

Abstract
To define the physiological role of IP33-kinase(A) in vivo, we have generated a mouse strain with a null mutation of the IP33-kinase(A) locus by gene targeting. Homozygous mutant mice were fully viable, fertile, apparently normal, and did not show any morphological anomaly in brain sections. In the mutant brain, the IP4 level was significantly decreased whereas the IP3 level did not change, demonstrating a major role of IP33-kinase(A) in the generation of IP4. Nevertheless, no significant difference was detected in the hippocampal neuronal cells of the wild-type and the mutant mice in the kinetics of Ca2+ regulation after glutamate stimulation. Electrophysiological analyses carried out in hippocampal slices showed that the mutation significantly enhanced the LTP in the hippocampal CA1 region, but had no effect on the LTP in dentate gyrus (DG). No difference was noted, however, between the mutant and the wild-type mice in the Morris water maze task. Our results indicate that IP33-kinase(A) may play an important role in the regulation of LTP in hippocampal CA1 region through the generation of IP4, but the enhanced LTP in the hippocampal CA1 does not affect spatial learning and memory.
External signals arriving at cell-surface receptors induce second messengers in the cell to transfer information to the final effector system. Inositol 1,4,5-trisphosphate (IP3) is one of the second messengers that is produced when phospholipase C (PLC) hydrolyzes phosphatidyl inositol bisphosphate (PIP2; Rhee et al. 1989). IP3 produced by PLC binds to IP3 receptor on the intracellular Ca2+ stores and induces calcium release into the cytosol (Irvine 1990; Furuichi and Mikoshiba 1995). IP3 is then metabolized to inositol 1,3,4,5-tetrakisphosphate (IP4) by IP33-kinase or to inositol 1,4-bisphosphate (IP2) by 5-phosphatase. Two isotypes of IP33-kinase, A and B, each encoded by a distinct gene, were cloned from rat and human (Choi et al. 1990; Mailleux et al. 1992). IP33-kinase(A) is expressed in testis and in neuronal cells in brain (Vanweyenberg et al. 1995). In brain, the IP33-kinase(A) gene is highly expressed in pyramidal cells in the hippocampal CA1 region and Purkinje cells in cerebellum (Mailleux et al. 1991b; Yamada et al. 1992). These cells have often been studied with respect to long-term potentiation (LTP) and long-term depression (LTD), which are believed to be involved in learning and memory (Artola and Singer 1993; Malenka 1994). Therefore, it has been assumed that IP33-kinase(A) has a role in the memory process (Mailleux et al. 1991a). IP33-kinase(B) is widely distributed in various organs including heart, lung, thymus, and astrocytes in brain (Vanweyenberg et al. 1995). Until now, the IP33-kinase reaction was believed to be the only known pathway to produce IP4 (Communi et al. 1995). However, the physiological role(s) of IP33-kinase(A) is not known yet.
There has been much effort over the last few years to envisage the possible physiological role(s) of IP4. As IP4 is rapidly produced in response to activation of PLC-coupled receptors in various cell types, it is believed that IP4 has a second messenger role in the cell (Batty et al. 1985; Challiss and Nahorski 1991). Experimental results suggested a possible role of IP4 in calcium homeostasis (Irvine 1991). IP4 was also implicated to be involved in the neurotransmitter release by binding the C2B domain of synaptotagmin II (Fukuda et al. 1995). Recently, high-affinity IP4-binding proteins were characterized to be GTPase-activating protein (GAP; Cullen et al. 1995; Fukuda and Mikoshiba 1996). However, the exact physiological role(s) of IP4 is not clear yet.
LTP is an activity-dependent modification of the synaptic efficacy (Bliss and Collingridge 1993). Induction of LTP requires activation of postsynaptic glutamate receptors by depolarization of postsynaptic neurons, which is generated by high-frequency afferent stimulation (Madison et al. 1991; Bliss and Collingridge 1993). Two types of receptors, ionotropic receptors and metabotropic receptors, mediate the increase of intracellular calcium concentration in neuronal cells and are believed to be necessary for induction of LTP (Malenka and Nicoll 1993; Bortolotto et al. 1994). Although NMDA receptors increase the intracellular Ca2+ concentration by opening their own channels, metabotropic glutamate receptors increase the intracellular Ca2+ concentration through activation of PLC enzymes, an event that produces IP3 and diacylglycerol (DAG) (Regher and Tank 1990; Nakanishi 1992). Many reports have shown that metabotropic glutamate receptors are necessary for synaptic function and induction of LTP or LTD (Bashir et al. 1993; Hayashi et al. 1993; Linden 1994). It is generally believed that LTP in hippocampus is a synaptic substrate of memory (Bliss and Collingridge 1993; Eichenbaum 1996). Gene targeting strategies have been successfully used for studying molecular and cellular mechanisms of learning, especially for understanding the relationship between LTP and spatial learning (Mayford et al. 1995). In some cases, an association between LTP and learning and memory was demonstrated (Grant et al. 1992; Silva et al. 1992a,b; Aiba et al. 1994; Sakimura et al. 1995). In other cases, a dissociation was observed between LTP and learning and memory (Abeliovich et al. 1993a,b; Huang et al. 1996).
Here, we have defined the role of IP33-kinase(A) in vivo by generating and analyzing a null mutation of the IP33-kinase(A) locus. Focus was given to the role of this enzyme in Ca2+ regulation, LTP, and learning and memory.
Materials and Methods
ANIMALS
All the mice used for analysis of the mutants were F2 progeny derived from matings of the heterozygotes in the F1 (129/Sv × C57BL/6J) genetic background. The mice had ad lib access to food and water. Mice were housed in standard plastic cages (length × width × height, 25 cm × 18 cm × 13 cm) in an animal facility and kept on a 14:10–hr light–dark cycle with the light onset at 6 a.m. All animal handling was done following the guidelines of the Animal Care and Use Committee.
GENERATION OF IP33-KINASE(A)-DEFICIENT MICE
The strategy for vector construction was as described previously (Kim et al. 1997). Genomic clones containing an exon that encodes the 5′ NTR and the nucleotide sequence 1–456 from the initiation codon of cDNA were isolated by use of the rat brain cDNA (pKK 233-2/IP3K, Choi et al. 1990) as a probe, mapped by various enzymes, and used to make the targeting vector. The neo cassette [PGK promoter–neo–PGK poly(A)] from pPNT (Tybulewicz et al. 1991) was inserted into nucleotide sequence 389 from the translation start codon for gene disruption and positive selection. The TK cassette from pPNT was attached to the end of the 3′ homology region for negative selection. The final targeting construct had a 3.7-kb 5′ homology and a 6.2-kb 3′ homology to the genomic locus (Fig. 1A). The targeting vector was linearized at the 5′ end and transfected into J1 ES cells (gift of E. Li, Massachusetts General Hospital/Harvard Medical School, Boston) at 270 V and 500 μF in the gene pulser (Bio-Rad). The transfected cells were plated on feeder layers of mitomycin C-treated (Sigma) embryonic fibroblast cells derived from 13.5-day embryos carrying the targeted mutation of β2-microglobulin (gift of R. Jaenisch, Whitehead Institute for Biomedical Research, Cambridge, MA). Selection with 0.3 mg/ml G418 (GIBCO) and 2 μm Gancyclovir (Syntax, CA) was started 48 hr after transfection and sustained for 10 days. Resistant colonies were picked and analyzed by Southern blotting. Three targeted ES cell clones were injected into C57BL/6J blastocysts to make chimeric mice. Male chimeras were crossed to C57BL/6J females to obtain the germ-line-transmitted offspring. The progeny were genotyped by Southern blotting.
Figure 1.


