

Abstract
BACKGROUND
The possible risk of iatrogenic transmissible spongiform encephalopathies (TSEs, prion diseases) from transplantation of bone marrow-derived mesenchymal stem cells (MSCs) is uncertain. While most cell lines resist infection, a few propagate TSE agents.
STUDY DESIGN AND METHODS
We generated MSC-like (MSC-L) cell cultures from bone marrow (BM) of mice inoculated with the human-derived Fukuoka-1 (Fu) strain of TSE agent. Cultured cells were characterized for various markers and cellular prion protein (PrPC) by FACS and for PrPC and its pathologic TSE-associated form (PrPTSE) by Western blotting (WB). Cell cultures were tested for their susceptibility to infection with Fu in vitro. Infectivity of one Fu-infected cell culture was assayed in mice.
RESULTS
BM cells from Fu-infected mice expressed neither PrPC nor PrPTSE after three days in culture as demonstrated by WB. Cells adherent to plastic and maintained under two different culture conditions became spontaneously immortalized and began to express PrPC at about the same time. One culture became transformed shortly after exposure to Fu in vitro and remained persistently infected, continuously generating PrPTSE through multiple passages; infectivity of cultured cells was confirmed by intracerebral inoculation of lysates into mice. Both persistently TSE-infected and uninfected cells expressed a number of typical MSC markers.
CONCLUSION
BM-derived MSC-L cells of mice became persistently infected with the Fu agent under certain conditions in culture—conditions that differ substantially from those currently used to develop investigational human stem cell therapies.
Keywords: Bone marrow, mesenchymal stem cells, transmissible spongiform encephalopathy, prion, PrP, Fukuoka-1, mouse
INTRODUCTION
Transmissible spongiform encephalopathies (TSEs) are a group of fatal neurodegenerative disorders of humans and animals that have certain features of infectious diseases. Examples include CJD, Gerstmann-Sträussler-Scheinker (GSS) disease, fatal insomnia, and kuru. Sporadic CJD (sCJD), of unknown origin, is the most common human TSE, occurring worldwide with the usual estimated frequency of about one case per million people per year (~ 300 cases diagnosed each year in the USA). Variant CJD (vCJD) is a relatively new disease first described in the United Kingdom (UK) in 1996 and later in other countries. vCJD presumably resulted from food-borne transmission of bovine spongiform encephalopathy (BSE) infection to humans.1 The total number of vCJD cases reported worldwide has risen to 222 cases of which 174 cases were identified in the UK [www.eurocjd.ed.ac.uk/vcjdworldeuro.htm, accessed on November 1, 2010]. Sources of iatrogenic CJD include transplantation of contaminated cadaveric dura mater allografts and corneas, treatments with contaminated human cadaveric pituitary-derived hormones and reuse of contaminated surgical instruments.2 More recently, four iatrogenic vCJD infections transmitted by transfusions of non-leukoreduced RBCs from donors incubating vCJD3 were reported, and an additional transmission has been attributed to injections of a plasma-derived clotting factor4 in the UK. In spite of years of accumulating epidemiological evidence failing to confirm transmission by blood in the US,5 concern remains that sCJD might also be transmitted through blood transfusions or treatments with blood products or cellular therapies under certain circumstances and a few reports described transmitting CJD from human blood to animals 6,7. No reliable test has yet been developed to detect pre-clinical infection or for early clinical diagnosis of TSEs8,9, and no effective therapy is available.10
A prevailing opinion—not universally accepted11—is that TSEs are caused by prions: proteinaceous agents,12 composed of conformationally altered “scrapie-type” prion protein (PrPSc) that converts a normal host- encoded cellular prion protein (PrPC) into a misfolded pathogen under conditions not fully understood. The general term PrPTSE, proposed by a WHO consultation,13 will be used here for any abnormal misfolded PrP associated with a TSE. PrPC is a glycoprotein, normally attached to the plasma membrane through a phosphatidylinositol moiety.14 PrPC occurs in all mammals and is widely expressed in various tissues, including organs of the lymphoreticular system and blood, with highest levels found in brain.15,16 Its physiological function is unknown, but the expression of PrPC is not essential for normal development and survival of mice17 and cattle.18
For years, cell cultures have offered a potentially useful tool to study the physiological functions of PrPC and its role in pathogenesis of TSEs, including susceptibility of cells to TSE infection, molecular mechanisms of PrPTSE propagation, biological properties of TSE agent strains, and basic pathogenic mechanisms leading to neurodegeneration.19 TSE-infected cell cultures have also served as experimental models to screen various potential therapies19,20 and to develop sensitive bioassays for TSE infectivity.21
We recently developed unique spontaneously transformed cell cultures from the splenic (SP) stroma22 and bone marrow (BM) stroma23 of a SJL/OlaHsd (Ola) mouse; those cells expressed certain features of mesenchymal stem cells (MSC) or multipotent mesenchymal stromal cells (the designation currently recommended by International Society for Cellular Therapy),24 so we termed them MSC-like (MSC-L) cell cultures. The SP-MSC-L cultures efficiently propagated two TSE agents, the Fukuoka-1 agent (Fu) derived from brain tissue of a patient with GSS and serially propagated in mice,25 and a mouse-adapted brain-derived vCJD agent (mo-vCJD).22 The BM-derived MSC-L cultures also propagated Fu.23
MSCs are generally defined by three criteria: (1) adherence to plastic, (2) expression of specific surface markers, and (3) differentiation in vitro to mesodermal cells such as osteoblasts, chondrocytes and adipocytes.26 MSCs can be derived from different organs of which BM is the most commonly used for experimental therapy. Attempts to treat various diseases with injections of viable MSCs have increased dramatically during the past few years. MSCs have already been used with some success in regenerative medicine and tissue engineering.27,28 The risk of transmitting TSEs by transplantation of MSCs is unknown.
TSEs might be accidentally transmitted through MSC transplantations in at least two ways: (1) infected MSCs might be obtained from a donor during the pre-clinical stages of a TSE, and (2) MSCs might be exposed to a TSE agent in vitro during propagation by some contaminated human-derived or animal-derived component of the culture medium. Cultures of high-yield clinical-grade MSCs from BM-derived mononuclear cells usually require propagation in media containing fetal bovine serum (FBS),29 and cells are often cryopreserved in media containing FBS.30 FBS, while thought to be a low-risk material when carefully obtained (http://edocket.access.gpo.gov/2007/pdf/E6-22329.pdf) and never implicated in accidental transmission of a TSE, represents a potential source of contamination with BSE agent, because the blood used to prepare FBS is obtained in slaughter houses by exsanguinating fetuses removed from the carcasses of older culled cows, neural tissues of which are considered [BSE] “specified risk materials.”
To address both potential modes of transmission, we aimed in this study to develop MSC from BM of conventional Ola mice infected with Fu and examine freshly isolated cells for the presence of PrPC and PrPTSE and then to propagate them long-term as primary adherent cell cultures assayed at intervals for PrPC and PrPTSE. We also explored susceptibility of spontaneously immortalized BM-MSC-L cultures to infection with Fu agent in culture.
MATERIALS AND METHODS
Experimental animals
Ola mice used to isolate BM cells were from Harlan Laboratories (Bicester, UK). RIIIS/J (RIII) mice, used to prepare a working stock of Fu agent, were from the Jackson Laboratory (Bar Harbor, ME), and FVB/NCr (FVB) mice used for infectivity bioassays were from the National Cancer Institute (Frederick, MD). The Institutional Animal Care and Use Committee of the American Red Cross Holland Laboratory approved all experiments in mice.
TSE inocula
The Fu strain of TSE agent, propagated through multiple passages in Swiss-Webster mice at NIH (Fu/Swiss), was a generous gift from Dr. P. Brown. Fu was further propagated in our laboratory in RIII mice (Fu/RIII) to prepare a stock of bacteria-free 10% w/v brain homogenate in 0.9% NaCl that was stored at −80°C and used to inoculate experimental animals and cell cultures.
