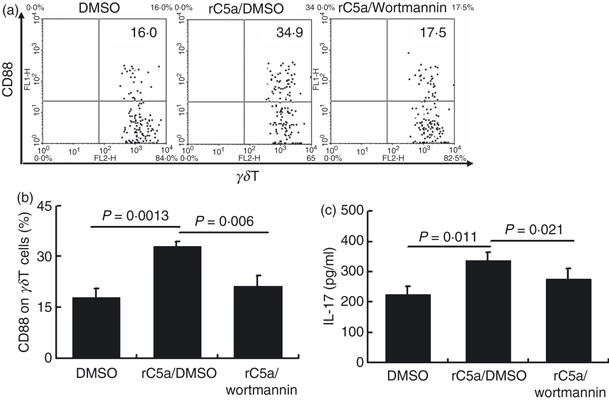
Inhibition of the phosphoinositide 3-kinase (PI3K)/Akt signalling pathway by wortmannin prevents the up-regulation of CD88 and interleukin-17 (IL-17) expression by C5a. (a) Spleen cells were pre-treated with vehicle (DMSO) or the PI3K/Akt inhibitor wortmannin (1 μm) for 1 hr and incubated for 12 hr with recombinant C5a (20 nm), then were analysed by flow cytometry for expression of CD88 on CD3+γδT+ cells. (b) Statistical analysis of CD88 and IL-17 expression by the above γδT cells. The data are representative of those for three independent experiments with at least four mice per group. (c) Purified γδT cells were pre-treated with wortmannin (1 μm) for 1 hr, then were stimulated with C5a and IL-23 (10 ng/ml) for 24 hr and the culture supernatant was collected and analysed for IL-17 by ELISA. The results shown are representative of those from three independent experiments.
Discussion
γδT cells are actively involved in both innate and adaptive immunity. However, the underlying mechanisms remain to be determined. Here, we showed, for the first time, that γδT cells constitutively expressed the C5aR and that its expression was up-regulated in mice following sepsis. Ligation of the C5aR by C5a directly promoted the activation of γδT cells in a PI3K-Akt signalling pathway-dependent manner. Our data suggest an important C5a–γδT–cytokine (IL-17) axis, which partially explains the pathogenic roles of C5a in inflammatory diseases. The observation that IL-17 secretion by γδT cells could also be modified by C5a provides further evidence supporting the existence of a cross-link between the complement system and T-cell immunity.
Excessive production of C5a and up-regulation of the C5aR has been found in different tissues in mice following injury or sepsis.19 These data suggest that excessive production of C5a might cause its pathogenic effects by binding to the over-expressed C5aR on different cells. Although recent studies have shown regulation of T-cell immunity by the C5a–C5aR axis,8–14 whether T cells express the C5aR and its implications in sepsis remained to be determined. In this study, we evaluated, for the first time, the cellular expression and kinetics of the C5aR on γδT cells. The observations that CD88 expression on γδT cells was up-regulated in mice following sepsis and that C5a neutralization in vivo down-regulated C5aR expression (Fig. 3) suggest that C5a is involved in the regulation of γδT cells in sepsis. γδT cells show increased expression of CD62L, CD44 and CD69 after activation.21,22 However, although CD62L, CD44 and CD69 expression on γδT cells was significantly up-regulated in mice following sepsis, C5a treatment alone did not affect the expression of these markers on γδT cells (data not shown). Here we also examined the absolute number of CD88+γδT cells in the spleen after anti-C5a administration. Consistent with our previous findings,15 anti-C5a antibody did not significantly down-regulate the absolute number of CD88+γδT cells in vivo, indicating that some other factors are involved in enhancing the activity of γδT cells in sepsis (data not shown).