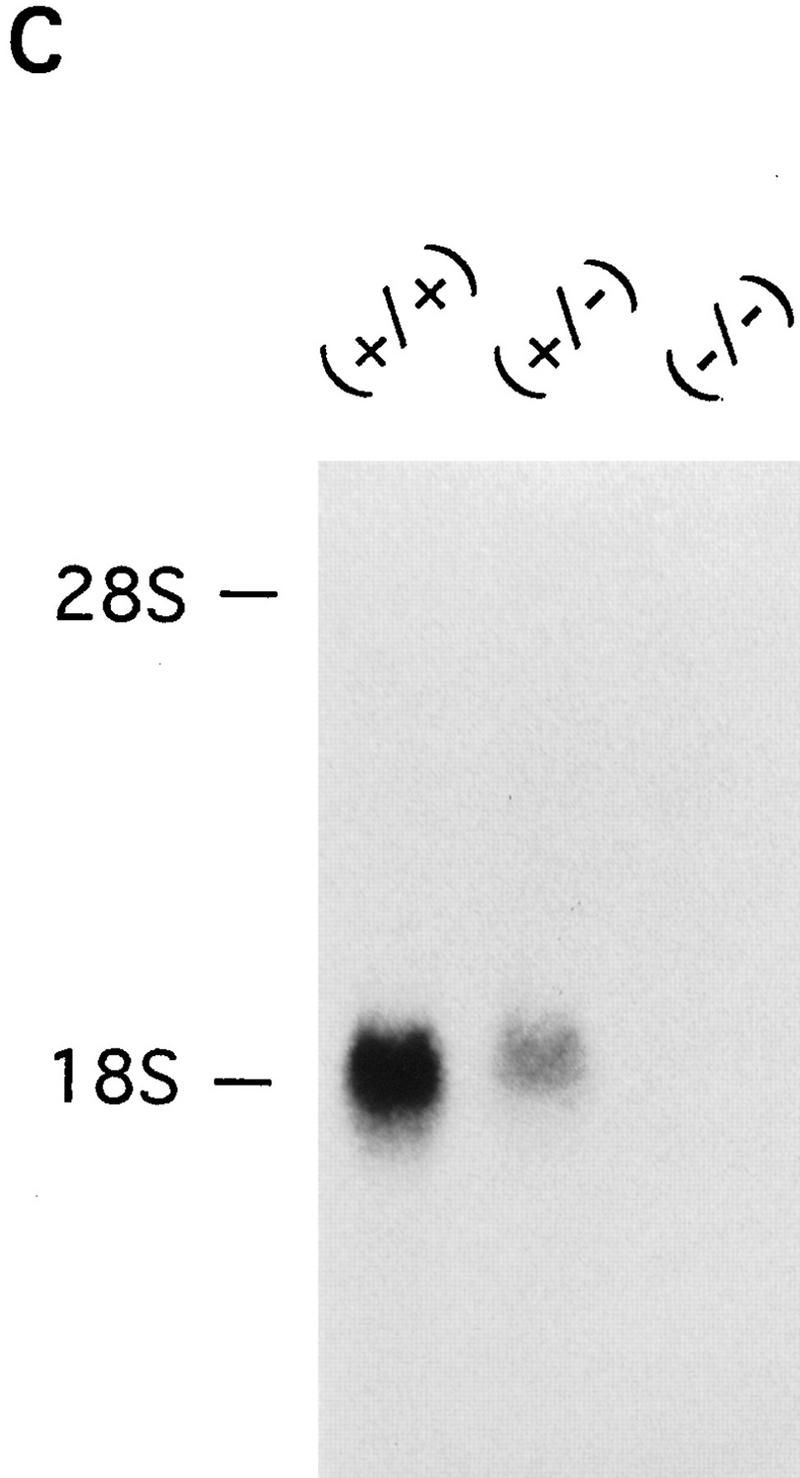

Disruption of the mouse IP33-kinase(A) locus by gene targeting. (A) Structure of the wild type IP33-kinase(A) gene locus, the targeting vector, and the targeted IP33-kinase(A) gene locus. Rules under the wild-type and the targeted loci represent expected fragments when digested with SacI and hybridized with the probe (hatched bar). (BI) BstBI; (E) EcoRI; (H) HindIII; (K) KpnI; (S) SacI; (XI) XhoI. (Horizontal arrows) Transcriptional direction of the pgk-neo and the pgk-tk cassettes. (B) Southern blot analysis of offspring derived from intercrosses of IP33-kinase (A) heterozygous parents. The 7.2-kb fragment is the wild-type allele and the 9.0-kb fragment, the targeted allele. (C) Northern blot analysis of the brain tissue. Ten micrograms of total brain RNA was isolated from a mouse of each genotype and hybridized with 32P-labeled rat IP33-kinase(A) cDNA. (D) Western blot analysis of brain tissue. Total brain protein was isolated from a mouse of each genotype and immunoblotted with anti-rat-IP33-kinase(A) monoclonal antibodies. (+/+) Wild type; (+/−), heterozygote; (−/−), homozygote mutant.
SOUTHERN BLOT ANALYSIS
Ten micrograms of DNA isolated from each tail tip was digested with SacI, electrophoresed through 0.6% agarose gel, transferred with 0.4 n NaOH onto a nylon membrane, and hybridized at 65°C with 32P-labeled probe, 1-kb XbaI/BstBI fragment that is 5′ to and not included in the targeting vector. Blots were washed in 0.1× SSC and 0.1% SDS at 65°C for 10 min and exposed to autoradiography.
NORTHERN BLOT ANALYSIS
Brain total RNA was isolated from adult mice, 9 weeks old, by use of the Trireagent (MRC). Ten micrograms of total RNA from each genotype was electrophoresed through 1% formaldehyde agarose gel, transferred onto a nylon membrane, and hybridized at 65°C with 32P-labeled rat IP33-kinase(A) cDNA probe. Blots were washed in 0.1× SSC and 0.1% SDS at 65°C for 10 min and exposed to autoradiography.
WESTERN BLOT ANALYSIS
Total protein (150 μg) derived from 9-week-old wild-type and mutant brain was separated by 10% SDS-PAGE and electrophoretically transferred onto a nitrocellulose membrane overnight at 0.5 A in TGM buffer [25 mm Tris-Cl at pH 8.3, 192 mm glycine, 20% (vol/vol) methanol]. Equal loading of protein per lane was confirmed by Coomassie staining of a duplicated gel. The blotted membrane was incubated with 1% BSA in TBST (50 mm Tris-Cl at pH 7.5, 0.15 m NaCl, 0.2% Tween 20) at room temperature and then probed with a mixture of three kinds of mouse anti-rat IP33-kinase(A) monoclonal antibodies for 1 hr (Lee et al. 1990). After washing three times with TBST for 10 min, the membrane was incubated with alkaline phosphatase-conjugated goat anti-mouse IgG antibodies (diluted 1:7500 in TBS, Promega) at 4°C for 1.5 hr and then washed three times with TBST. Finally, the membrane was incubated with NBT/BCIP substrate of alkaline phosphatase to develop the color.
IMMUNOHISTOCHEMISTRY
Eight-week old mice were anesthetized and perfused transcardially with 4% paraformaldehyde in PBS. Brain sections with thickness of 50 μm were obtained by use of a Vibratome. Mouse anti-rat IP33-kinase(A) monoclonal antibody (1:100 dilution) was used as a primary antibody. Sections were incubated for 2 hr at room temperature in goat anti-mouse biotinylated secondary antibody (1:200) in PBS plus 3% goat serum. Sections were incubated with the avidin–biotin complex for 40 min at room temperature and developed with a diaminobenzydine solution (Vector).
RENATURATION OF IP33-KINASE AFTER SDS-PAGE
The crude IP33-kinase from 9-week-old mouse brain homogenates was denatured in the presence of 3% SDS and 5% 2-mercaptoethanol without boiling and was immediately separated in a 10% SDS–polyacrylamide slab gel at 4°C. After electrophoresis, the gel was cut into 3-mm slices and each slice was homogenized in 400 μl of a solution containing 84 mm HEPES/NaOH (pH 7.5), 0.1% Triton X-100, 12 mm 2-mercaptoethanol, and 25% sucrose. After 12 hr of incubation at 4°C, fractions were centrifuged (12,000g at 4°C for 2 min) and the supernatants were assayed for IP33-kinase activity in the presence of 0.1% Chaps. Twenty microliters of the supernatant was used in the assay with a reaction time of 1 hr.