Generation of BM-derived cell cultures
BM cells were isolated from two six-month-old Ola mice, a strain known to develop spontaneous B cell lymphomas.31,32 Mice were inoculated intracerebrally (i.c.) with a 1% suspension of the Fu/RIII brain homogenate. At the time of euthanasia (141 days after inoculation) mice showed early clinical signs of TSE such as weight loss and lethargy; at necropsy they had a few enlarged peritoneal lymph nodes and spleens typical of early B cell lymphoma. Lymph nodes, spleens and brains were removed aseptically from mice for detection of PrPTSE by Western blotting (WB). We also isolated adherent cells from BM stroma as described elsewhere23 but with modification in cell culture media used for generation of cell cultures. The BM cells were isolated and maintained in growth medium (GM) comprised of Iscove’s Modified Dulbecco’s Medium (IMDM) supplemented with HEPES, L-Glutamine (Lonza, Walkersville, MD), 10% heat-inactivated FBS (Lonza), 50 µM 2-ME (Invitrogen, Carlsbad, CA) and 1% penicillin-streptomycin (Invitrogen) plus 5% conditioned medium containing mouse recombinant IL-3 produced by X63Ag8-653 myeloma cells.33 Next we removed cells from the original flasks and split them into six-well plates, where they were maintained under several different culture conditions. Two of these separately-maintained cell cultures were used in this study. The first cell culture (O1BM) was initially maintained in GM supplemented with 2.5% medium conditioned by CHO cells expressing kit soluble ligand (KLS, a generous gift from Dr. G. Keller, McEwen Centre for Regenerative Medicine, Toronto, Canada) — a medium designated here as GM-KLS. The second cell culture (O2BM) was maintained in GM containing 10% BIT 9500 (StemCell Technologies Inc., Vancouver, Canada) and 5 ng/mL sodium selenite (Sigma-Aldrich, St. Louis, MO) instead of KLS; that medium is designated here as GM-BIT/SS. On day nine after explanting, adherent and non-adherent cells in both cell cultures were separated, and adherent cell cultures were further propagated. Because O2BM cells maintained in GM-BIT/SS contained fewer dead cells and showed more proliferation, we transferred O1BM cells into that medium and used it for all subsequent cell propagations. Propagated cells were harvested periodically to test for PrP by WB. Immortalized cells were frozen in GM supplemented with 20% FBS and 10% DMSO (Sigma-Aldrich).
Infection of cell cultures with Fu and subsequent propagation
The O1BM and O2BM cells (2×105) were plated into 25-cm2 culture flasks and maintained for one day before exposure to Fu agent as described elsewhere34 with minor modifications. Ten-percent Fu/RIII brain homogenate was thawed on wet ice and diluted a further 100-fold (to a final 0.1% w/v homogenate) with Opti-MEM medium (Invitrogen) and centrifuged for 10 minutes at 3,000 rpm; clarified supernatant was used to inoculate cells. We typically applied 1.2 mL of 0.1% Fu/RIII brain supernatant, incubated cells for 4 hours at 37°C and 95% humidity in 5% CO2, and then added 5 mL of fresh GM-BIT/SS to each flask. We maintained cultures under the same conditions for 96 hr after inoculation. Cells were harvested to test for PrPTSE at 96 hours and then at every 5th passage through at least 15 passages after inoculation. The O1BM and O2BM cell cultures were named OF1BM and OF2BM, respectively, after being inoculated with Fu agent.
Differentiation of cells
Differentiation of cells was performed as described elsewhere35 with some modifications. The OF1BM cells and OF2BM cell cultures frozen at passage 5 after inoculation with Fu agent were revitalized and incubated overnight in complete expansion medium (CEM) consisting of IMDM (Invitrogen) supplemented with 9% FBS (Invitrogen), 9% horse serum (Invitrogen), 100 U/mL penicillin, 100 µg/mL streptomycin, and 12 µM L-glutamine (Invitrogen). The next day, cells were plated at a density of 50 cells per cm2 into 78.5-cm2 culture dishes and incubated for 7 days. CEM was changed twice during the incubation of cells. Next, to induce adipogenesis, the experimental cell cultures were incubated in CEM medium supplemented with 5 µg/mL insulin (Sigma), 50 µM indomethacin (Sigma), 1 µM dexamethasone, and 0.5µM 3-isobutyl-1-methylxanthine (Sigma). Control cells were maintained in CEM only. The medium was changed twice a week for 3 weeks. Then OF2BM and OF1BM incubated in differentiation medium and control cultures were fixed with 10% formalin for 20 minutes and stained with 0.5% Oil Red O (Sigma) solution prepared in methanol (Sigma) for 20 minutes.
To induce osteogenesis, cultures were incubated in CEM medium containing 20 mM β-glycerol phosphate (Sigma), 50 ng/ml thyroxine (Sigma), 1nM dexamethasone (Sigma), and 0.5 µM ascorbate 2-phosphate (Sigma). Control cells were maintained in CEM medium only. The medium was changed twice a week for 3 weeks. Cells were fixed with 70% chilled ethanol for one hour, washed twice with water and stained for calcium deposits with Alizarin Red S (Fisher Scientific, Pittsburgh, PA), pH 4.5 for 20 minutes.
Detection of surface antigens on OF1BMS and OF2BMS cells by FACS
We performed FACS analysis using antibodies listed in Table 1 on a FACSCanto™ flow cytometer with FACSDiva software (Becton Dickinson, San Jose, CA) as previously described.23
TABLE 1.
Immunophenotyping by FACS analysis of spontaneously immortalized cell cultures derived from bone marrow of Ola mice
| Cell-surface marker | Analyzed cell culture, % of positive cell (mean ± SD) | |
|---|---|---|
| OF2BM (untransformed, not generating PrPTSE) |
OF1BM (transformed, generating PrPTSE) |
|
| Ly-6A/E (Sca-1) | 99.0 ± 0.5 | 71.9 ± 4.0 |
| CD90 (Thy1.1) | 4.1 ± 1.2 | 6.1 ± 1.6 |
| CD34 | 7.4 ± 1.0 | 4.8 ± 0.9 |
| CD106 (VCAM-1) | 90.0 ± 1.9 | 3.8 ± 1.1 |
| CD31 (PECAM) | 1.9 ± 0.3 | 2.8 ± 1.7 |
| CD44 (H-CAM) | 100 | 100 |
| CD45R/B220 | 15.1 ± 3.5 | 8.2 ± 5.6 |
| 6D11 (PrP) | 97.9 ± 1.9 | 96.2 ± 2.5 |
Ly-6A/E (Sca-1: stem cell antigen-1), CD34 (expressed on a small subset of bone marrow cells in mouse), CD106 (VCAM-1: vascular cell adhesion molecule-1), CD31 (PECAM-1: platelet/endothelial cell adhesion molecule), CD45 (leukocyte common antigen), CD45R/B220 (B220 antigens on B lymphocytes), CD11b (Mac-1: β2-integrin family adhesion molecule), Ly-6G/Ly-6C (myeloid and monocyte linage markers: Gr-1) and 6D11 antibodies were FITC-conjugated; the CD90 (Thy1.1) and CD44 (H-CAM: homing-associated cell adhesion molecule) antibodies were phycoerythrin-conjugated. All antibodies were from BD Pharmingen except CD34 (AbD Serotec) and anti-PrP specific monoclonal 6D11 (SIGNET). Analysis was performed by FACSCanto™ flow cytometer using FACSDiva software (BD Biosciences). Note: cell cultures were negative for CD45, CD11b and Gr-1 markers.
