

Abstract
γ-Melanocyte Stimulating Hormone (γ-MSH) regulates sodium (Na+) balance and blood pressure through activation of the melanocortin receptor 3 (MC3-R). The mechanism of the natriuretic effect is proposed to involve binding of MC3-R either in the kidney to directly inhibit tubular Na+ transport or in the brain to inhibit central neural pathways that control renal tubular Na+ absorption. This study aimed to clarify the mechanism involved in the natriuretic effect of γ-MSH on MC3-R in kidney cells. In Ussing chamber studies, we observed no effects of γ-MSH on NaCl transport in the mouse inner medullary collecting duct cell line (mIMCD-K2). We also found that neither MC3-R protein nor mRNA was expressed in mouse kidney, suggesting that renal γ-MSH action may not be mediated through direct effects on tubular Na+ transport but rather through effects on central neural pathways that innervate the kidney.
Keywords: γ-MSH, MC3-R, natriuresis, IMCD
1. Introduction
γ-Melanocyte Stimulating Hormone (γ-MSH) regulates a multitude of metabolic functions including energy homeostasis, food intake, sodium (Na+) balance, and blood pressure regulation. γ-MSH is processed from the precursor hormone pro-opio-melanocortin (POMC) in the pituitary gland, where component peptides are released into the cerebral spinal fluid and systemic circulation [11]. The natural receptor for γ-MSH is the melanocortin receptor 3 (MC3-R), a G-protein-coupled receptor that activates adenylate cyclase and is expressed in brain (hypothalamus, cortex, and thalamus), gut, placenta, and kidney [5, 6, 9, 17, 18, 23].
There is growing evidence that γ-MSH signals through MC3-R to regulate Na+ balance and blood pressure. For example, rats ingesting a high Na+ diet exhibit an increase in expression of the POMC precursor and the processing enzymes, pro-convertase 1 and 2 (PC1, PC2), in the pituitary gland, which lead to increases in γ-MSH content in pituitary tissue [3] and a doubling of γ-MSH concentration in plasma [15]. Furthermore, induction of γ-MSH deficiency by bromocriptine infusion in rats or by PC2 gene knockout (KO) in mice results in salt-sensitive hypertension [16, 20]. Interestingly, administration of γ-MSH reverses salt-sensitive hypertension in PC2 KO but not in MC3-R KO mice, suggesting that γ-MSH can regulate Na+ balance and blood pressure by acting through MC3-R [20].
γ-MSH regulates Na+ balance by inducing urinary Na+ excretion (natriuresis) under conditions of high dietary salt intake. The mechanisms by which γ-MSH induces natriuresis are not well established, but two general mechanisms have been proposed [11]. One involves direct activation of MC3-R in kidney, where circulating γ-MSH binds to MC3-R to modulate renal tubular Na+ transport, and the other involves binding of MC3-R in brain, which inhibits central neural pathways that regulate sympathetic stimulation of renal tubular Na+ absorption. In support of the direct effect of γ-MSH on renal tubular Na+ transport, infusion of γ-MSH into the renal artery of rats leads to an increase in urinary Na+ excretion in the infused kidney without altering the urine output from the contralateral kidney [4]. Moreover, the natriuretic effect of γ-MSH can be blocked with intra-renal infusion of MC3-R antagonists SHU9005 and SHU9119 [19]. A more recent study has demonstrated that high Na+ feeding in rats induces an increase in MC3-R mRNA and protein expression in isolated kidney inner medullary collecting duct (IMCD) cells [18]. Moreover, incubation of IMCD cell suspensions with increasing concentrations of γ-MSH increases cAMP levels, suggesting that γ-MSH directly signals through MC3-R in IMCD cells [18].
The purpose of this study was to identify the molecular mechanisms underlying the natriuretic effect of γ-MSH in IMCD cells. Given the prior evidence showing that γ-MSH signals through MC3-R in isolated IMCD cells, we asked whether γ-MSH modulates Na+ or chloride (Cl−) transport in a mouse inner medullary collecting duct cell line (mIMCD-K2). The mIMCD-K2 cell line is an attractive model system because it retains characteristic features of the IMCD, including the signaling and ion transport machinery that support NaCl transport [12, 24]. In Ussing chamber studies, we observed no effects of γ-MSH on NaCl transport in mIMCD-K2 cells. Unexpectedly, we also found no MC3-R to be expressed in mouse kidney, suggesting that γ-MSH action in the kidney may not be mediated through direct effects on renal tubular Na+ transport but rather through effects on central neural pathways that innervate the kidney.