MEASUREMENT OF IP3
Freshly isolated mouse brains, 7–11 weeks old, were frozen rapidly in dry ice. Samples were weighed and sonicated in 1 ml of 6% trichloroacetic acid (TCA). The samples were left on ice for 30 min to extract the water-soluble inositol phosphates. After centrifugation of the samples at 12,000g at 4°C for 10 min, the supernatant was extracted with diethyl ether to remove the TCA. Then, this extract was neutralized with 200 mm Trizma base and used as the IP3 source. The protein concentration of the tissue pellet was determined for each sample to normalize the assay. IP3 levels were determined by a competition assay (Challiss and Nahorski 1991). Assays were performed at 4°C in a final volume of 80 μl. A 20-μl aliquot of each sample was added to 20 μl of a buffer solution containing 100 mm Tris-HCl, 4 mm EDTA (pH 8.0) and 20 μl of [3H]IP3 (0.15 μCi/ml). The binding assay was initiated by adding to each sample 300 μg of bovine adrenal cortex membrane preparation containing IP3-binding proteins, and the mixtures were vortex-mixed intermittently for 15 min. Bound and free [3H]IP3 was separated by centrifugation (12,000g, 4°C for 10 min), followed by aspiration of the supernatant. Water (100 μl) and 1 ml of scintillation cocktail were added to the pellet for the measurement of the radioactivity. A standard curve obtained from using varying amounts of IP3 (0.15–1.5 pmole) was used for the calibration of the measurements in the binding assay. Student’s t-test was used for data analysis.
METAL DYE DETECTION HPLC ANALYSIS OF INOSITOL PHOSPHATES
The whole procedure for metal dye detection HPLC is described in detail (Mayr 1988) and HPLC conditions and detection procedure were as described (Guse et al. 1995). Briefly, brains from mice, 9–11 weeks old, killed rapidly by cervical dislocation, were removed and dissected within 2 min into cortex, cerebellum, midbrain, hippocampus, and brain stem. They were frozen in liquid nitrogen and weighed prior to extraction. Samples up to 150 mg wet weight were added to 1 ml of ice-cold 8% TCA (wt/vol) containing 15 μl of EDTA (0.2 m) and 15 μl of NaF (0.1 m). After homogenization [four times with a Mini-Ultra-Turrax (tubes and rotor were precooled in ice) at 13,500 rpm for 10 sec] the samples were frozen and thawed two times. The precipitates containing acid-labile phosphoinositides were removed by centrifugation at 14,000g for 5 min at 4°C, and then the supernatant was incubated for 30 min at 37°C to destroy creatine phosphate. Then, TCA was removed by extraction with 3 ml of ice-cold diethyl ether three times. Extracts were adjusted to pH 6.0 with 1 m triethanolamine and subsequently charcoal treated by addition of 50 μl of a 20% (wt/vol) suspension of Norit A in 0.1 m NaCl and 50 mm NaOAc (pH 4.0) per sample corresponding to 100 mg of brain tissue. Each charcoal pellet was washed with 100 μl of 0.1 m NaCl, 25 mm NaF, and 0.5 mm EDTA at pH 7.0, which subsequently combined with the supernatant. The supernatants were concentrated to 1 ml by lyophilization in a vacuum centrifuge and applied to a column of Mini Q3,2/3 for HPLC with metal dye detection. Student t-test was used for data analysis.
ELECTROPHYSIOLOGY
Mouse brains, 7–9 weeks old, were separated and placed in an ice-cold (4°C) artificial cerebrospinal fluid (ACSF) containing 124 mm NaCl, 4.4 mm KCl, 25 mm NaHCO3, 1 mm Na2H2PO4, 3 mm CaCl2, 3 mm MgSO4, and 10 mm glucose. Hippocampal slices (400 μm thick) were prepared by use of the McIlwain chopper (Mickle Laboratory Engineering) and transferred to a submersion recording chamber containing ACSF with 1 μm bicuculine methiodide (Sigma). The temperature in the recording chamber was maintained at 30°C. Electrophysiological experiments were begun after waiting at least 2 hr for the slice to recover. A bipolar stimulating electrode (Master-8, AMPI) was used to stimulate Schaffer collateral and commisural fibers in the stratum radiatum of the CA1 region. To stimulate the perforant pathway, a bipolar stimulating electrode was placed along the margin of the hippocampal fissure between the dentate molecular layer and the subiculum. In field potential recordings, synaptic responses were elicited at 0.03 Hz and recorded with a low-resistance glass microelectrode filled with ACSF. Tetanus-induced LTP was elicited with trains (1 sec) of 100 Hz stimulation delivered three times with an intertrain interval of 20 sec. Field potential was recorded with the DAM 80 differential amplifier (WPI). Student t-test was used for data analysis.
CELL CULTURE AND CALCIUM IMAGING ANALYSIS
The hippocampi were dissected from newborn pups (1 day old; F2 progeny from 129/Sv × C57BL/6J background) and stored in DMKM solution containing 82 mm Na2SO4, 30 mm K2SO4, 16 mm MgCl2, 0.25 mm CaCl2, 1 mm kynurenic acid, 20 mm glucose, 0.001% phenol red, and 1.5 mm HEPES in ddH2O at pH 7.4. The hippocampi were digested at 37°C for 20 min with papain solution containing 10 units of papain (Worthington) and 0.45 mg of cysteine (Sigma) in 1 ml of DMKM. The supernatant was discarded and the tissues were washed with DMKM three times. Then, the tissues were triturated with DMKM by pipetting up and down. After trituration, the supernatant was plated onto a laminin-coated (0.5 μg/cm2, GIBCO) and lysine-coated (100 μg/ml, Collaborative Biomedical Product) coverslip and incubated at 37°C with 5% CO2. After 1–2 hr, the media were replaced with growth media consisting of Neurobasal Medium (GIBCO) supplemented with 1× B27 (GIBCO), 1% penicillin/streptomycin, and 0.5 mm glutamine. These cultures were incubated for 7–10 days and then used for calcium imaging. The calcium imaging was carried out as described previously with some modifications (Wayman et al. 1995). Before imaging, cells were incubated for 30 min at room temperature in the dark with 5 μm fura-2 actoxymethylester (Molecular Probes) in a calcium imaging buffer (140 mm NaCl, 10 mm HEPES, 5 mm KCl, 0.5 mm MgCl2, 1.5 mm CaCl2, 10 mm glucose in ddH2O at pH 7.4). Cells were then washed with the imaging buffer and allowed to rest in the original culture media in a CO2 incubator for 1 hr before imaging. Excitation light was provided by a 75-W xenon lamp and was attenuated by 75% to minimize photobleaching. A G.W. Ellis fiber optic light scrambler (Technical Video Ltd.) was used to tranmit the light to the microscope. Excitation, emission, and neutral density filters were purchased from Omega Optical. The objective used was a Nikon Fluor 20/1.3 NA. The images were intensified with the GenIIsys image intensifier (Dage-MTI) and acquired with the Dage-MTI CCD-72 series camera. All image acquisitions were obtained at 1-sec intervals and recorded by use of the FLUOR software (Universal Imaging, West Chester, PA). Images were viewed on the Sony Trinitron color video monitor (PVM-1343MD) and printed.
MORRIS WATER MAZE TEST
The water maze was a circular plastic pool 120 cm in diameter and 50 cm in height. The pool was centrally located in a room (2.5 × 2.5 m) in which four different distal ques were hung on the four walls high enough to be seen in the pool. The water temperature was maintained at 24°C–26°C. Nontoxic water soluble paint was added to make the water opaque. A 10-cm diam. escape platform was submerged 1 cm below the water surface at a fixed location during the test. The day before training, each mouse was allowed to swim freely for 60 sec, allowed to climb the platform three times, and rest on the platform for 30 sec. On each trial, the mouse was placed facing along the edge of the wall and randomly released at four different start points. Each trial was ended when either the mouse had climbed onto the platform or 60 sec had passed before the mouse located the platform. After locating the platform, each mouse was allowed to rest for 30 sec on the platform. Mice were given one block of three trials a day for 9 consecutive days with a 1-hr intertrial interval each day. During each trial, the time elapsed for locating the platform was determined. A single probe test was carried out either 3 or 7 days after the last trial. Mice were placed and released at the location opposite the site where the platform had been located and allowed to swim for 60 sec. Probe trials were recorded by a video cassette recorder that was fixed on the ceiling where the entire surface area of the pool could be viewed. The time spent in each quadrant and the number of crossings of the platform were analyzed by double-blind test. Both male and female mice, aged 7–11 weeks, were used for the Morris water maze task. No significant gender difference was detected. The data were analyzed by use of one-way analysis of variance (ANOVA) and Newman–Keuls post hoc analysis.