Sample preparation for PrP detection in cell cultures
Cells were grown in 25-cm2 flasks to confluent density and lysed on wet ice with 1 mL of lysis buffer (100 mM NaCl, 10 mM EDTA, 0.5% Triton X-100, 0.5% sodium deoxycholate, 10 mM Tris, pH 7.5) for 1 hour with constant shaking. Cells were harvested and insoluble material removed by centrifugation at 10,000 g for 5 minutes. Two 400-µL aliquots of supernatant were incubated with 5 µg/mL proteinase K (PK) (Invitrogen) at 37°C for 30 minutes to reveal the presence of core PrPTSE protein. One 150-µL aliquot was left PK-untreated. Next, proteins were precipitated with methanol overnight at −80°C. Precipitates were centrifuged at 10,000 g for 30 min and pellets dissolved in Laemmli Sample Buffer (Bio-Rad, Hercules, CA) containing 5% 2-ME (Sigma-Aldrich) and then heated at 95°C for 10 minutes before loading into electrophoresis gels.
Sample preparation for PrPTSE detection in brain
Samples from brains of infected mice were prepared as previously described with minor modifications.36 Briefly, pieces of frozen brain were thawed on wet ice, and a 10% w/v suspension was prepared in chilled lysis buffer (described above). Samples were clarified by centrifugation at 3,000 g for 30 minutes. Supernatants were treated with 100 µg/mL PK following by incubation at 37°C for 1 hour and then centrifuged at 50,000 g for 45 minutes at 4°C. The resulting pellets were dissolved as described for cell cultures to a final concentration of 1 mg of the original tissue per mL and then heated at 95°C for 10 minutes before loading into electrophoresis gel.
PrP detection by Western blotting
Proteins extracted either from cell cultures or from brain tissue were separated on 4 –12% Bis-Tris gradient gels (Invitrogen) and transferred to nitrocellulose membranes. PrP was detected using mouse monoclonal antibody 6D11 (SIGNET, Dedham, MA) as previously described.37
Infectivity bioassays of Fu/Swiss brain homogenate and OF1BMS cell culture
FVB mice were used to test infectivity of Fu/Swiss brain homogenate and OF1BM cell culture. A frozen stock of 10% homogenate from brains of sick Fu/Swiss mice was thawed on wet ice, and samples were prepared by 10-fold serial dilution in 0.9% NaCl. Confluent OF1BM monolayers were harvested at passage 30 after exposure to the Fu agent. Cells were counted, diluted to 4×108 cells per mL, and stored in liquid nitrogen before testing. On the day of infectivity assay, cells were thawed on wet ice and homogenized in 0.5 mL of sterile 0.9% NaCl by successively forcing the suspensions through syringes with needles of decreasing gauge from 18 to 27. The original suspensions were serially diluted in 0.9% NaCl to obtain the following cell equivalents: 8×106, 1.6×106, 3.2×105, and 6×104 cells per mL in the first experiment and 3.2×105, 6×104, 1.2 ×104, 2.5 ×103, 5×102, 1×102, 20 and 4 cells per mL in the second experiment.
Mice were injected i.c. with 30 µL of appropriate samples while deeply anesthetized by Isofluorane (Abbott Laboratories, Abbott Park, IL) inhalation. Animal health status was monitored at least once a day. Mice showing clinical signs of TSE, such as lethargy, poor grooming, incoordination and incontinence, were euthanized. Immunohistochemistry and WB confirmed the final TSE status of each mouse. Infectivity titers of the brain inoculum and cell lysates were calculated by the method of Reed and Muench.38
Neuropathology
Clinically ill and control mice were euthanized by cardiac bleeding while under deep Isoflurane anesthesia. Brains were removed using separate disposable instruments for each mouse, and a small sample consisting of parts of the cerebrum and cerebellum was frozen for molecular studies. The remaining sample was immersion fixed in 10% formalin for 72 hours followed by decontamination in 96% formic acid and then dehydration through graded alcohols and embedding in paraffin. Microtome sections (5 µm thick) were hematoxylin-eosin stained. Adjacent sections were immunostained to detect PrPTSE using monoclonal antibody 6H4 following previously described protocols.39 The same observer, blinded to the experimental design, interpreted all stained sections.
RESULTS
Detection of PrP in primary BM-derived cell cultures after propagation in vitro
We attempted to generate long-term cell cultures from BM of Ola mice that spontaneously develop B cell lymphomas31,32 and, following i.c. inoculation with Fu agent, accumulate PrPTSE in spleen and tumor-affected lymph nodes.37 No PrP of any kind was detected by WB in cells explanted from BM of two Fu-infected mice on day three in culture (data not shown). However, extracts from brains of the same mice used to explant the BM cells were positive by WB for total PrP and PrPTSE (Fig. 1A). Expression of PrPC, but not PrPTSE, was first observed in both adherent BM-derived cell cultures O1BM and O2BM (maintained in GM-KLS and GM-BIT/SS, respectively) on day 21 after explanting, remaining at high levels thereafter (Fig. 1B). After propagation for approximately six weeks, cells in both cultures escaped from proliferative crisis and showed stable continuing growth, indicating spontaneous immortalization. In confluent cultures, a few cells contained cytoplasmic vacuoles suggestive of lipid-containing vacuoles typically seen in adipocytes.
Fig. 1. Immunoblots for PrP.

(A). Total PrP (lanes 1 and 3) and PrPTSE (lanes 2 and 4) in samples of 0.1 % brain homogenates (10 µL loaded into the gel) of two SJL/Ola mice infected with Fukuoka-1 agent: lanes 1 and 3 and lanes 2 and 4 – samples untreated and treated with 100 µg/mL proteinase K (PK), respectively. The same two mice were used as sources of bone marrow (BM) described below. (B). BM-derived stromal cell cultures O1BM and O2BM (day 57 following isolation) maintained in two different growth media: GM-KLS or GM-BIT/SS media (see “Materials and Methods”). The presence of PrPC is evidenced by strong signals in PK-untreated samples (−) (lanes 1 and 3), and its absence in PK-treated samples (+) (lanes 2 and 4). The membranes were developed using the 6D11 antibody to PrP. A molecular mass standard in kilodaltons is shown on the left.
Exposure of BM-derived cell cultures to Fu
Twenty-two weeks after explantation, we exposed immortalized O1BM and O2BM cell cultures, generated under slightly different conditions, to 0.1 % Fu/RIII brain homogenate. (The cell cultures exposed to Fu we subsequently designated OF1BM and OF2BM). We successfully infected the OF1BM cell culture but not the OF2BM cell culture. OF1BM cells became spontaneously transformed during the second passage after in vitro exposure to the Fu agent confirmed by a soft-agar assay (data not shown)]. Similar transformation of cells after exposure in vitro to TSE agents has been previously described by others.40,41,42 We observed an approximately six-fold increase in cell proliferation rate of OF1BM cells with loss of contact inhibition of cell proliferation. OF1BM cells also underwent a striking change in morphology and a noticeable decrease in spreading on plastic surfaces compared with non-transformed OF2BM cells which remained well spread with fibroblast-like polygonal shapes and many well-developed filopodia (Fig. 2A).
Fig. 2. Characterization of BM stromal cell cultures.

(A). Morphology. Untransformed OF2BM cells with fibroblast-like polygonal shapes, some visible stress fibers, and many well-developed filopodia (arrows). Transformed OF1BM cells showed morphological changes including a marked decrease in cell spreading, disappearance of stress fibers, and a smaller number of filopodia. Scale bar, 200 mm. (B). Immunoblot for PrP. PrPC (lane 1) but not PrPTSE (lane 2) is present in OF2BM cell lysate but both forms of PrP are present in OF1BM cell lysate (lane 3 and 4, respectively). Proteinase K (PK)-untreated samples (−) (lanes 1 and 3) and samples treated with 10 µg/mL PK (+) (lanes 2 and 4) Three glycoforms of PrP are displayed as bands corresponding to di-, mono-, and non-glycosylated isoforms. Note that the PK-treated samples of cell lysates were five-fold more concentrated than PK-untreated samples. The membrane was developed using 6D11 antibody. Molecular mass standard in kilodaltons is shown on the left.