Although γδT cells constitute only a small proportion (1–5%) of the lymphocytes in the blood and peripheral organs, they play important roles in both innate and adaptive immunity.1 Recently, several groups, including our own, suggested that γδ T cells, which are a major source of IL-17, are involved in the pathogenesis of sepsis.15,27,28 In addition, it is clear that, in septic mice, there is excessive production of C5a and IL-17 and that blockade of either is beneficial for the outcome in sepsis.17,29 However, whether there are interactions between these two important pro-inflammatory cytokines remained unclear. Previously, we found that C5a is involved in the regulation of IL-17 production in septic mice and that C5a neutralization in vivo increases the survival rate and decreases IL-17 production by γδT cells.15 These data indicate a link between IL-17-producing γδT cells and C5a. However, whether C5a could directly regulate the activity of IL-17-producing γδT cells was unclear. In this study, we provided evidence that, by binding to C5aRs expressed on γδT cells; C5a directly takes part in the regulation of γδT-cell immune responses. Hence, we have identified a new axis, in which C5a acts directly on γδT cells and increases C5aR expression on γδT cells, enhances γδT-cell activity (such as promoting IL-17 production), and promotes the inflammatory response in sepsis.
After demonstrating expression of C5aR on γδT cells in vivo, we investigated the effects of C5aR ligation on the surface markers and function of γδT cells. As shown in Figs 4 and 5, incubation of recombinant complement C5a increased CD88 and IL-17 expression by γδT cells. Blockade of CD88 with antibody abolished the effects of recombinant C5a-induced CD88 and IL-17 up-regulation, indicating that the effects of C5a on γδT cells were mediated by CD88 but not another receptor of C5a–C5L2. γδT cells from septic mice show up-regulated CD88 expression, which raised the possibility that these γδT cells might show an enhanced response to C5a stimulation compared with cells from control mice. As expected, C5a stimulation did induce a significant increase in IL-17 production by γδT cells from septic mice compared with those from sham control mice (Fig. 5d). Although recombinant C5a alone did not induce significant IL-17 secretion, it facilitated the activity of IL-23. We also examined the effects of C5a ligation on interferon-γ production by γδT cells. We found that C5a alone could not induce the production of interferon-γ. However, C5a enhanced IL-23-mediated interferon-γ production by γδT cells (data not shown). These data again indicate that complement components are involved in the regulation of γδT cells through the up-regulation of CD88 expression on γδT cells. In this scenario, excessive production of C5a itself is directly or indirectly involved in the up-regulation of C5aR expression, then binds to the up-regulated C5aR, and promotes the pathogenic roles of γδT cells in septic mice.
Another important finding in this study was the identification of the intracellular signalling pathways by which C5a modulates γδT-cell function. The PI3Ks are a conserved family of signal transduction enzymes that are involved in regulating cellular proliferation and survival.30 Akt, the downstream serine/threonine kinase of PI3Ks (also known as protein kinase B), which was first described as an oncogene, regulates cellular activation, inflammatory responses, chemotaxis and apoptosis.31,32 Our data clearly demonstrated that the PI3K-Akt signalling pathway was involved in the activation of γδT cells by C5a, as (i) C5a ligation increased Akt phosphorylation and (ii) a PI3K inhibitor, which inhibits the PI3K/Akt pathway, prevented the C5a-induced increase in CD88/IL-17 expression by γδT cells. Bommhardt et al.33 reported that over-expression of Akt by lymphocytes results in decreased lymphocyte apoptosis and a better survival in mice in response to CLP-induced sepsis. These data indicate that the PI3K-Akt pathway is involved in maintaining the homeostasis of T cells. Our data showing that ligation of C5aR activates the Akt pathway in γδT cells suggest that C5a might also be involved in maintaining the homeostasis of γδT cells in inflammatory diseases, such as sepsis.
Nuclear factor-κB is a ubiquitous inducible transcription factor that stimulates gene expression, in particular of those genes that promote immune and inflammatory responses.34 A variety of extracellular signals can activate NF-κB.35 To examine the effect of C5a on NF-κB activation in γδT cells, we measured levels of phosphor-NF-κB-p65 in response to C5a stimulation. Unexpectedly, we found that C5a engagement resulted in NF-κB suppression (Fig. 6b,c). Although there are data indicating that the PI3K-Akt signalling pathway is involved in cell survival by inhibiting the activation of pro-apoptotic proteins and transcription factors,36–38 the underlying mechanisms by which C5a signalling inhibits NF-κB activation remain to be determined.