Results
CHARACTERIZATION OF THE IP33-KINASE(A) NULL MUTATION
Mice heterozygous for the IP33-kinase(A) deletion showed no apparent abnormality. These heterozygous mice were intercrossed, and the mutant offspring were analyzed by Southern blotting (Fig. 1B). Of the 135 offspring analyzed, 37 (27%) were wild type, 63 (47%) were heterozygous, and 35 (26%) were homozygous for the mutation, indicating a normal development of IP33-kinase(A) mutant embryos. The mutation of the IP33-kinase(A) gene in the targeted mice was verified by Northern analysis and Western blotting. No RNA or protein for IP33-kinase(A) was detectable in the mutant brain (Fig. 1C,D). Immunohistochemical analysis also confirmed that the mutant homozygotes did not express any IP33-kinase(A) protein in the brain (Fig. 2A–D). The IP33-kinase(A)-deficient mice were fully viable and fertile, and showed no overt behavioral or motor abnormality. The gross or histological examination of the mutant brain did not reveal any abnormality (data not shown).
Figure 2.
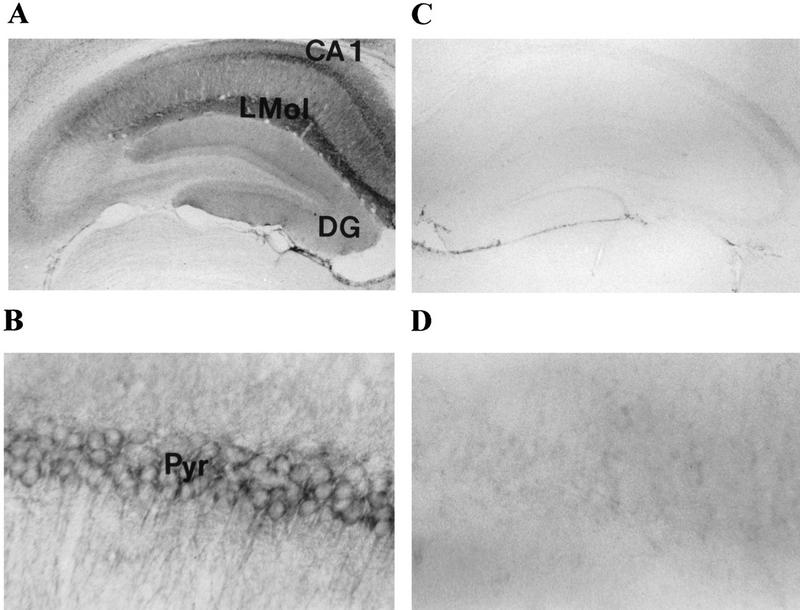
Distribution of IP33-kinase(A) in the hippocampus of wild-type and mutant mouse. (A) Coronal section of the hippocampus of a wild-type mouse stained with anti-rat-IP33-kinase(A) monoclonal antibodies. Magnification, 40×. (B) CA1 area of the hippocampus of a wild-type mouse. Magnification, 400×. (C) Coronal section of the hippocampus of a mutant mouse stained with anti-rat-IP33-kinase(A) monoclonal antibodies. Magnification, 40×. (D) CA1 area of the hippocampus of a mutant mouse. Magnification, 400×. (CA1) CA1 area of hippocampus; (DG) dentate gyrus of hippocampus; (Pyr) pyramidal cells; (LMol) Stratum lacunosum-moleculare.
BIOCHEMICAL ANALYSIS OF IP33-KINASE(A)-DEFICIENT MICE
The extent and selectivity of the IP33-kinase(A) deficiency in the IP33-kinase(A) mutant mice was examined biochemically. Crude extracts of the whole brain from the wild-type and the mutant mice were used as the enzyme source. Enzymatic assay using [3H]IP3 as a substrate showed a >80% decrease in IP33-kinase activity in the mutant mice (data not shown). As IP33-kinase can be easily renatured after SDS-PAGE, we have used this technique to identify the nature of the residual IP33-kinase activity in the IP33-kinase(A) mutant mice. Two peaks of the IP33-kinase activity were identified in the SDS–gel analysis of the wild-type brain: A small peak, IP33-kinase(B) migrating at 70–80 kD, and a large peak, IP33-kinase(A) migrating at 53 kD (Fig. 3A). In the mutant brain samples, the IP33-kinase(A) peak was totally absent, whereas the IP33-kinase(B) peak was intact, indicating that the residual enzyme activity in the mutant was attributable to the IP33-kinase(B) isotype, which was known to be expressed in astrocytes.
Figure 3.


(A) Characterization of the residual IP33-kinase activity after renaturation from SDS-PAGE. (a) IP33-kinase(A) activity; (b) IP33-kinase(B) activity. (Solid bars) +/+, wild-type; (open bars) −/−, mutant. (B) Comparison of the basal levels of IP4 in wild-type and mutant hippocampi (n = 6 mice).
The effect of the IP33-kinase(A) deficiency on the level of IP3 and IP4 was examined. We predicted that the level of IP3 in the mutant mice would be higher than that in the wild-type mice on the basis of a previous suggestion that IP33kinase(A) was involved in the regulation of the IP3 level in the brain (Communi et al. 1995). But the whole brain of the wild-type and the mutant mice contained the same quantity of IP3 [wild type (n = 7 independent measurements from three mice), 43.9 ± 4.8 pmoles/mg; mutant (n = 7 independent measurements from three mice), 45.8 ± 5.7 pmoles/mg; Student t-test, P =0.47] when determined by the radioreceptor assay as described in Materials and Methods. Next, the level of IP4 in the hippocampus was measured by HPLC analysis of brain homogenates as explained in Materials and Methods. As expected, the IP4 level was significantly reduced in the mutant brain compared to that of wild-type [wild type (n = 7 mice), 0.617 ± 0.159 nmole/gram wet weight; mutant (n = 7 mice), 0.187 ± 0.088 nmole/gram wet weight; Student t-test, P = 0.04] (Fig. 3B). As IP3 can be dephosphorylated by IP35-phosphatase to become IP2, we examined the IP35-phosphatase activity. The IP35-phosphatase activity was not up-regulated in the mutant mice (data not shown).
LTP
In an attempt to study the role of IP33-kinase(A) in synaptic plasticity, LTP was examined in the CA1 and the dentate gyrus (DG) area of the hippocampal slices from wild-type and mutant mice at 5–9 weeks of age. Because the immunostaining with anti-IP33-kinase(A) antibodies revealed that IP33-kinase(A) was highly localized in the CA1 pyramidal cell layer with a weak staining in the DG area, we thought it would be informative to compare the LTP in CA1 with that of the DG. We used seven mice (five males and two females) for each genotype, mutant and wild-type. When tetanic stimulation was delivered to the Schaffer collaterals in the presence of bicuculine, LTP was observed in the CA1 area in the slices from wild-type and mutant mice. The normalized fEPSPs slope for the wild type (n = 7 slices, seven mice) at 60 min after tetanus was 141 ± 11% and that of the mutant (n = 7 slices, seven mice) at 60 min after tetanus was 309 ± 35%, which was a twofold enhancement from that of the wild type (Student’s t-test, P = 0.0002; Fig. 4A). Contrary to the increased LTP in the CA1 of the mutant mice, however, no difference in the degree of LTP was observed in the DG area between wild-type and mutant mice (Fig. 4A). The increase of the fEPSPs slope at 60 min after tetanus was 169 ± 13% and 160 ± 25% in wild type (n = 7 slices, seven mice) and mutant (n = 7 slices, seven mice), respectively (Student’s t-test, P = 0.7783).