2. Materials and methods
2.1. Animal tissue preparation
Animal protocols were reviewed and approved by the Animal Care and Use Committee at Stanford University. C57Bl/6 and MC3-R KO mice (deposited by Dr. Roger Cone [2] to Jackson Laboratory, Bar Harbor, ME and characterized recently by Dr. Michael Humphreys [20]) at 6 weeks of age were anesthetized with pentobarbital (40–60 mg/kg) and perfused with cold PBS. Kidneys and brains were excised and snap frozen in liquid N2 and stored at −80°C for future immunoblot studies. For immunohistochemical studies, kidneys were fixed with 4% paraformadehyde in PBS overnight at 4°C, incubated in 30% sucrose in PBS, and then frozen in optimal cutting temperature (OCT) compound.
2.2. Western Blotting
Kidney and brain tissue were homogenized in ice-cold RIPA lysis buffer containing protease inhibitors and 10 μg/ml EDTA (1g tissue/1 ml buffer). Lysates were cleared by centrifugation, resolved by 10% SDS-PAGE and then analyzed by immunoblotting with the following antisera against mouse MC3-R: 1) rabbit IgG anti-MC3-R antibody (Product M4937, Sigma, St. Louis, MO, 1:2000); 2) goat polyclonal IgG anti-MC3-R antibody (Product SC-6878, Santa Cruz Biotechnology, Santa Cruz, CA, 1:100); 3) rabbit polyclonal IgG anti-MC3-R antibody (Product SC-8990, Santa Cruz Biotechnology, 1:100); and 4) Prestige polyclonal rabbit anti-MC3-R antibody (Product HPA017431, Sigma, 1:200). Membranes were incubated with species-specific horseradish peroxide-conjugated (HRP) secondary antibodies and processed, as described [22].
2.3. Immunohistochemistry
Five micron sections were cut from mouse kidney and then blocked for 1 hour in buffer composed of PBS with 10% chicken or donkey serum and 0.2% Triton X-100. Sections were then incubated with the following primary antibodies: 1) rabbit anti-MC3-R antibody (Product M4937, Sigma, 1:1000); 2) goat polyclonal IgG anti-MC3-R antibody (Product SC-6878, Santa Cruz Biotechnology, 1:100); 3) rabbit polyclonal IgG anti-MC3-R antibody (Product SC-8990, Santa Cruz Biotechnology, 1:100); 4) Prestige polyclonal rabbit anti-MC3-R antibody (Product HPA017431, Sigma, 1:20). Slides were washed with PBS + 0.2% Triton X-100 (PBST) and then incubated with secondary antibodies including Alexa Fluor 488 Chicken anti-rabbit IgG (Invitrogen, Carlsbad, CA, 1:1000) and donkey anti-goat IgG Texas Red IgG (Jackson Immunoresearch, West Grove, PA, 1:100). Sections were washed with PBST and mounted in Vectamount with DAPI (Vector Labs, Burlingame, CA) prior to visualization with a Leica DM4000 epifluorescence microscope, Leica DFC500 digital camera, and Leica Application Suite imaging software (Leica Microsystems, Bannockburn, IL).
2.4. Reverse transcriptase-PCR
Mouse brain and kidney total RNA were obtained from Zyagen (San Diego, CA). Reverse transcriptase (RT) reactions were performed according to manufacturer instructions (New England BioLabs, Ipswich, MA). Thermal cycling parameters were the following: incubation at 98°C for 30 s followed by 35 cycles (or 50 cycles for extended reactions): at 98°C for 10 s, 55°C for 10 s, 72°C for 50 s. The following PCR primers were designed and used for detecting gene amplification: 5’-TCA TGA AGT GTG ACG TTG ACA TCC G (mouse actin); 3’-CCT AGA AGC ATT TGC GGT GCA CGA T (mouse actin); 5’-ATG AAC TCT TCC TGC TGC CTG TC (mouse MC3-R); 3’-TTT CCT CAC TGT CAT GAT GCT GTG G (mouse MC3-R). Specificity of each set of primers was confirmed by BLAST search against GenBank.