We detected PrPTSE, presumably originating from residual brain inoculum, in samples from the first few passages after exposure of cell cultures to Fu agent (Fig. 3, lane 1) derived from the brain (Fig.3, lane 9); PrPTSE then decreased to levels undetectable by WB. Newly-formed PrPTSE appeared in the OF1BM cell culture at passage 10 and was repeatedly detected for 85 passages (Fig. 2B, lane 4 and Fig. 3, lanes 3, 5, 7)—the highest passage level examined. However, we found no PrPTSE in samples from any passage during propagation of OF2BM through 15 passages (Fig. 2B, lane 2). One subpassaged OF1BM culture stopped generating PrPTSE after 65 passages. Remarkably, the loss of PrPTSE in the OF1BM cell subculture was accompanied by a subtle change in cell morphology to a less transformed phenotype with increased cell spreading, although the cell proliferation rate remained unchanged; that subpassaged cell culture resisted a further attempt to re-infect with Fu (data not shown).
Fig. 3. Immunoblots for PrP from OF1BM cell cultures infected with Fukuoka-1 (Fu) agent.

Cell lysates were prepared from cells collected at 96 hr (lanes 1 and 2) after in vitro exposure to Fu agent and from several subsequent serial passages (lanes 3 – 8). Presence of total PrP and PrPTSE is evidenced in samples untreated (−) or treated (+) with 10 µg/mL Proteinase K (PK), respectively. Note that the PK-treated samples (+) were five-fold more concentrated than samples not treated with PK (−). Control (lane 9) – sample of 0.1 % brain homogenate (10 µL loaded into the gel) treated with 100 µg/mL of PK. Three glycoforms of PrP are displayed as bands corresponding to di-, mono-, and non-glycosylated isoforms. The membranes were developed using 6D11 antibody. A molecular mass standard in kilodaltons is shown on the left.
Differentiation of BM-derived cells into mesodermal cells
In order to elucidate whether, in addition to their adherence to plastic, BM-derived cells exhibited essential defining feature of MSC—the ability to differentiate in vitro to mesodermal cells26—we cultured OF1BM and OF2BM cells in media that stimulate adipogenesis and osteogenesis and then assayed them for evidence of differentiation. Both cell cultures differentiated into adipocytes and mineralized cells (Fig.4). The OF2BM cells readily differentiated into adipocytes within 10 days (Fig. 4, panel B), while in OF1BM cell culture only some cells differentiated into adipocytes after 21 days (Fig. 4, panel F). Similarly, OF2BM cells differentiated into mineralized cells with high efficiency, producing large clusters of newly growing cells (Fig. 4, panel D). On the other hand, the OF1BM cells formed much smaller clusters of mineralized cells (Fig. 4, panel H). The control OF1BM and OF2BM cells maintained in medium without differentiation stimulation did not differentiate, as shown by the absence of staining with appropriate stain (Fig.4, panels A, E, C, G).
Fig. 4. Differentiation of OF1BM and OF2BM cells in cultures.
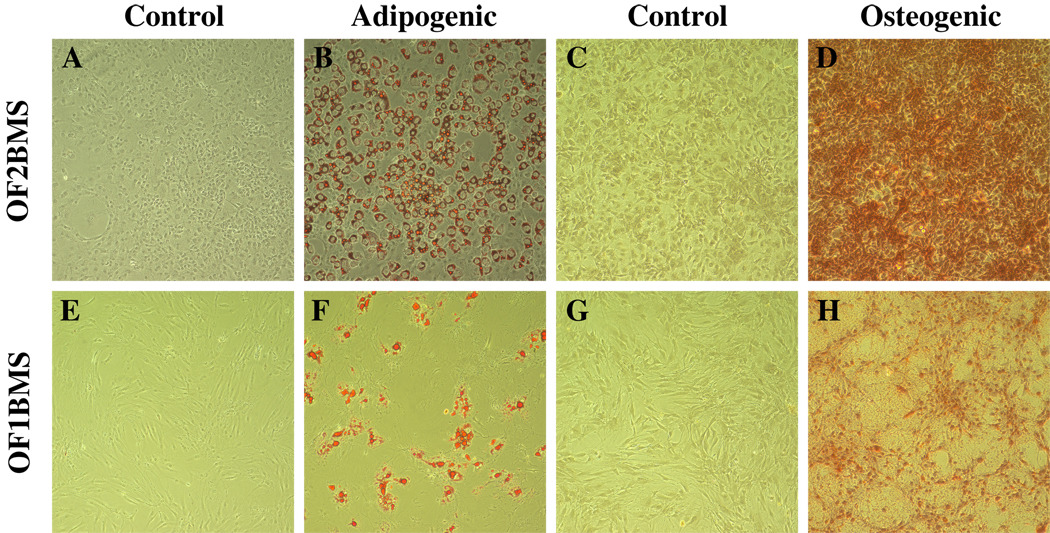
OF1BM and OF2BM cell cultures frozen at passages 5 were recovered for the differentiation study. Controls represent cells maintained in medium without differentiation factors (Panels A, E, C, G). Adipogenic and osteogenic cultures were maintained in media with appropriate differentiation factors for 21 days (see “Materials and Methods”) (Panels B and F and D and H, respectively). Panels A, B, E, F – cells stained with 0.5% Oil Red O; panels C, D, G, H – cells stained with Alizarin Red S. Cells were fixed with either 10% formalin (panels A, B, I,J) or 70% ethanol (panels C, D, K, L) before staining. Original magnification, ×10.
Immunophenotypic characterization of cell cultures
In order to demonstrate a third cardinal feature of stem cells—expression of a variety of typical markers on the cell surface—and to characterize the lineages of BM-derived stromal cell cultures OF1BM and OF2BM, we immunophenotyped the cells using a variety of antibodies and FACS analysis as summarized in Tab. 1. Many cells in both cultures displayed stem cell antigen-1 (Sca-1), although a somewhat smaller proportion of OF1BM cells were positive for Sca-1. Both cell cultures contained small subpopulations (less than 8 %) of cells positive for two other stem cell markers: Thy1.1 (thymocyte differentiation antigen-1) and CD34. All cells tested were positive for the homing-associated cell adhesion molecule (H-CAM). We also observed a striking difference in expression of the vascular cell adhesion molecule-1 (VCAM-1)—a typical marker of BM stromal cells—detected on over 90 % of untransformed OF2BM cells but on only a small percentage of transformed OF1BM cells. More than 80 % of cells in all cultures were positive for surface PrP. Cells of both types were either negative or contained only a very few positive cells for all other markers tested (Tab.1).
Infectivity bioassays of Fu/Swiss mouse brain homogenate and the Fu-infected OF1BM cells
Table 2 shows the mortality of FVB mice inoculated with various dilutions of OF1BM cells or with the Fu/Swiss mouse brain homogenates as well as incubation periods of disease. The infectivity titer of Fu/Swiss brain homogenate was 9.2 log10 LD50 units/g of brain; the mouse i.c. LD50 for OF1BM was approximately 103.6 cells. The infectivity titer of 106 cells was 5.4 log10 LD50 units. There were no differences in signs of disease between groups of mice inoculated with Fu/Swiss brain homogenate and Fu-infected cells. Signs of disease were typical for TSEs in mice: slowness, kyphosis, weight loss, reduced responsiveness to sound and touch, weakness of hind limbs, poor grooming and incontinence (manifest as urine-stained fur).
TABLE 2.