Figure 4.
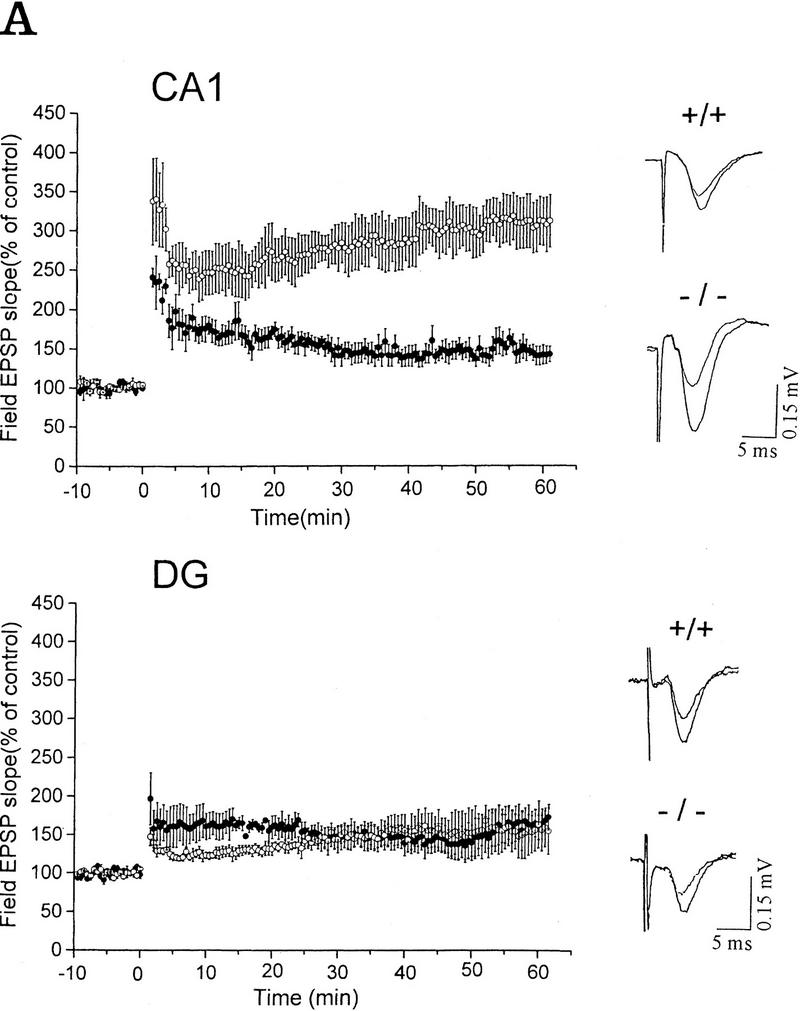

Synaptic responses of wild-type and mutant mice in the hippocampal CA1 area and the dentate gyrus. (A) LTP in wild-type and mutant mice. LTP was expressed as the percentage of the mean ± s.e.m. of the value before tetanic stimulation. (CA1) Hippocampal CA1 area; (DG) dentate gyrus. Representative recordings are shown at right. (B) Paired pulse facilitation in wild-type and mutant mice. The size of the peak field potential for the second pulse of a pair as a function of the interpulse interval (○) −/−; (•) +/+.
PAIRED PULSE FACILITATION
To test for any modification of the presynaptic function, we investigated the difference in the paired pulse facilitation (PPF) in the CA1 and the DG when two identical stimuli were delivered in three different interstimulus intervals (ISI) to the Schaffer collaterals and the perforant pathway, respectively. PPF is the presynaptic form of the short-term plasticity in which the synaptic response to the second paired stimulus in rapid succession is increased because of residual Ca2+ in the presynaptic terminal from the first stimulus. Therefore, the extent of facilitation is increased with shorter ISI. We could not observe any significant difference in the extent of the PPF over three ISIs (50, 100, and 200 msec) between the slices obtained from wild type (n = 5 slices, three male mice) and mutant (n = 5 slices, three male mice) in the CA1 and the DG region (CA1, 50-msec delay: Wild type, 177 ± 5.6%; mutant, 173 ± 9.3%; Student’s t-test, P = 0.9825; DG, 50-msec delay: wild type, 149 ± 6.1%; mutant, 151 ± 13.4%; Student t-test, P = 0.9024; Fig. 4B). These results suggest that IP33-kinase(A) may not be essential in the mobilization of Ca2+ and presynaptic neurotransmitter release.
CALCIUM IMAGING ANALYSIS
As the IP4 was suggested to be involved in calcium homeostasis in cells and the hippocampal CA1 LTP is significantly increased in IP33-kinase(A)-mutant mice, we analyzed the intracellular calcium change in hippocampal neuron cell cultures of wild-type and mutant mice. Unexpectedly, the relative calcium change, when treated with 50 μm glutamate followed by a 5-min perfusion with the calcium imaging buffer, showed no significant difference between wild-type and mutant mice (Fig. 5). Therefore, the increase of the hippocampal CA1 LTP in IP33-kinase(A)-mutant mice is not coupled to a global intracellular Ca2+ increase induced by glutamate.
Figure 5.

Glutamate-induced Ca2+ change in wild-type and mutant hippocampal neurons. Representative hippocampal neuronal cells from wild-type and mutant mice showing Ca2+ response. Glutamate (50 μm) was applied during the underlined time periods (30 sec) and washed off by perfusion with Ca2+ imaging buffer for 5 min. Each value represents the mean ± s.e.m.. (•) +/+, wild type (n = 27); (○)−/−, mutant (n = 29). (Glu) Glutamate.
WATER MAZE TEST
To test the spatial learning ability of the IP33-kinase(A)-mutant mice, we used the Morris water maze task as described in Materials and Methods. For the hidden-platform test, mice were trained by one block of three trials for 9 consecutive days. During the 9-day test, both wild type (n = 15 mice) and mutant (n = 17 mice) showed a significant reduction in the time taken to find the hidden platform, and the two groups did not show any difference in this test. No difference between wild-type and mutant mice was detected [F(1,30) = 0.03, P = 0.8618, one-way ANOVA; Fig. 6A]. In the probe test carried out 3 days after the last training block, both wild-type and mutant mice spent significantly more time in the quadrant where the platform had been located during training than in the other quadrants. Wild-type mice spent more time in the trained quadrant than in the other quadrants [F(3,56) = 185.61, P < 0.0001, one-way ANOVA; Newman-Keuls post hoc comparison: trained greater than all other quadrants, P < 0.01]. Similarly, the mutant mice spent more time in the trained quadrant than in the other quadrants [F(3,64) = 126.32, P < 0.0001, one-way ANOVA; Newman-Keuls post hoc comparison: trained greater than all other quadrants, P < 0.01; Fig. 6B]. In addition, both wild-type and mutant mice crossed the exact position where the platform had been located more frequently than any other corresponding position of other quadrants. Wild-type mice crossed the exact location of the platform in the trained quadrant more than the other alternative locations in other quadrants [F(3,56) = 103.49, P < 0.0001, one-way ANOVA; Newman-Keuls post hoc comparison: trained greater than all other quadrants, P < 0.01]. Similarly, mutant mice crossed the exact location of the platform in the trained quadrant more often than the other alternative locations in the other quadrants [F(3,64) = 114.64, P < 0.0001, one-way ANOVA; Newman-Keuls post hoc comparison: trained greater than all other quadrants, P < 0.01; Fig. 6C]. Taken together, these data indicate that the IP33-kinase(A)-mutant mice are not different from the wild-type mice in spatial learning and memory. As the hippocampal CA1 LTP in the mutant mice was much greater than in the wild-type mice, we also tested whether memory was maintained longer in the mutant mice. For this test, we have carried out the probe test 7 days after the last training block. Both wild type (n = 11 mice) and mutant (n = 8 mice) showed a slight decrease of memory retention, and no significant difference in memory retention was detected between wild-type and mutant mice (data not shown).