2.5. Cell Culture
The following murine collecting duct cell lines were maintained and expanded, as described previously, for electrophysiological studies: mpkCCDc14 [1] (Dr. Alain Vandewalle, INSERM, Paris, France), mOMCDis [10] (Dr. Thomas DuBose, Jr., Wake Forest University, Winston-Salem, NC) and mIMCD-K2 [12] (Dr. Bruce Stanton, Dartmouth Medical School, Hanover, NH). Cells were plated on polycarbonate Snapwell inserts (Corning Costar, Lowell, MA) and grown in defined medium until transepithelial resistance (Rte) reached values between 800–1200 Ω·cm2, as measured with an EVOM “chopstick voltmeter” (World Precision Instruments, Sarasota, FL).
2.6. Ussing Chamber Measurements
Cell sheets were mounted between the Lucite half chambers of the Ussing chamber apparatus (Physiological Instruments, San Diego, CA) and bathed in Krebs-Henseleit solution (in mmol/L: 115 NaCl, 25 NaHCO3, 4.7 KCl, 1.2 MgSO4·7H2O, 2.5 CaCl2, 1.2 KH2PO4, 11.1 glucose and 0.01 Na2EDTA) and gassed with a mixture of 95% O2 and 5% CO2. Transepithelial voltage (Vte) across cell sheets was clamped to 0 mV, and a set voltage pulse of 1 mV was applied across cell sheets for 200 ms every 20 s. The short-circuit current (Isc) and Rte across cell sheets were continuously recorded using Acquire and Analyze Software (Physiological Instruments) as previously described [22].
Once Isc and Rte stabilized, cell sheets were exposed to a series of pharmacological agents. In some experiments, the apical side of some cell sheets was treated with 10−5 M of amiloride (Sigma), an inhibitor of ENaC, to verify that ENaC currents were intact in cultured cells. Both apical and basal surfaces were exposed to increasing concentrations of γ-MSH (Product H-4400, BaChem, Belmont, CA, 10−8 to 10−5 M). The γ-MSH utilized was from the same preparation as a prior study showing the stimulatory effects of γ-MSH on natriuresis in rat kidney and MC3-R signaling in IMCD cells [18]. Cell sheets were also treated directly with apical cAMP (Sigma, 10−8 M to 10−5 M) and/or apical and basal forskolin (Sigma, 10−5 M) as controls to verify that anion secretion was present in cultured cells.
2.7. cAMP Assay
Intracellular cAMP levels were measured using the Parameter cAMP ELISA assay kit (R & D Systems, Minneapolis, MN) following addition of γ-MSH (10−8 M to 10−6 M), forskolin (10−5 M), or vehicle control to both sides of cell monolayers. Cell monolayers in each treatment condition were pooled, and the intracellular cAMP concentration in each treatment group was measured per manufacturer instructions.
3. Results
3.1. γ-MSH does not modulate Isc or MC3-R signaling in mouse collecting duct cells
To assess whether γ-MSH has a direct effect on ion transport in IMCD cells, we used the Ussing chamber system to assess Isc across mIMCD-K2 cell monolayers. Addition of γ-MSH from concentrations of 10−8 M to 10−5 M to either side of cell monolayers did not modulate Isc in mIMCD-K2 cells. Similar findings were observed in two other model cell lines derived from the cortical and outer medullary collecting duct, mpkCCDc14 and mOMCDis, respectively (data not shown).
To determine whether γ-MSH signals through MC3-R to activate adenylate cyclase activity, we measured intracellular cAMP levels in response to γ-MSH treatment in mIMCD-K2 cells. Addition of 10−5 M forskolin to mIMCD-K2 cells induced a robust increase in cAMP accumulation; however, addition of γ-MSH from 10−8 M to 10−6 M to similar mIMCD-K2 cells failed to enhance intracellular cAMP levels (data not shown). Together, these findings indicate that the γ-MSH/MC3-R signaling pathway is not present in mIMCD-K2 cells.