Cyclic AMP is a cyclic nucleotide that functions as an intracellular second messenger26 and regulates a wide range of important cellular processes. As the cAMP pathway is a key signalling pathway for G protein-coupled receptors, such as the C5aR, we examined whether the C5aR/cAMP signalling pathway was also involved in the regulation of γδT cells. Different from those found in dendritic cells, in which C5aR, a G protein-coupled receptor, decreases cAMP production,7 we found that ligation of C5aR increased cAMP levels in γδT cells (data not shown). As increased cAMP levels may contribute to the inhibition of NF-κB,35 our data suggest a dual effect of C5a signalling on the activity of γδT cells, on the one hand activating the PI3K signalling pathway and enhancing the activity of γδT cells and, on the other, being involved in maintaining γδT-cell homeostasis by inhibiting the activation of NF-κB (Fig. 7). The underlying mechanisms require further investigation.
In summary, our data demonstrate, for the first time, that γδT cells constitutively express the C5aR and that, following sepsis, C5aR expression is significantly up-regulated. Excessively produced C5a is involved in the regulation of CD88 expression on γδT cells. Ligation of the C5aR by C5a enhances CD88 and IL-17 expression by γδT cells in a PI3-K/Akt signalling pathway-dependent manner. C5a engagement increases cAMP levels and inhibits NF-κB activation in γδT cells. The C5a/C5aR–γδT–IL-17 axis provides further evidence for the involvement of C5a in the regulation of T cells and a new mechanism by which C5a influences inflammatory responses. Further studies on cross-talk between the complement system and γδT cells and its biochemical basis will provide insight into the pathogenesis of, and therapeutic approaches to, inflammatory diseases.
Acknowledgments
We are grateful to Professor Ren-feng Guo (Department of Pathology, University of Michigan, Ann Arbor, MI) for manuscript revision. This work was supported by grants from the National Key Basic Research Program of China (2007CB512406) and the National Natural Science Foundation of China (81072475, and 30801029).
Glossary
Abbreviations
- C5aR
complement C5a receptor
- CD62L
CD62 ligand
- CLP
caecal ligation and puncture
- IL-17
interleukin-17
- NF-κB
nuclear factor-k-gene binding
- PE
phycoerythrin
- PI3K/Akt
phosphoinositide 3-kinase/Akt
Disclosures
The authors have no financial conflict of interest.
References
- 1.Carding SR, Egan PJ. γδT cells: functional plasticity and heterogeneity. Nat Rev Immunol. 2002;2:336–45. doi: 10.1038/nri797. [DOI] [PubMed] [Google Scholar]
- 2.Lockhart E, Green AM, Flynn JL. IL-17 production is dominated by γδT cells rather than CD4 T cells during Mycobacterium tuberculosis infection. J Immunol. 2006;177:4662–9. doi: 10.4049/jimmunol.177.7.4662. [DOI] [PubMed] [Google Scholar]
- 3.Roark CL, French JD, Taylor MA, Bendele AM, Born WK, O'Brien RL. Exacerbation of collagen-induced arthritis by oligoclonal, IL-17-producing gamma delta T cells. J Immunol. 2007;179:5576–83. doi: 10.4049/jimmunol.179.8.5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheng L, Cui Y, Shao H, et al. Mouse γδ T cells are capable of expressing MHC class II molecules, and of functioning as antigen-presenting cells. J Neuroimmunol. 2008;203:3–11. doi: 10.1016/j.jneuroim.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barrington R, Zhang M, Fischer M, Carroll MC. The role of complement in inflammation and adaptive immunity. Immunol Rev. 2001;180:5–15. doi: 10.1034/j.1600-065x.2001.1800101.x. [DOI] [PubMed] [Google Scholar]