Mortality and incubation periods of FVB mice inoculated with Fukuoka-1 infected brain or with OF1BM cell culture lysates
| Type of inoculum | Dilution | Mortality (positive/total) |
Incubation period days (mean ± SD) |
|---|---|---|---|
| Brain homogenate Fu/Swiss* | 10−3 | 2/2 | 110 ± 0 |
| 10−4 | 4/4 | 123 ± 8.0 | |
| 10−5 | 4/4 | 124.5 ± 8.0 | |
| 10−6 | 4/4 | 162 ± 0 | |
| 10−7 | 3/3 | 180.3 ± 23 | |
| 10−8 | 0/5 | 414 ± 0 | |
| 10−9 | 0/4 | 414 ± 0 | |
| OF1BMS cell lysate † | 18.0×106 | 4/4 | 113 ± 0 |
| 11.6×106 | 3/3 | 122.3 ± 8.1 | |
| 1,23.2×105 | 4/41; 4/42 | 127.8 ± 7.51; 154.8 ± 42.32 | |
| 1,26.0×104 | 4/41; 4/52 | 142.8 ± 3.51; 152.8 ± 4.02 | |
| 21.2×104 | 5/5 | 162.2 ± 34.1 | |
| 22.4×103 | 0/5 | NA‡ | |
| 25.0×102 | 0/5 | NA‡ | |
| 21.0×102 | 0/5 | NA‡ |
Fu/Swiss brain homogenate from Swiss-Webster mice inoculated with Fukuoka-1 agent;
Cells were diluted in 1 mL of 0.9%NaCl before inoculation.
Two experiments on inoculating of OF1BM cells were performed: 1 - First experiment; 2 -Second experiment.
Surviving animals were euthanized at end of experiment, more than 500 days after inoculation.
TSE was confirmed by histopathological examination and immunohistochemical demonstration of PrP in selected brains and by the finding of PrPTSE in brains of all sick mice by WB. Representative PrPTSE profiles of WBs from brain tissue extracts and from an OF1BM cell lysate are shown in Fig. 5. The glycosylation patterns (relative abundance of di-glycosylated, mono-glycosylated and non-glycosylated forms of PrP and their electrophoretic mobility) of PrPTSE extracted from brains of mice injected with Fu-infected brain suspension were identical to those of PrPTSE from brains of mice injected with OF1BM cells (Figure 5, lanes 1 and 3). However, the glycosylation profile of PrPTSE extracted from OF1BM cells differed from that of PrPTSE from mouse brain extracts in the electrophoretic mobility of the three glycoforms (non-glycosylated cell-derived PrP slightly faster); cell-derived PrP also yielded a more heterogeneous di-glycosylated band than PrP of mouse brain origin (Fig.5, lane 2).
Fig. 5. Immunoblot comparing PrPTSE extracted from brain tissue of mice and from OF1BM cell lysate.

Lane 1 – extract from the pooled brains of Swiss-Webster mice inoculated with Fukuoka-1 (Fu) agent; lane 2 – extract from lysate of an OF1BM cell culture persistently infected with Fu agent; lane 3 – extract from the brain of an FVB mouse inoculated with OF1BM cell lysate. All samples were treated with 100 µg/mL Proteinase K (PK). Three glycoforms of PrP are displayed as bands corresponding to di-, mono-, and non-glycosylated isoforms. The membranes were developed using 6D11 antibody. Molecular mass standard in kilodaltons is on the left.
Neuropathologic changes in selected groups of FVB mice inoculated with OF1BM cells were similar to those observed in mice inoculated with Fu/Swiss mouse brain homogenates in that similar areas of the brain were affected, including cerebral cortex, hippocampus and thalamus, and the patterns and intensity of spongiform degeneration were similar (Fig. 6). In contrast to the uniformity in distribution and intensity of spongiform degeneration, some variation was evident in PrP deposits in the cerebral cortex, septum, hippocampus, thalamus, hypothalamus and brain stem of the mice; however, PrP deposits were consistently found in thalamus of mice inoculated with both materials. Brains of animals inoculated with Fu/Swiss mouse brain homogenates showed extensive accumulations of fine punctuate, coarse and plaque-like deposits of PrP (Fig. 6, panels F and I), while those of mice inoculated with OF1BMS cells contained only fine punctate deposits of PrP (Fig.6, panels E and H); that difference was not related either to infectious dose of inoculum or to the incubation period of TSE in the mouse.
Fig. 6. Comparative histological features in brain tissues of FVB mice inoculated with OF1BM cell lysate or with brain homogenate infected with Fu agent.
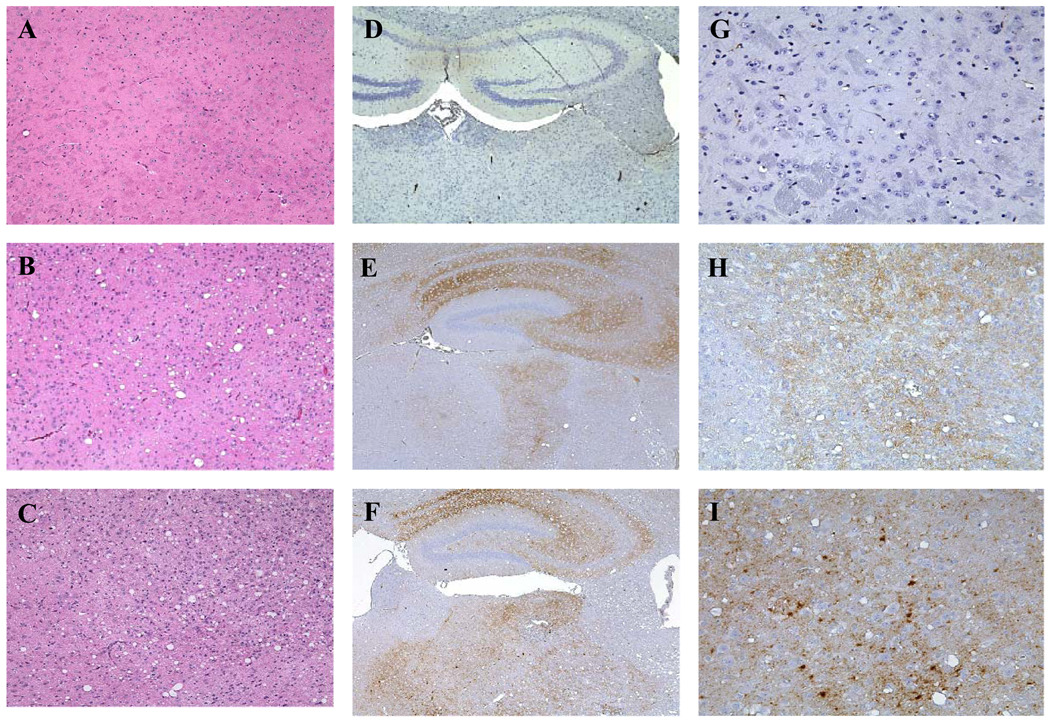
Panels A, D, G — representative sections of the cerebrum of a healthy mouse inoculated 570 days earlier with cells not exposed to Fukuoka-1 (Fu) agent: note absence of spongiform degeneration (A) and lack of PrP accumulation by immunostaining (D, G). Panels B, E, H — representative sections of the cerebrum of a sick mouse inoculated 114 days earlier with 4 × 108 Fu-infected OF1BM cells, showing severe spongiform degeneration (B) and fine-punctate PrP-positive deposits (E, H). Panels C, F, I — representative sections of the cerebrum of a sick mouse 162 days after inoculation with a mouse brain homogenate (10−6 dilution) containing Fu agent, showing severe spongiform degeneration (C) and fine punctate and coarse PrP-positive deposits (F, I). Sections in panels A, B, C were stained with hematoxylin and eosin (original magnification, ×10). Sections in panels D, G, E, H, F, I were immunostained with anti-PrP antibody 6H4; D, E and F (original magnification, ×4). Sections in panels G, H, and I (original magnification, ×20).