3.2. MC3-R protein is not specifically detectable in mouse brain and kidney
Because γ-MSH treatment of mIMCD-K2 cells did not elicit any Isc response, we asked whether MC3-R was expressed in mouse kidney. We used 4 previously characterized antibodies [8, 13, 18, 25] to detect MC3-R protein by Western blot analysis in mIMCD-K2 cells (data not shown) and mouse kidney homogenates. We also tested the specificity of anti-MC3-R antibodies against brain and kidney lysates from MC3-R KO mice as negative controls. Using the Sigma anti-MC3-R antibody, we detected an immunoreactive band at ~40 KDa in wild-type (WT) mouse brain lysates, and a smaller ~28 KDa band in WT mouse kidney lysates; however the same patterns of staining were also observed in MC3-R KO brain and kidney lysates (Figure 1a). Western blotting with 3 other anti-MC3-R antibodies demonstrated the same patterns of immunoreactive bands in brain and kidney lysates from WT and KO mice. These results indicate that 4 previously characterized antibodies used for analyzing MC3-R protein expression fail to specifically detect MC3-R protein.
Figure 1. MC3-R protein is not specifically detectable in mouse brain and kidney.

a) Western blot analysis of brain (Brain) and kidney (Kidney) homogenates from wild type (+/+) and MC3-R knockout (−/−) mice. Blots were probed with a rabbit anti-MC3-R antibody (Sigma Aldrich M4937). A ~40Kda band (**) is seen both in the +/+ and −/− samples of mouse brain tissue. A smaller ~28 kDa band (*) is seen in the +/+ and −/− kidney lanes. b) Immunohistochemistry of MC3-R wild-type (WT) and knockout (KO) mouse kidney sections. Sample sections from WT (A–C) and KO (D–F) mouse kidney stained with a rabbit anti-MC3-R antibody (Sigma M4937) and a goat anti-AQP2 antibody (Santa Cruz Biotechnology C-17). Images stained for MC3-R (A and D), AQP2 (B and E), and merge with DAPI staining (C and F).
To further characterize MC3-R protein expression in mouse kidney and simultaneously assess the specificity of anti-MC3-R antibodies, we used these 4 antibodies for immunohistochemical staining of WT and KO mouse kidney (Figure 1b). Sample sections of mouse kidney stained with the Sigma anti-MC3-R antibody demonstrated immunoreactivity in kidney tubules of the inner medulla. Co-staining of kidney sections with goat anti-aquaporin 2 (AQP2) antibody, which detects AQP2 as a marker for the principal cell of the collecting duct, revealed that apparent MC3-R immunoreactivity was present in medullary collecting duct cells, as has been previously described in rat kidney [18]. However, we found that immunostaining with the anti-MC3-R antibody of kidneys from MC3-R KO mice showed an identical pattern. Figure 1b is also representative of patterns of immunostaining detected with the other antibodies for MC3-R, again indicating that these antibodies are not specific for MC3-R protein.
3.3. MC3-R gene expression is not detectable by RT-PCR in mouse kidney
Since the existing anti-MC3-R antibodies could not specifically detect MC3-R protein, we used RT-PCR to determine whether MC3-R mRNA is present in mouse kidney. Using sequence-specific primers for mouse MC3-R, we detected PCR products of the predicted size for MC3-R in mouse brain but not in mouse kidney (Figure 2). With extended cycles of PCR amplification, we did observe a faint band of expression, but we also detected a faint band in the negative RT control sample. We conclude that this PCR product from extended cycles of PCR amplification could represent a low level of MC3-R mRNA in mouse kidney or a low level of genomic contamination. The latter is a theoretical possibility given that we could not design primers that span sequences across exon-intron junctions because MC3-R is a single exon gene [9].
Figure 2. MC3-R gene expression is not detectable by RT-PCR in mouse kidney.
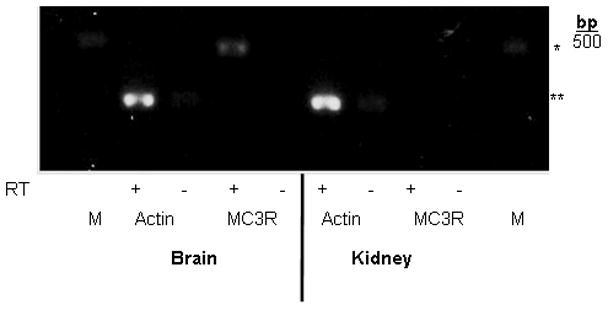
Photograph of gel showing PCR amplification products from wild-type mouse brain and kidney tissue after reverse transcription of actin (loading control) and MC3-R. Samples containing reverse transcriptase (+) or negative controls lacking reverse transcriptase (−) are included for each tissue/primer combination. The end lane on each side contains a 500 base pair marker (M). *denotes the predicted product size for MC3-R (480 bp), and **denotes the product size for actin (285 bp).