- 6.Kohl J. The role of complement in danger sensing and transmission. Immunol Res. 2006;34:157–76. doi: 10.1385/IR:34:2:157. [DOI] [PubMed] [Google Scholar]
- 7.Peng Q, Li K, Wang N, et al. Dendritic cell function in allostimulation is modulated by C5aR signaling. J Immunol. 2009;183:6058–68. doi: 10.4049/jimmunol.0804186. [DOI] [PubMed] [Google Scholar]
- 8.Strainic MG, Liu J, Huang D, et al. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity. 2008;28:425–35. doi: 10.1016/j.immuni.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nataf S, Davoust N, Ames RS, Barnum SR. Human T cells express the C5a receptor and are chemoattracted to C5a. J Immunol. 1999;162:4018–23. [PubMed] [Google Scholar]
- 10.Lalli PN, Strainic MG, Yang M, Lin F, Medof ME, Heeger PS. Locally produced C5a binds to T cell-expressed C5aR to enhance effector T-cell expansion by limiting antigen-induced apoptosis. Blood. 2008;112:1759–66. doi: 10.1182/blood-2008-04-151068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim AH, Dimitriou ID, Holland MC, Mastellos D, Mueller YM, Altman JD, Lambris JD, Katsikis PD. Complement C5a receptor is essential for the optimal generation of antiviral CD8+ T cell responses. J Immunol. 2004;173:2524–9. doi: 10.4049/jimmunol.173.4.2524. [DOI] [PubMed] [Google Scholar]
- 12.Carroll MC. The complement system in regulation of adaptive immunity. Nat Immunol. 2004;5:981–6. doi: 10.1038/ni1113. [DOI] [PubMed] [Google Scholar]
- 13.Kopf M, Abel B, Gallimore A, Carroll M, Bachmann MF. Complement component C3 promotes T-cell priming and lung migration to control acute influenza virus infection. Nat Med. 2002;8:373–8. doi: 10.1038/nm0402-373. [DOI] [PubMed] [Google Scholar]
- 14.Hawlisch H, Köhl J. Complement and toll-like receptors: key regulators of adaptive immune responses. Mol Immunol. 2006;43:13–21. doi: 10.1016/j.molimm.2005.06.028. [DOI] [PubMed] [Google Scholar]
- 15.Xu R, Wang R, Han G, et al. Complement C5a regulates IL-17 by affecting the crosstalk between DC and γδ T cells in CLP-induced sepsis. Eur J Immunol. 2010;40:1079–88. doi: 10.1002/eji.200940015. [DOI] [PubMed] [Google Scholar]
- 16.Niederbichler AD, Hoesel LM, Westfall MV, et al. An essential role for complement C5a in the pathogenesis of septic cardiac dysfunction. J Exp Med. 2005;203:53–61. doi: 10.1084/jem.20051207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo RF, Huber-Lang MS, Wang X, et al. Protective effects of anti-C5a in sepsis-induced thymocyte apoptosis. J Clin Invest. 2000;106:1271–80. doi: 10.1172/JCI10793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo RF, Ward PA. Role of C5a in inflammatory responses. Annu Rev Immunol. 2005;23:821–52. doi: 10.1146/annurev.immunol.23.021704.115835. [DOI] [PubMed] [Google Scholar]
- 19.Riedemann NC, Guo RF, Neff TA, et al. Increased C5a receptor expression in sepsis. J Clin Invest. 2002;110:101–8. doi: 10.1172/JCI15409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rittirsch D, Flierl MA, Nadeau BA, et al. Functional roles for C5a receptors in sepsis. Nat Med. 2008;14:551–7. doi: 10.1038/nm1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao YL, Rajan AJ, Raine CS, Brosnan CF. γδT cells express activation markers in the central nervous system of mice with chronic-relapsing experimental autoimmune encephalomyelitis. J Autoimmun. 2001;17:261–71. doi: 10.1006/jaut.2001.0547. [DOI] [PubMed] [Google Scholar]