Figure 6.



Behavioral analysis of wild-type and mutant mice in the Morris water maze task. (A) Mean escape latency during the hidden platform test. Mice were trained in three trials a day for 9 consecutive days. (█) +/+; n = 15, (□) −/−, n = 17. (B) The mean search time spent in each quadrant during the probe test 3 days after the last training. (C) The mean platform crossings in each quadrant during the probe test 3 days after the last training. (B,C) (Solid bars) +/+, n = 15; (open bars) −/−, n = 17. (T) Trained quadrant; (AL) adjacent left quadrant; (AR) adjacent right quadrant; (OP) opposite quadrant.
Discussion
IP3 mediates various cellular functions by releasing Ca2+ from internal Ca2+ pools in response to various hormones or neurotransmitters in a variety of cell types (Berridge 1993). As IP3 induces various cellular responses, the amount of IP3 was assumed to be tightly regulated by IP33-kinase and 5-phosphatase (Woodring and Garrison 1997). While IP35-phosphatase provides the inositol moiety for recycling, IP33-kinase produces IP4, which is thought to be involved in controlling Ca2+ homeostasis (Communi et al. 1995). Therefore, IP33-kinase has a crucial role in inositol metabolism. However, many aspects of inositol phosphate metabolism are not yet determined. In our IP33-kinase(A)-null mutant study, biochemical analysis revealed that the generation of IP4 was severely impaired in the mutant brain, whereas the amount of IP3 was comparable to that of wild-type mice. These data indicate that the IP33-kinase is not essential for the regulation of the IP3 level but is important for generation of IP4.
Many IP4-binding proteins were purified and the binding motif was characterized, although the real IP4 receptor has not been yet determined (Fukuda et al. 1994; Cullen et al. 1995). There are many suggestions concerning the possible role(s) of IP4. The first proposed role of IP4 is calcium mobilization through binding to the IP3 receptor or to the yet-unidentified IP4 receptor (Wilcox et al. 1993; Irvine and Cullen 1993). Our Ca2+ imaging data suggest that the global calcium mobilization in response to glutamate was normal in the hippocampal neuronal cells of the IP33-kinase(A)-mutant mice. However, this result does not exclude the possibility that Ca2+ release from intracellular Ca2+ stores might be altered in the mutant when treated with metabotropic glutamate receptor specific agonists. The second possible role of IP4 is the regulation of neurotransmitter release. It has been reported that binding of IP4 to the C2B domain of synaptotagmin II blocks neurotransmitter release from the presynaptic terminal of giant axon of squids (Fukuda et al. 1995). Interestingly, our mutant mice, which have a reduced amount of IP4 compared to that of wild-type mice, showed an increased hippocampal CA1 LTP as discussed below. However, the PPF measurement in the hippocampus of the IP33-kinase(A)-mutant mice was normal, suggesting that Ca2+ mobilization and presynaptic neurotransmitter release were normal in mutant mice. This finding does not exclude the possibility that changes in presynaptic function in the mutant could be altered during higher frequencies or longer durations of presynaptic fiber stimulation. Finally, the third possible role of IP4 may be through the stimulation of one of the GAPs (Cullen et al. 1995). Recently, a targeted disruption of the guanine nucleotide exchange factor (Ras–GRF), which activates Ras signaling, caused a significant decrease in amygdala LTP and an impairment of amygdala-dependent learning and memory without affecting hippocampal CA1 LTP or spatial learning and memory (Brambilla et al. 1997). Therefore, the investigators suggested that the Ras signaling pathway was involved in the synaptic transmission and amygdala-dependent long-term memory. As the Ras signaling is inactivated by GAP (Lowy and Willumsen 1993), which is activated by IP4 (Cullen et al. 1995), it is plausible to speculate that the enhanced hippocampal CA1 LTP in the IP33-kinase(A) mutant mice might have been caused by the continuous presence of an active Ras signaling because GAP is not activated in the absence of IP4.
The hippocampus, which expresses LTP, is thought to be an important structure for contextual, spatial, and declarative learning and memory (Kim and Fanselow 1992; Eichenbaum 1997). However, the correlation of LTP and learning and memory is still controversial. Some mutant mice showed that an absence or decrease of hippocampal LTP impaired learning and memory. Studies with CaM kinase II-deficient mice revealed deficient hippocampal LTP and impaired spatial learning (Silva et al. 1992a,b). The NMDA receptor ε1-deficient mice also showed a decrease of hippocampal CA1 LTP and reduced spatial learning and memory (Sakimura et al. 1995). Recently, a conditional deletion of the NMDAR1 revealed a close relationship between CA1 LTP and spatial learning (Tsien et al. 1996). On the other hand, other mutant mice showed that an absence or decrease of hippocampal LTP was dissociated from learning and memory. Mice with the protein kinase C-γ null mutation showed an absence of hippocampal CA1 LTP but retained normal spatial learning and memory (Abeliovich et al. 1993a,b). Mice lacking tissue-type plasminogen activator (t-PA) also revealed that a reduction of L-LTP in the hippocampal CA1 and CA3 regions did not affect hippocampus-dependent learning and memory (Huang et al. 1996). Interestingly, targeted disruption of the AMPA receptor subunit GluR2 showed enhanced hippocampal CA1 LTP, which was caused by an increased Ca2+ permeability in the CA1 pyramidal neurons (Jia et al. 1996). Unfortunately, however, a behavioral test for learning and memory was not possible with these mutant mice because of high mortality and defective motor coordination. As the IP33-kinase(A)-deficient mice were healthy, showed no morphological abnormalities in the brain, and expressed a significantly enhanced LTP in the hippocampal CA1 region, these mutant mice provided an excellent opportunity to examine whether increased synaptic plasticity in the hippocampal CA1 affects spatial learning and memory. Perhaps contrary to our expectation, the IP33kinase(A)-deficient mice showed no enhancement or other abnormality of learning and memory in the Morris water maze task. The dissociation of increased LTP and spatial learning and memory in the IP33-kinase(A)-deficient mice may be explained as follows. First, the test paradigms that we used for learning and memory were not sensitive enough to quantify a minor change in learning and memory in the mutant. Second, hippocampal LTP is just an on/off switch system for learning and memory, and, therefore, the amplitude of LTP is not important for learning and memory (Bortolotto et al. 1994). Third, it is possible that low-frequency-stimulated LTP such as υ burst stimulation may be normal in mutant mice, and it could be the low-frequency-stimulated LTP that is more closely associated with spatial learning and memory. Finally, hippocampal LTP is actually dissociated from spatial learning and memory as in the case of PKC-γ- or t-PA-deficient mice (Abeliovich et al. 1993a,b; Huang et al. 1996).
The increase of intracellular Ca2+ concentration by both ionotropic and metabotropic receptors is an essential factor for LTP induction (Bliss and Collingridge 1993). In this study, the deficiency of IP33-kinase(A) showed a significantly increased LTP in the hippocampal CA1 region, but not in the DG. However, our Ca2+ imaging analysis suggested that the increase in the hippocampal CA1 LTP of the IP33-kinase(A)-mutant mice was not caused by a global increase of the intracellular Ca2+ in the neuronal cells induced by glutamate. The discrepancy here may be explained as follows. As the LTP measurements were carried out with slices derived from young mice (5–9 weeks), neuronal cell cultures derived from newborn mice (1 day old) might not represent the actual Ca2+ concentration change in the neurons of the young mice. In fact, the IP33-kinase(A) activity from the rat brain is very low until 1 week of age, increases by 13-fold between 2 and 3 weeks after birth, and reaches a plateau at 13 weeks (Moon et al. 1989). To answer this question, it will be necessary to examine the Ca2+ regulation in hippocampal slices of the young mutant mice.
In conclusion, IP33-kinase activity is the major pathway to generate IP4, and IP4 or IP4-derived inositol phosphates may be necessary for induction and maintenance of hippocampal CA1 LTP, and the high-frequency-induced enhancement of hippocampal CA1 LTP does not affect spatial learning and memory.