4. Discussion
There has been growing interest in the role of the γ-MSH/MC3-R pathway in regulating NaCl balance and blood pressure. The molecular and cellular mechanisms underlying how this pathway controls NaCl transport in the kidney are virtually unknown. MC3-R expression in the kidney has previously been localized to rat IMCD cells [18]. As a first step towards characterizing MC3-R function in kidney cells, we used the mIMCD-K2 cell line as a model system to explore the possibility that γ-MSH directly modulates NaCl transport. We found that γ-MSH does not directly alter ion transport or cAMP signaling in mouse collecting duct cells, including mIMCD-K2 cells. Our data further demonstrate that anti-MC3-R antibodies used in previous studies for characterizing MC3-R protein expression could not specifically detect MC3-R in mouse brain or kidney. Finally, we could not find definitive evidence by RT-PCR that MC3-R is expressed in mouse kidney.
At first glance, our results conflict with prior reports showing that MC3-R mRNA is expressed in mouse or rat kidney. Although MC3-R mRNA has been detected in isolated rat IMCD cells and kidney homogenates, these studies were performed without demonstrating a negative RT control [18], which leaves open the possibility that genomic MC3-R was being detected. Use of this negative control is particularly important because the MC3-R gene consists of a single exon. Similar to results from this study, a recent report also failed to detect MC3-R expression in both human kidney homogenates and specific kidney cell types (podocytes, glomerular endothelial cells, mesangial cells, and tubular epithelial cells) [14].
MC3-R protein has also been detected in rat kidney homogenates and IMCD cells by Western blot analysis [18]. Using kidneys from MC3-R KO mice as controls, we tested the specificity of an array of anti-MC3-R antibodies to characterize MC3-R protein expression in mouse brain and kidney. We did not see any differences in the pattern of staining by Western blot or immunohistochemistry in brain and kidney samples from wild type or MC3-R KO mice. These data suggest that currently available anti-MC3-R antibodies do not specifically detect MC3-R and call into question prior MC3-R localization studies involving these particular antibodies.
While the possibility remains that the collecting duct cells studied here have lost endogenous MC3-R expression and/or that MC3-R is expressed in a small population of cells not detectable in kidney homogenates, another possibility is that the mechanism for γ-MSH action on urinary NaCl excretion involves suppression of central neural pathways that regulate renal NaCl absorption, first proposed by Humphreys [11]. In support of this mechanism, the natriuretic effect of renal arterial infusion of γ-MSH is eliminated if the kidneys of rats are first surgically denervated [4]. Salt-sensitive hypertension in PC2 KO mice can be corrected with intra-cerebral γ-MSH infusion at doses in the picomolar range, a concentration too low to account for hormonal action via the systemic circulation [20]. The blood pressure response to γ-MSH infusion in these animals is also rapid, on the time scale of minutes, which makes the vasodepressor effect of γ-MSH unlikely to involve changes in urinary salt excretion. Renal salt retention, however, could still contribute to long-term maintenance of salt-sensitive hypertension in mice with disrupted γ-MSH signaling. The findings presented here are consistent with the notion that the natriuretic effect of γ-MSH, like the vasodepressor effect, involves suppression of central neural pathways, possibly limiting sympathetic outflow to kidney tubules, which normally maintain a tonic level of renal salt absorption [7]. Indeed, recent evidence demonstrates that intravenous infusion of γ-MSH normalizes the elevated plasma noradrenaline levels in γ-MSH-deficient rats [21], consistent with the role of the γ-MSH/MC3-R pathway serving as an adrenergic brake mechanism.
5. Conclusions
In summary, our study demonstrates the absence of a direct effect of γ-MSH on MC3-R signaling or ion transport in mouse IMCD cells and the absence of MC3-R expression in mouse kidney. These data support the notion that the mechanism for γ-MSH-induced natriuresis may not involve circulating γ-MSH directly activating MC3-R in renal tubular cells, but rather may include inhibiting central neural pathways that tonically maintain renal tubular Na+ absorption. These findings also highlight the difficulties in assessing MC3-R mRNA and protein expression and should provide guidance for future studies regarding which antibodies to avoid in the characterization of MC3-R protein expression in mice and rats.