- 22.Rakasz E, Rigby S, de Andres B, Mueller A, Hagen M, Dailey MO, Sandor M, Lynch RG. Homing of transgenic γδT cells into murine vaginal epithelium. Int Immunol. 1998;10:1509–17. doi: 10.1093/intimm/10.10.1509. [DOI] [PubMed] [Google Scholar]
- 23.Kastl SP, Speidl WS, Kaun C, et al. The complement component C5a induces the expression of plasminogen activator inhibitor-1 in human macrophages via NF-kB activation. J Thromb Haemost. 2006;4:1790–7. doi: 10.1111/j.1538-7836.2006.02046.x. [DOI] [PubMed] [Google Scholar]
- 24.Sayah S, Jauneau AC, Patte C, Tonon MC, Vaudry H, Fontaine M. Two different transduction pathways are activated by C3a and C5a anaphylatoxins on astrocytes. Brain Res Mol Brain Res. 2003;112:53–60. doi: 10.1016/s0169-328x(03)00046-9. [DOI] [PubMed] [Google Scholar]
- 25.Marinissen MJ, Gutkind JS. G-protein-coupled receptors and signaling networks: emerging paradigms. Trends Pharmacol Sci. 2001;22:368–76. doi: 10.1016/s0165-6147(00)01678-3. [DOI] [PubMed] [Google Scholar]
- 26.Kambayashi T, Wallin RP, Ljunggren HG. cAMP-elevating agents suppress dendritic cell function. J Leukoc Biol. 2001;70:903–10. [PubMed] [Google Scholar]
- 27.Roark CL, Simonian PL, Fontenot AP, Born WK, O'Brien RL. γδT cells: an important source of IL-17. Curr Opin Immunol. 2008;20:1–5. doi: 10.1016/j.coi.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tschop J, Martignoni A, Goetzman HS, Choi LG, Wang Q, Noel JG, Ogle CK, et al. γδT cells mitigate the organ injury and mortality of sepsis. J Leukoc Biol. 2007;83:581–9. doi: 10.1189/jlb.0707507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fierl MA, Rittirech D, Gao HW, Hoesel LM, Nadeau BA, Day DE, Zetoune FS, et al. Adverse functions of IL-17A in experimental sepsis. FASEB J. 2008;22:2198–205. doi: 10.1096/fj.07-105221. [DOI] [PubMed] [Google Scholar]
- 30.Fruman DA, Cantley LC. Phosphoinositide 3-kinase in immunological systems. Semin Immunol. 2002;14:7–18. doi: 10.1006/smim.2001.0337. [DOI] [PubMed] [Google Scholar]
- 31.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–7. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 32.Hill MM, Hemmings BA. Inhibition of protein kinase B/Akt: implications for cancer therapy. Pharmacol Ther. 2005;93:243–51. doi: 10.1016/s0163-7258(02)00193-6. [DOI] [PubMed] [Google Scholar]
- 33.Bommhardt U, Chang KC, Swanson PE, Wagner TH, Tinsley KW, Karl IE, Hotchkiss RS. Akt decreases lymphocyte apoptosis and improves survival in sepsis. J Immunol. 2004;172:7583–91. doi: 10.4049/jimmunol.172.12.7583. [DOI] [PubMed] [Google Scholar]
- 34.Baeuerle PA, Baltimore D. NF-KB: ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 35.Ollivier V, Parry GC, Cobb RR, de Prost D, Mackman N. Elevated cyclic AMP inhibits NF-kB-mediated transcription in human monocytic cells and endothelial cells. J Biol Chem. 1996;271:20828–35. doi: 10.1074/jbc.271.34.20828. [DOI] [PubMed] [Google Scholar]
- 36.Brazil DP, Yang ZZ, Hemmings BA. Advances in protein kinase B signalling: AKTion on multiple fronts. Trends Biochem Sci. 2004;29:233–42. doi: 10.1016/j.tibs.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 37.Guha M, Mackman N. The PI3K-Akt pathway limits LPS activation of signaling pathways and expression of inflammatory mediators in human monocytic cells. J Biol Chem. 2002;277:32124–32. doi: 10.1074/jbc.M203298200. [DOI] [PubMed] [Google Scholar]
- 38.Fukao T, Koyasu S. PI3K and negative regulation of TLR signaling. Trends Immunol. 2005;24:358–63. doi: 10.1016/s1471-4906(03)00139-x. [DOI] [PubMed] [Google Scholar]