Acknowledgments
We thank Y. Namkung, D. Kim, and N.-G. Kang for ES cell culture and embryo manipulation, Y.-H. Kim, and Y.H. Park for help with behavioral test and statistics, and M.P. Kong for caring for the mice. This work was supported by a grant from the Creative Research Initiative Program of the Ministry of Science and Technology, Korea, and Korea Genome Project of the Ministry of Science and Technology, Korea, and in part by a genetic engineering grant from the Ministry of Education and a medical science grant from the Ministry of Health and Welfare of Korea.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
References
- Abeliovich A, Chen C, Goda Y, Silva AJ, Stevens CF, Tonegawa S. Modified hippocampal long-term potentiation in PKC γ-mutant mice. Cell. 1993a;75:1253–1262. doi: 10.1016/0092-8674(93)90613-u. [DOI] [PubMed] [Google Scholar]
- Abeliovich A, Paylor R, Chen C, Kim JJ, Wehner JM, Tonegawa S. PKC γ-mutant mice exhibit mild deficits in spatial and contextual learning. Cell. 1993b;75:1263–1271. doi: 10.1016/0092-8674(93)90614-v. [DOI] [PubMed] [Google Scholar]
- Aiba A, Chen C, Herrup K, Rosenmund C, Stevens CF, Tonegawa S. Reduced hippocampal long-term potentiation and context-specific deficit in associative learning in mGluR1 mutant mice. Cell. 1994a;79:365–375. doi: 10.1016/0092-8674(94)90204-6. [DOI] [PubMed] [Google Scholar]
- Artola A, Singer W. Long-term depression of excitatory synaptic transmission and its relationship to long-term potentiation. Trends Neurosci. 1993;16:480–487. doi: 10.1016/0166-2236(93)90081-v. [DOI] [PubMed] [Google Scholar]
- Bashir ZI, Bortolotto ZA, Davies CH, Berretta N, Irving AJ, Seal AJ, Henley JM, Jane DE, Watkins JC, Collingridge GL. Induction of LTP in the hippocampus needs synaptic activation of glutamate metabotropic receptors. Nature. 1993;363:347–350. doi: 10.1038/363347a0. [DOI] [PubMed] [Google Scholar]
- Batty IR, Nahorski SR, Irvine RF. Rapid formation of inositol 1,3,4,5-tetrakisphosphate following muscarinic receptor stimulation of rat cerebral cortical slices. Biochem J. 1985;232:211–215. doi: 10.1042/bj2320211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- Bliss TVP, Collingridge GL. A synaptic model of memory: Long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Bortolotto ZA, Bashir ZI, Davies CH, Collingridge GL. A molecular switch activated by metabotropic glutamate receptors regulates induction of long-term potentiation. Nature. 1994;368:740–743. doi: 10.1038/368740a0. [DOI] [PubMed] [Google Scholar]
- Brambilla R, Gnesutta N, Minichiello L, White G, Roylance AJ, Herron CE, Ramsey M, Wolfer DP, Cestari V, Rossi-Arnaud C, Grant SGN, Chapman PF, Lipp H-P, Sturani E, Klein R. A role for the Ras signalling pathway in synaptic transmission and long-term memory. Nature. 1997;390:281–285. doi: 10.1038/36849. [DOI] [PubMed] [Google Scholar]
- Challiss RAJ, Nahorski SR. Depolarization and agonist-stimulated changes in inositol 1,4,5-trisphosphate and 1,3,4,5-tetrakisphosphate mass accumulation in rat cerebral cortex. J Neurochem. 1991;57:1042–1051. doi: 10.1111/j.1471-4159.1991.tb08255.x. [DOI] [PubMed] [Google Scholar]
- Choi KY, Kim HK, Lee SY, Moon KH, Sim SS, Kim JW, Chung HK, Rhee SG. Molecular cloning and expression of a complementary DNA for inositol 1,4,5-trisphosphate 3-kinase. Science. 1990;248:64–66. doi: 10.1126/science.2157285. [DOI] [PubMed] [Google Scholar]
- Communi D, Vanweyenberg V, Erneux C. Molecular study and regulation of D-myo-inositol 1,4,5-trisphosphate 3-kinase. Cell Signal. 1995;7:643–650. doi: 10.1016/0898-6568(95)00035-n. [DOI] [PubMed] [Google Scholar]
- Cullen PJ, Hsuan JJ, Truong O, Letcher AJ, Jackson TR, Dawson AP, Irvine RF. Identification of a specific Ins(1,3,4,5)P4-binding protein as a member of the GAP1 family. Nature. 1995;376:527–530. doi: 10.1038/376527a0. [DOI] [PubMed] [Google Scholar]
- Eichenbaum H. Learning from LTP: A comment on recent attempts to identify cellular and molecular mechanisms of memory. Learn & Mem. 1996;3:61–73. doi: 10.1101/lm.3.2-3.61. [DOI] [PubMed] [Google Scholar]
- Eichenbaum H. Declarative memory: Insights from cognitive neurobiology. Annu Rev Psychol. 1997;48:547–572. doi: 10.1146/annurev.psych.48.1.547. [DOI] [PubMed] [Google Scholar]
- Fukuda M, Mikoshiba K. Structure-function relationships of the mouse Gap1m. J Biol Chem. 1996;271:18838–18842. doi: 10.1074/jbc.271.31.18838. [DOI] [PubMed] [Google Scholar]
- Fukuda M, Moreira JE, Lewis FMT, Sugimori M, Niinobe M, Mikoshiba K, Llinas R. Role of the C2B domain of synaptotagmin in vesicular release and recycling as determined by specific antibody injection into the squid giant synapse preterminal. Proc Natl Acad Sci. 1995;92:10708–10712. doi: 10.1073/pnas.92.23.10708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuichi T, Mikoshiba K. Inositol 1, 4, 5-trisphosphate receptor mediated Ca2+ signaling in the brain. J Neurochem. 1995;64:953–960. doi: 10.1046/j.1471-4159.1995.64030953.x. [DOI] [PubMed] [Google Scholar]
- Grant SG, O’Dell TJ, Karl KA, Stein PL, Soriano P, Kandel ER. Impaired long-term potentiation, spatial learning, and hippocampal development in fyn mutant mice. Science. 1992;258:1903–1910. doi: 10.1126/science.1361685. [DOI] [PubMed] [Google Scholar]
- Guse AH, Goldwich A, Weber K, Mayr GW. Non-radioactive, isomer-specific inositol phosphate mass determination: Micro metal-dye detection-H.P.L.C. strongly improves speed and sensitivity of analyses from cells, tissue specimens, and micro-enzyme assays. J Chromatogr. 1995;B672:189–198. doi: 10.1016/0378-4347(95)00219-9. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Momiyama A, Takahashi T, Ohishi H, Ogawa-Meguro R, Shigemoto R, Mizuno N, Nakanishi S. Role of a metabotropic glutamate receptor in synaptic modulation in the accessory olfactory bulb. Nature. 1993;366:687–690. doi: 10.1038/366687a0. [DOI] [PubMed] [Google Scholar]
- Huang YY, Bach ME, Lipp HP, Zhou M, Wolfer DP, Hawkins RD, Schoonjans L, Kandel ER, Godfraind JM, Mulligan R, Collen D, Carmeliet P. Mice lacking the gene encoding tissue-type plasminogen activator show a selective interference with late-phase long-term potentiation in both Schaffer collateral and mossy fiber pathways. Proc Natl Acad Sci. 1996;93:8699–8704. doi: 10.1073/pnas.93.16.8699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine RF. “Quantal” Ca2+ release and the control of Ca2+ entry by inositol phosphates—a possible mechanism. FEBS Lett. 1990;263:5–9. doi: 10.1016/0014-5793(90)80692-c. [DOI] [PubMed] [Google Scholar]