Acknowledgments
We are grateful to Drs. Bruce Stanton (Dartmouth), Alain Vandewalle (INSERM), and Thomas DuBose, Jr. (Wake Forest) for providing mIMCD-K2, mpkCCDc14, and mOMCDis cells, respectively. We also thank Dr. Michael Humphreys (University of California, San Francisco), Dr. John Stokes (University of Iowa) and Drs. Vivek Bhalla and Glenn Chertow (Stanford University) for valuable discussion on the manuscript. This work was supported by grants from National Institutes of Health (K08-DK-073487 to A.C.P. and T32-DK07357-26A1 to P.K.), Amgen (2009 Amgen Nephrology Junior Faculty Research Support Program to A.C.P), Satellite Healthcare (2008 Norman S. Coplon Extramural Grant to A.C.P.), and the Forest Research Institute (2009 Emerging Leaders in Hypertension Program to P.K.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bens M, Vallet V, Cluzeaud F, Pascual-Letallec L, Kahn A, Rafestin-Oblin ME, et al. Corticosteroid-dependent sodium transport in a novel immortalized mouse collecting duct principal cell line. J Am Soc Nephrol. 1999;10:923–34. doi: 10.1681/ASN.V105923. [DOI] [PubMed] [Google Scholar]
- 2.Butler AA, Kesterson RA, Khong K, Cullen MJ, Pelleymounter MA, Dekoning J, et al. A unique metabolic syndrome causes obesity in the melanocortin-3 receptor-deficient mouse. Endocrinology. 2000;141:3518–21. doi: 10.1210/endo.141.9.7791. [DOI] [PubMed] [Google Scholar]
- 3.Chandramohan G, Ni XP, Kalinyak JE, Humphreys MH. Dietary sodium modulates mRNA abundance of enzymes involved in pituitary processing of proopiomelanocortin. Pituitary. 2001;4:231–7. doi: 10.1023/a:1020746414046. [DOI] [PubMed] [Google Scholar]
- 4.Chen XW, Ying WZ, Valentin JP, Ling KT, Lin SY, Wiedemann E, et al. Mechanism of the natriuretic action of gamma-melanocyte-stimulating hormone. Am J Physiol. 1997;272:R1946–53. doi: 10.1152/ajpregu.1997.272.6.R1946. [DOI] [PubMed] [Google Scholar]
- 5.Chhajlani V. Distribution of cDNA for melanocortin receptor subtypes in human tissues. Biochem Mol Biol Int. 1996;38:73–80. [PubMed] [Google Scholar]
- 6.Desarnaud F, Labbe O, Eggerickx D, Vassart G, Parmentier M. Molecular cloning, functional expression and pharmacological characterization of a mouse melanocortin receptor gene. Biochem J. 1994;299 ( Pt 2):367–73. doi: 10.1042/bj2990367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DiBona GF, Kopp UC. Neural control of renal function. Physiol Rev. 1997;77:75–197. doi: 10.1152/physrev.1997.77.1.75. [DOI] [PubMed] [Google Scholar]
- 8.Evans JF, Niu QT, Canas JA, Shen CL, Aloia JF, Yeh JK. ACTH enhances chondrogenesis in multipotential progenitor cells and matrix production in chondrocytes. Bone. 2004;35:96–107. doi: 10.1016/j.bone.2004.03.015. [DOI] [PubMed] [Google Scholar]
- 9.Gantz I, Konda Y, Tashiro T, Shimoto Y, Miwa H, Munzert G, et al. Molecular cloning of a novel melanocortin receptor. J Biol Chem. 1993;268:8246–50. [PubMed] [Google Scholar]
- 10.Guntupalli J, Onuigbo M, Wall S, Alpern RJ, DuBose TD., Jr Adaptation to low-K+ media increases H(+)-K(+)-ATPase but not H(+)-ATPase-mediated pHi recovery in OMCD1 cells. Am J Physiol. 1997;273:C558–71. doi: 10.1152/ajpcell.1997.273.2.C558. [DOI] [PubMed] [Google Scholar]
- 11.Humphreys MH. Gamma-MSH, sodium metabolism, and salt-sensitive hypertension. Am J Physiol Regul Integr Comp Physiol. 2004;286:R417–30. doi: 10.1152/ajpregu.00365.2003. [DOI] [PubMed] [Google Scholar]
- 12.Kizer NL, Lewis B, Stanton BA. Electrogenic sodium absorption and chloride secretion by an inner medullary collecting duct cell line (mIMCD-K2) Am J Physiol. 1995;268:F347–55. doi: 10.1152/ajprenal.1995.268.2.F347. [DOI] [PubMed] [Google Scholar]