DISCUSSION
The results of this study and our previous studies23 revealed that murine BM-derived stromal cell cultures with certain features of MSCs26 such as adherence to plastic, ability to differentiate into adipocytes and osteoblasts and expression of certain stem-cell-specific surface markers, can be persistently infected by exposure in vitro to Fu, a mouse-adapted TSE agent of human origin. We must emphasize that the conditions under which we were able to infect these murine cell cultures differed from those generally used to isolate and expand human stem cells for experimental therapies. During the course of this study and similar studies previously performed in the same laboratory, we never detected any intrinsic PrPTSE by WB in freshly isolated BM cells from mice infected with either the Fu agent, mo-vCJD agent, and mouse-adapted BSE (mo-BSE) agent or with primary human-derived vCJD or sCJD agents (unpublished data), when tested both on the third day of propagation in vitro and after many serial passages. However, we successfully detected PrPTSE in most cell cultures derived from spleens of the same mice on the third day after explantation (unpublished data) but not after further propagation. Our results differ from those in a recently published report describing PrPTSE in BM-derived MSCs both from terminally sick mice infected with either mo-BSE agent or Fu agent and from rats infected with the Nu-1 isolate, a TSE agent strain derived from a GSS patient, propagated in mice and then adapted to Wistar rats.43
We were not able to test the infectivity of BM-derived cells from Fu-infected mice at early stages following initial explantation and propagation, because the numbers of cells were insufficient for bioassays in mice. According to a recent report, one research group successfully demonstrated infectivity in MSCs derived from terminally sick mice infected with mo-BSE agent.43 We know of only one published attempt—unsuccessful—to demonstrate infectivity in BM of a human with TSE.44 However, recently Takakura et al. reported detecting PrPTSE in BM cells of sCJD patients, even at early stages of cell propagation,43 although infectivity of the cells was apparently not tested. BM from cattle naturally infected with BSE has failed to transmit disease to mice.45 In addition, infectivity has not been demonstrated in either blood or—with one exception—bone marrow of experimentally BSE-infected cattle, though efforts to do that were limited;46,47,48 infectivity was detected in one pooled sternal bone marrow sample from a cow experimentally infected with BSE, however the authors suspected that the sternum might have been contaminated with material from other tissues during necropsy.49 A classic experimental pathogenesis study in mice infected with mouse-adapted scrapie agent by the subcutaneous route found that whole femurs contained small amounts of infectivity both at onset of overt illness and during the course of the disease.50 Infectivity was found in BM from one of nine sheep with clinical scrapie,51 though not in BM of goats with either natural scrapie52 or experimental scrapie.53 In recent studies, PrPTSE was not detected, using immunohistochemistry, in BM of sheep with natural scrapie.54 Based on that modest scientific evidence, bone marrow of sheep naturally infected with scrapie has been considered to be potentially infectious.13 Taken together, these findings provide only limited evidence of TSE infectivity in BM.
Iatrogenic transmission of TSE in humans through transplantation of BM, while theoretically possible, has not been reported. However, infectivity has been repeatedly demonstrated in blood of animals with natural TSE agents and experimentally infected with various TSE agents (guinea pigs experimentally infected with CJD,55 mice infected with mouse-adapted GSS56,57,58 and mo-vCJD,36 sheep naturally infected and experimentally infected with scrapie and BSE,59,60,61 mice infected with mo-BSE62 and hamsters infected with rodent-adapted scrapie63,64). Reports from the UK convincingly attribute cases of vCJD to infected RBCs and plasma.3,4,65 BM must therefore be considered as a potential source of TSE infectivity originating from blood, especially from blood of humans infected with BSE (vCJD) agent, if not from infected BM cells themselves.
After we failed to develop PrPTSE-generating adherent primary cell cultures from BM-derived cells of Fu-infected mice that had PrPTSE in spleens, tumor-affected lymph nodes and brains, we continued to propagate some of those cell cultures until several became spontaneously immortalized. At the time of immortalization, PrPC, required for propagation of TSE agents66—absent earlier—was first detected in whole-cell lysates by WB and confirmed by FACS analysis to be on cell surfaces. Further characterization by FACS revealed that the immortalized cells had an immunophenotype similar to that of murine BM-derived stromal cells with features of MSCs23,35,67 and to the murine spleen-derived stromal cell culture that we previously derived under similar conditions.22 Specifically, a high proportion of BM-derived cells expressed the hematopoietic marker Sca-1, and all cells were positive for H-CAM. Others have found a significant variation in populations of Sca-1-positive cells in murine MSCs depending on strain of mice.35 In contrast to human MSCs, the Thy1.1-positive cells were not reported previously in murine BM-MSC,35 but we detected very small subpopulations of cells positive for that marker in our cultures both in this and in an earlier study.23 Spleen-derived MSC-L cultures were also positive for Thy1.1.22 The presence of a significant subpopulation of VCAM-1-positive cells in untransformed uninfected OF2BM cultures compared with transformed Fu-infected OF1BM cells deserves further investigation. We observed a similar difference in VCAM-1 expression in our previous study of BM cell cultures derived from mice not exposed to Fu agent.23 We also found that all BM-derived cultures expressed nestin (data not shown), a marker of neuronal progenitors found in immature MSCs.68,69
Each TSE agent strain generally transmits to experimental animals a disease with characteristic features. Both clinical signs—length of incubation period, certain physical findings—and histopathological changes—involvement of various brain areas, degree of spongiform change, amounts and distribution of PrPTSE (sometimes forming plaques)—are usually reproduced faithfully once an agent has adapted to a new host. Some features of TSE in the animals to which disease was first transmitted experimentally can differ substantially from those in the naturally infected host. One factor influencing change in biological properties of a TSE agent appears to be the magnitude of difference in genotypes between the prion-protein-encoding genes of the donor and the recipient animals.70 We compared the biological properties of Fu agent propagated in OF1BM cell culturse to those of a Fu/Swiss mouse-brain-derived strain by assaying both in FVB mice. We saw no obvious clinical or histopathological differences between mice inoculated with Fu/Swiss brain homogenate or with OF1BM cell lysates. The biochemical profiles of PrPTSE extracted from the brains were identical in both groups of mice. Our findings were very similar in that regard to those reported by others, 71 who compared neuropathological profiles and PrPTSE biochemical patterns in brains of mice inoculated either with Fu brain homogenates or with Fu-infected GT1–7 cells. We observed only subtle differences in accumulation of PrP in the brains of mice inoculated with Fu-infected mouse brain homogenates and OF1BMS cells. Those differences were not related to either infectious dose of inoculum, length of incubation period, or the ages of animals at death.
In conclusion, we failed to produce Fu-infected cell cultures of BM taken from clinically sick mice infected with Fu agent. However, some cells derived from BM of those mice, cells having three typical features of MSCs (adherence to plastic, differentiation to mesodermal cells in stimulating media and expression of several surface markers typical of MSCs) became persistently infected after exposure to the Fu agent under specific conditions in vitro. That finding demonstrates that the resistance of MSC cells to experimental infection with a TSE agent observed in vivo was not maintained after propagation of the cells in culture.
The conditions under which we infected the mouse MSCs in vitro differ substantially from procedures currently used to develop investigational human stem cell therapies. Nonetheless, because most protocols for in vitro culture of human MSCs use media containing FBS, a complex bovine-derived material obtained from fetuses of gravid slaughter cows, bone-marrow-derived cultures must be considered potentially infectable with a human-derived TSE agent in vitro and hence to carry some theoretical risk—albeit remote—of exposing humans to the BSE agent. Even though murine BM-MSCs derived from terminally ill Fu-infected mice contained no detectable PrPTSE, the possible iatrogenic transmission of sCJD or vCJD by MSCs infected in culture raises a concern. Previous studies suggested that PrPC can be a marker for long-lived BM HSCs of mice and may play a role in supporting their self-renewal;72 furthermore, BM HSCs change phenotype during expansion in culture.73 We continue to wonder if certain changes during prolonged cultivation in vitro might sometimes render BM MSCs more susceptible to TSE infections than are native BM cells. In an era of increasing research with human stem cells and in anticipation of their eventual use to treat patients with a variety of ailments, including neurodegenerative diseases, further studies might be of value to elucidate the mechanisms causing cultured BM-derived cells to become susceptible to TSE infection. To minimize potential exposure of cells used in the manufacture of biologics to BSE agent, careful sourcing of all culture reagents remains important.13,74
ACKNOWLEDGEMENT
Research in this report was supported in part by the National Institute of Allergy and Infectious Diseases (NIAID), NIH, under NIH Agreement No.Y1-AI-4893-02 (Assessing Safety of Cell Substrates and Vaccine Components) with the FDA (FDA Agreement No. 224-05-1307).