- ————— Inositol tetrakisphosphate as a second messenger: Confusions, contradictions, and a potential resolution. BioEssays. 1991;13:419–427. doi: 10.1002/bies.950130810. [DOI] [PubMed] [Google Scholar]
- Irvine RF, Cullen PJ. Will the real IP4 receptor please stand up? Curr Biol. 1993;3:540–543. doi: 10.1016/0960-9822(93)90052-p. [DOI] [PubMed] [Google Scholar]
- Jia Z, Agopyan N, Miu P, Xiong Z, Henderson J, Gerlai R, Taverna FA, Velumian A, MacDonald J, Carlen P, Abramow-Newerly W, Roder J. Enhanced LTP in mice deficient in the AMPA receptor GluR2. Neuron. 1996;17:945–956. doi: 10.1016/s0896-6273(00)80225-1. [DOI] [PubMed] [Google Scholar]
- Kim D, Jun KS, Lee SB, Kang N-G, Min DS, Kim Y-H, Ryu SH, Suh P-G, Shin H-S. Phospholipase C isozymes selectively couple to specific neurotransmitter receptors. Nature. 1997;389:290–293. doi: 10.1038/38508. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Fanselow MS. Modality-specific retrograde amnesia of fear. Science. 1992;256:675–677. doi: 10.1126/science.1585183. [DOI] [PubMed] [Google Scholar]
- Lee SY, Sim SS, Kim JW, Moon KH, Kim JH, Rhee SG. Purification and properties of D-myo-inositol 1,4,5-trisphosphate 3-kinase from rat brain. J Biol Chem. 1990;265:9434–9440. [PubMed] [Google Scholar]
- Linden DJ. Long-term synaptic depression in the mammalian brain. Neuron. 1994;12:457–472. doi: 10.1016/0896-6273(94)90205-4. [DOI] [PubMed] [Google Scholar]
- Lowy DR, Willumsen BM. Function and regulation of Ras. Annu Rev Biochem. 1993;62:851–891. doi: 10.1146/annurev.bi.62.070193.004223. [DOI] [PubMed] [Google Scholar]
- Madison DV, Malenka RC, Nicoll RA. Mechanisms underlying long term potentiation of synaptic transmission. Annu Rev Neurosci. 1991;14:379–397. doi: 10.1146/annurev.ne.14.030191.002115. [DOI] [PubMed] [Google Scholar]
- Mailleux P, Takazawa K, Erneux C, Vanderhaeghen J-J. Inositol 1,4,5-trisphosphate 3-kinase distribution in the rat brain. High levels in the hippocampal CA1 pyramidal and cerebellar Purkinje cells suggest its involvement in some memory processes. Brain Res. 1991a;539:203–210. doi: 10.1016/0006-8993(91)91622-8. [DOI] [PubMed] [Google Scholar]
- Mailleux P, Takazawa K, Erneux C, Vanderhaeghen J-J. Inositol 1,4,5-trisphosphate 3-kinase mRNA: High levels in the rat hippocampal CA1 pyramidal and dentate gyrus granule cells and in cerebellar Purkinje cells. J Neurochem. 1991b;56:345–347. doi: 10.1111/j.1471-4159.1991.tb02601.x. [DOI] [PubMed] [Google Scholar]
- Mailleux P, Takazawa K, Albala N, Erneux C, Vanderhaeghen J-J. Astrocytic localization of the messenger RNA encoding the isoenzyme B of inositol (1,4,5) 3-kinase in the human brain. Neurosci Lett. 1992;148:177–180. doi: 10.1016/0304-3940(92)90833-s. [DOI] [PubMed] [Google Scholar]
- Malenka RC. Synaptic plasticity in the hippocampus: LTP and LTD. Cell. 1994;78:535–538. doi: 10.1016/0092-8674(94)90517-7. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. NMDA-receptor-dependent synaptic plasticity: Multiple forms and mechanisms. Trends Neurosci. 1993;16:521–527. doi: 10.1016/0166-2236(93)90197-t. [DOI] [PubMed] [Google Scholar]
- Mayford M, Abel T, Kandel ER. Transgenic approaches to cognition. Curr Opin Neurobiol. 1995;5:141–148. doi: 10.1016/0959-4388(95)80019-0. [DOI] [PubMed] [Google Scholar]
- Mayr GW. A novel metal-dye detection system permits picomolar-range H.P.L.C. analysis of inositol polyphosphates from non-radioactively labelled cell or tissue specimens. Biochem J. 1988;254:585–591. doi: 10.1042/bj2540585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon KH, Lee SY, Rhee SG. Developmental changes in the activities of phospholipase C, 3-kinase, and 5-phosphatase in rat brain. Biochem Biophys Res Commun. 1989;164:370–374. doi: 10.1016/0006-291x(89)91728-2. [DOI] [PubMed] [Google Scholar]
- Nakanishi S. Molecular diversity of glutamate receptors and implications for brain function. Science. 1992;258:597–603. doi: 10.1126/science.1329206. [DOI] [PubMed] [Google Scholar]
- Regehr WG, Tank DW. Postsynaptic NMDA receptor-mediated calcium accumulation in hippocampal CA1 pyramidal cell dendrites. Nature. 1990;345:807–810. doi: 10.1038/345807a0. [DOI] [PubMed] [Google Scholar]
- Rhee SG, Suh PG, Ryu SH, Lee SY. Studies of inositol phospholipid-specific phospholipase C. Science. 1989;244:546–550. doi: 10.1126/science.2541501. [DOI] [PubMed] [Google Scholar]
- Sakimura K, Tatsuya T, Ito I, Manabe T, Takayama C, Kushiya E, Yagi T, Aizawa S, Inoue Y, Sugiyama H, Mishina M. Reduced hippocampal LTP and spatial learning in mice lacking NMDA receptor ε1 subunit. Nature. 1995;373:151–155. doi: 10.1038/373151a0. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Stevens CF, Tonegawa S, Wang Y. Deficient hippocampal long-term potentiation in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992a;257:201–206. doi: 10.1126/science.1378648. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Paylor R, Wehner JM, Tonegawa S. Impaired spatial learning in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992b;257:206–211. doi: 10.1126/science.1321493. [DOI] [PubMed] [Google Scholar]
- Tsien JZ, Huerta PT, Tonegawa S. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell. 1996;87:1327–1338. doi: 10.1016/s0092-8674(00)81827-9. [DOI] [PubMed] [Google Scholar]
- Tybulewicz VLJ, Crawford CE, Jackson PK, Bronson RT, Mulligan RC. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- Vanweyenberg V, Communi D, D’Santos CS, Erneux C. Tissue- and cell-specific expression of Ins(1,4,5)P3 3-kinase isoenzymes. Biochem J. 1995;306:429–435. doi: 10.1042/bj3060429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman GA, Hinds TR, Storm DR. Hormone stimulation of type III adenylate cyclase induces Ca2+ oscillations in HEK-293 cells. J Biol Chem. 1995;270:24108–24115. doi: 10.1074/jbc.270.41.24108. [DOI] [PubMed] [Google Scholar]
- Wilcox RA, Whitham EM, Liu C, Potter BVL, Nahorski SR. Myo-inositol 1,3,4,5-tetrakisphosphate can independently mobilise intracellular calcium, via the inositol 1,4,5-trisphosphate receptor: Studies with myo-inositol 1,4,5-trisphosphate-3-phosphorothioate and myo-inositol hexakisphosphate. FEBS Lett. 1993;336:267–271. doi: 10.1016/0014-5793(93)80817-e. [DOI] [PubMed] [Google Scholar]
- Woodring PJ, Garrison JC. Expression, purification, and regulation of two isoforms of the Inositol 1,4,5-trisphosphate 3-kinase. J Biol Chem. 1997;272:30447–30454. doi: 10.1074/jbc.272.48.30447. [DOI] [PubMed] [Google Scholar]
- Yamada M, Kakita A, Mizuguchi M, Rhee SG, Kim SU, Ikuta F. Ultrastructural localization of inositol 1,4,5-trisphosphate 3-kinase in rat cerebellar cortex. Brain Res. 1992;578:41–48. doi: 10.1016/0006-8993(92)90227-z. [DOI] [PubMed] [Google Scholar]