- 13.Leoni G, Patel HB, Sampaio AL, Gavins FN, Murray JF, Grieco P, et al. Inflamed phenotype of the mesenteric microcirculation of melanocortin type 3 receptor-null mice after ischemia-reperfusion. FASEB J. 2008;22:4228–38. doi: 10.1096/fj.08-113886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lindskog A, Ebefors K, Johansson ME, Stefansson B, Granqvist A, Arnadottir M, et al. Melanocortin 1 receptor agonists reduce proteinuria. J Am Soc Nephrol. 2010;21:1290–8. doi: 10.1681/ASN.2009101025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mayan H, Ling KT, Lee EY, Wiedemann E, Kalinyak JE, Humphreys MH. Dietary sodium intake modulates pituitary proopiomelanocortin mRNA abundance. Hypertension. 1996;28:244–9. doi: 10.1161/01.hyp.28.2.244. [DOI] [PubMed] [Google Scholar]
- 16.Mayan H, Ni XP, Almog S, Humphreys MH. Suppression of gamma-melanocyte-stimulating hormone secretion is accompanied by salt-sensitive hypertension in the rat. Hypertension. 2003;42:962–7. doi: 10.1161/01.HYP.0000097601.83235.F8. [DOI] [PubMed] [Google Scholar]
- 17.Ni XP, Bhargava A, Pearce D, Humphreys MH. Dietary sodium intake modulates renal expression of melanocortin 3 receptor (MC3-R) mRNA and protein abundance in the rat. Journal of the American Society of Nephrology. 2002;13:81a-a. [Google Scholar]
- 18.Ni XP, Bhargava A, Pearce D, Humphreys MH. Modulation by dietary sodium intake of melanocortin 3 receptor mRNA and protein abundance in the rat kidney. Am J Physiol Regul Integr Comp Physiol. 2006;290:R560–7. doi: 10.1152/ajpregu.00279.2005. [DOI] [PubMed] [Google Scholar]
- 19.Ni XP, Kesterson RA, Sharma SD, Hruby VJ, Cone RD, Wiedemann E, et al. Prevention of reflex natriuresis after acute unilateral nephrectomy by melanocortin receptor antagonists. Am J Physiol. 1998;274:R931–8. doi: 10.1152/ajpregu.1998.274.4.R931. [DOI] [PubMed] [Google Scholar]
- 20.Ni XP, Pearce D, Butler AA, Cone RD, Humphreys MH. Genetic disruption of gamma-melanocyte-stimulating hormone signaling leads to salt-sensitive hypertension in the mouse. J Clin Invest. 2003;111:1251–8. doi: 10.1172/JCI16993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ni XP, van Dijk C, Pearce D, Humphreys MH. Evidence for a noradrenergic mechanism causing hypertension and abnormal glucose metabolism in rats with relative deficiency of gamma-melanocyte-stimulating hormone. Exp Physiol. 2009;94:867–76. doi: 10.1113/expphysiol.2009.046748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rajagopal M, Pao AC. Adenosine activates a2b receptors and enhances chloride secretion in kidney inner medullary collecting duct cells. Hypertension. 2010;55:1123–8. doi: 10.1161/HYPERTENSIONAHA.109.143404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roselli-Rehfuss L, Mountjoy KG, Robbins LS, Mortrud MT, Low MJ, Tatro JB, et al. Identification of a receptor for gamma melanotropin and other proopiomelanocortin peptides in the hypothalamus and limbic system. Proc Natl Acad Sci U S A. 1993;90:8856–60. doi: 10.1073/pnas.90.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vandorpe D, Kizer N, Ciampollilo F, Moyer B, Karlson K, Guggino WB, et al. CFTR mediates electrogenic chloride secretion in mouse inner medullary collecting duct (mIMCD-K2) cells. Am J Physiol. 1995;269:C683–9. doi: 10.1152/ajpcell.1995.269.3.C683. [DOI] [PubMed] [Google Scholar]
- 25.Wachira SJ, Guruswamy B, Uradu L, Hughes-Darden CA, Denaro FJ. Activation and endocytic internalization of melanocortin 3 receptor in neuronal cells. Ann N Y Acad Sci. 2007;1096:271–86. doi: 10.1196/annals.1397.093. [DOI] [PubMed] [Google Scholar]