Footnotes
Disclaimer: The findings and conclusions in this article have not been formally disseminated by the United States Food and Drug Administration and should not be construed to represent any Agency determination or policy.
Conflict of interest: The authors declare that they have no conflicts of interest relevant to the manuscript submitted to TRANSFUSION.
REFERENCES
- 1.Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A, McCardle L, Chree A, Hope J, Birkett C, Cousens S, Fraser H, Bostock CJ. Transmissions to mice indicate that 'new variant' CJD is caused by the BSE agent. Nature. 1997;389:498–501. doi: 10.1038/39057. [DOI] [PubMed] [Google Scholar]
- 2.Brown P, Brandel JP, Preece M, Sato M. Iatrogenic Creutzfeldt-Jakob disease: the waning of an era. Neurology. 2006;67:389–393. doi: 10.1212/01.wnl.0000231528.65069.3f. Epub (2006 Jul 19). Erratum in: Neurology 2006; 67:1528. Preese, Michael [corrected to Preece, Michael] [DOI] [PubMed] [Google Scholar]
- 3.Hewitt PE, Llewelyn CA, Mackenzie J, Will RG. Creutzfeldt-Jakob disease and blood transfusion: results of the UK Transfusion Medicine Epidemiological Review study. Vox Sang. 2006;91:221–230. doi: 10.1111/j.1423-0410.2006.00833.x. [DOI] [PubMed] [Google Scholar]
- 4.Peden A, McCardle L, Head MW, Love S, Ward HJ, Cousens SN, Keeling DM, Millar CM, Hill FG, Ironside JW. Variant CJD infection in the spleen of a neurologically asymptomatic UK adult patient with haemophilia. Haemophilia. 2010;16:296–304. doi: 10.1111/j.1365-2516.2009.02181.x. Epub 2010 Jan 12. [DOI] [PubMed] [Google Scholar]
- 5.Dorsey K, Zou S, Schonberger KB, Sullivan M, Kessler D, Notari E, 4th, Fang CT, Dodd RY. Lack of evidence of transfusion transmission of Creutzfeldt-Jakob disease in a US surveillance study. Transfusion. 2009;49:977–984. doi: 10.1111/j.1537-2995.2008.02056.x. Epub 2009 Jan 5. [DOI] [PubMed] [Google Scholar]
- 6.Manuelidis EE, Kim JH, Mericangas JR, Manuelidis L. Transmission to animals of Creutzfeldt-Jakob disease from human blood. Lancet. 1985;2:896–897. doi: 10.1016/s0140-6736(85)90165-5. [DOI] [PubMed] [Google Scholar]
- 7.Tamai Y, Kojima H, Kitajima R, Taguchi F, Ohtani Y, Kawaguchi T, Miura S, Sato M, Ishihara Y. Demonstration of the transmissible agent in tissue from a pregnant woman with Creutzfeldt-Jakob disease. New Engl J Med. 1992;327:649. doi: 10.1056/NEJM199208273270918. [DOI] [PubMed] [Google Scholar]
- 8.Brown P, Cervenakova L, Diringer H. Blood infectivity and the prospects for a diagnostic screening test in Creutzfeldt-Jakob disease. J Lab Clin Med. 2001;137:5–15. doi: 10.1067/mlc.2001.111951. [DOI] [PubMed] [Google Scholar]
- 9.Grassi J, Maillet S, Simon S, Morel N. Progress and limits of TSE diagnostic tools. Vet Res. 2008;39:3. doi: 10.1051/vetres:2008009. [DOI] [PubMed] [Google Scholar]
- 10.Stewart LA, Rydzewska LH, Keogh GF, Knight RS. Systematic review of therapeutic interventions in human prion disease. Neurology. 2008;70:1272–1281. doi: 10.1212/01.wnl.0000308955.25760.c2. Review. [DOI] [PubMed] [Google Scholar]
- 11.Manuelidis L, Liu Y, Mullins B. Strain-specific viral properties of variant Creutzfeldt-Jakob disease (vCJD) are encoded by the agent and not by host prion protein. J Cell Biochem. 2009;106:220–231. doi: 10.1002/jcb.21988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prusiner SB. Molecular biology of prion diseases. Science. 1991;252:1515–1522. doi: 10.1126/science.1675487. [DOI] [PubMed] [Google Scholar]
- 13.WHO Guidelines on Tissue Infectivity Distribution in Transmissible Spongiform Encephalopathies. WHO Press, World Health Organization; 2006:26. http://www.who.int/bloodproducts/cs/TSEPUBLISHEDREPORT.pdf.
- 14.Stahl N, Borchelt DR, Hsiao K, Prusiner SB. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell. 1987;51:229–240. doi: 10.1016/0092-8674(87)90150-4. [DOI] [PubMed] [Google Scholar]
- 15.Bendheim PE, Brown HR, Rudelli RD, Scala LJ, Goller NL, Wen GY, Kascsak RJ, Cashman NR, Bolton DC. Nearly ubiquitous tissue distribution of the scrapie agent precursor protein. Neurology. 1992;42:149–156. doi: 10.1212/wnl.42.1.149. [DOI] [PubMed] [Google Scholar]
- 16.Horiuchi M, Yamazaki N, Ikeda T, Ishiguro N, Shinagawa M. A cellular form of prion protein (PrPC) exists in many non-neuronal tissues of sheep. J Gen Virol. 1995;76:2583–2587. doi: 10.1099/0022-1317-76-10-2583. [DOI] [PubMed] [Google Scholar]
- 17.Weissmann C, Flechsig E. PrP knock-out and PrP transgenic mice in prion research. Br Med Bull. 2003;66:43–60. doi: 10.1093/bmb/66.1.43. Review. [DOI] [PubMed] [Google Scholar]
- 18.Richt JA, Kasinathan P, Hamir AN, Castilla J, Sathiyaseelan T, Vargas F, Sathiyaseelan J, Wu H, Matsushita H, Koster J, Kato S, Ishida I, Soto C, Robl JM, Kuroiwa Y. Production of cattle lacking prion protein. Nat Biotechnol. 2007;25:132–138. doi: 10.1038/nbt1271. Epub 2006 Dec 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vilette D. Cell models of prion infection. Vet Res. 2008;39:10. doi: 10.1051/vetres:2007049. [DOI] [PubMed] [Google Scholar]
- 20.Kocisko DA, Caughey B. Searching for anti-prion compounds: cell-based high-throughput in vitro assays and animal testing strategies. Methods Enzymol. 2006;412:223–234. doi: 10.1016/S0076-6879(06)12014-5. [DOI] [PubMed] [Google Scholar]
- 21.Mahal SP, Demczyk CA, Smith EW, Jr, Klohn PC, Weissmann C. Assaying prions in cell culture: the standard scrapie cell assay (SSCA) and the scrapie cell assay in end point format (SCEPA) Methods Mol Biol. 2008;459:49–68. doi: 10.1007/978-1-59745-234-2_4. [DOI] [PubMed] [Google Scholar]
- 22.Akimov S, Yakovleva O, Vasilyeva I, McKenzie C, Cervenakova L. Persistent propagation of variant Creutzfeldt-Jakob disease agent in murine spleen stromal cell culture with features of mesenchymal stem cells. J Virol. 2008;82:10959–10962. doi: 10.1128/JVI.01085-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Akimov S, Vasilyeva I, Yakovleva O, McKenzie C, Cervenakova L. Murine bone marrow stromal cell culture with features of mesenchymal stem cells susceptible to mouse-adapted human TSE agent, Fukuoka-1. Folia Neuropathol. 2009;47:205–214. [PubMed] [Google Scholar]
- 24.Horwitz EM, Le Blanc K, Dominici M, Mueller I, Slaper-Cortenbach I, Marini FC, Deans RJ, Krause DS, Keating A International Society for Cellular Therapy. Clarification of the nomenclature for MSC: The International Society for Cellular Therapy position statement. Cytotherapy. 2005;7:393–395. doi: 10.1080/14653240500319234. [DOI] [PubMed] [Google Scholar]
- 25.Tateishi J, Ohta M, Koga M, Sato Y, Kuroiwa Y. Transmission of chronic spongiform encephalopathy with kuru plaques from humans to small rodents. Ann Neurol. 1979;5:581–584. doi: 10.1002/ana.410050616. [DOI] [PubMed] [Google Scholar]
- 26.Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A, Prockop DJ, Horwitz E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. 2006;8:315–317. doi: 10.1080/14653240600855905. [DOI] [PubMed] [Google Scholar]
- 27.Dazzi F, Horwood NJ. Potential of mesenchymal stem cell therapy. Curr Opin Oncol. 2007;19:650–655. doi: 10.1097/CCO.0b013e3282f0e116. [DOI] [PubMed] [Google Scholar]
- 28.Granero-Molto F, Weis JA, Longobardi L, Spagnoli A. Role of mesenchymal stem cells in regenerative medicine: application to bone and cartilage repair. Expert Opin Biol Ther. 2008;8:255–268. doi: 10.1517/14712598.8.3.255. [DOI] [PubMed] [Google Scholar]
- 29.Gastens MH, Goltry K, Prohaska W, Tschöpe D, Stratmann B, Lammers D, Kirana S, Götting C, Kleesiek K. Good manufacturing practice-compliant expansion of marrow-derived stem and progenitor cells for cell therapy. Cell Transplant. 2007;16:685–696. doi: 10.3727/000000007783465172. [DOI] [PubMed] [Google Scholar]
- 30.Dennis JE, Esterly K, Awadallah A, Parrish CR, Poynter GM, Goltry KL. Clinical-scale expansion of a mixed population of bone-marrow-derived stem and progenitor cells for potential use in bone-tissue regeneration. Stem Cells. 2007;25:2575–2582. doi: 10.1634/stemcells.2007-0204. [DOI] [PubMed] [Google Scholar]
- 31.Ponzio NM, Brown PH, Thorbecke GJ. Host-tumor interactions in the SJL lymphoma model. Intern Rev Immunol. 1986;1:273–301. doi: 10.3109/08830188609056610. [DOI] [PubMed] [Google Scholar]
- 32.Tang JC, Ho FC, Chan AC, Srivastava G. Clonality of lymphomas at multiple sites in SJL mice. Lab Invest. 1998;78:205–212. [PubMed] [Google Scholar]
- 33.Karasuyama H, Melchers F. Establishment of mouse cell lines which constitutively secrete large quantities of interleukin 2, 3, 4 or 5, using modified cDNA expression vectors. Eur J Immunol. 1988;18:97–104. doi: 10.1002/eji.1830180115. [DOI] [PubMed] [Google Scholar]
- 34.Vorberg I, Priola SA. Molecular basis of scrapie strain glycoform variation. J Biol Chem. 2002;277:36775–36781. doi: 10.1074/jbc.M206865200. [DOI] [PubMed] [Google Scholar]
- 35.Peister A, Mellad JA, Larson BL, Hall BM, Gibson LF, Prockop DJ. Adult stem cells from bone marrow (MSCs) isolated from different strains of inbred mice vary in surface epitopes, rates of proliferation, and differentiation potential. Blood. 2004;103:1662–1668. doi: 10.1182/blood-2003-09-3070. [DOI] [PubMed] [Google Scholar]
- 36.Cervenakova L, Yakovleva O, McKenzie C, Kolchinsky S, McShane L, Drohan WN, Brown P. Similar levels of infectivity in the blood of mice infected with human-derived vCJD and GSS strains of transmissible spongiform encephalopathy. Transfusion. 2003;43:1687–1694. doi: 10.1046/j.0041-1132.2003.00586.x. [DOI] [PubMed] [Google Scholar]
- 37.Cervenakova L, Yakovleva O, McKenzie C. Protease-resistant prion protein in lymphoreticular tumors of variant Creutzfeldt-Jakob disease mice. Emerg Infect Dis. 2006;12:511–513. doi: 10.3201/eid1203.051348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reed LJ, Muench H. A simple method of estimating fifty percent end points. Amer J Hyg. 1938;27:493–497. [Google Scholar]
- 39.Piccardo P, Manson JC, King D, Ghetti B, Barron RM. Accumulation of prion protein in the brain that is not associated with transmissible disease. Proc Natl Acad Sci USA. 2007;104:4712–4717. doi: 10.1073/pnas.0609241104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Markovits P, Dautheville C, Dormont D, Dianoux L, Latarjet R. In vitro propagation of the scrapie agent. I. Transformation of mouse glia and neuroblastoma cells after infection with the mouse-adapted scrapie strain c-506. Acta Neuropathol. 1983;60:75–80. doi: 10.1007/BF00685350. [DOI] [PubMed] [Google Scholar]
- 41.Oleszak EL, Manuelidis L, Manuelidis EE. In vitro transformation elicited by Creutzfeldt-Jakob-infected brain material. J Neuropathol Exp Neurol. 1986;45:489–502. doi: 10.1097/00005072-198609000-00001. [DOI] [PubMed] [Google Scholar]
- 42.Manuelidis EE, Fritch WW, Kim JH, Manuelidis L. Immortality of cell cultures derived from brains of mice and hamsters infected with Creutzfeldt-Jakob disease agent. Proc Natl Acad Sci U S A. 1987;84:871–875. doi: 10.1073/pnas.84.3.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takakura Y, Yamaguchi N, Nakagaki T, Satoh K, Kira J, Nishida N. Bone marrow stroma cells are susceptible to prion infection. Biochem Biophys Res Commun. 2008;377:957–961. doi: 10.1016/j.bbrc.2008.10.099. [DOI] [PubMed] [Google Scholar]
- 44.Brown P, Gibbs CJ, Jr, Rodgers-Johnson P, Asher DM, Sulima MP, Bacote A, Goldfarb LG, Gajdusek DC. Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol. 1994;35:513–529. doi: 10.1002/ana.410350504. [DOI] [PubMed] [Google Scholar]
- 45.Frazer H, Foster J. Transmission to mice, sheep and goats and bioassay of bovine tissues. In Transmissible Spongiform Encephalopathies. A consultation on BSE with the Scientific Veterinary Committee of the Comission of the European Communities held in Brussels, September 14–15, 1993. In: Bradley R, Marchant B, editors. Brussels: European Commision Agriculture; 1994. pp. 145–159. Document VI/4131/94-EN. [Google Scholar]
- 46.Bradley R. The research programme on transmissible spongiform encephalopathies in Britain with special reference to bovine spongiform encephalopathy. Dev Biol Stand. 1993;80:157–170. [PubMed] [Google Scholar]
- 47.Bradley R. BSE transmission studies with particular reference to blood. Dev Biol Stand. 1999;99:35–40. [PubMed] [Google Scholar]
- 48.Sohn HJ, Lee YH, Green RB, Spencer YI, Hawkins SA, Stack MJ, Konold T, Wells GA, Matthews D, Cho IS, Joo YS. Bone marrow infectivity in cattle exposed to the bovine spongiform encephalopathy agent. Vet Rec. 2009;164:272–273. doi: 10.1136/vr.164.9.272. [DOI] [PubMed] [Google Scholar]
- 49.Wells GA, Hawkins SA, Green RB, Spencer YI, Dexter I, Dawson M. Limited detection of sternal bone marrow infectivity in the clinical phase of experimental bovine spongiform encephalopathy (BSE) Vet Rec. 1999;144:292–294. doi: 10.1136/vr.144.11.292. [DOI] [PubMed] [Google Scholar]
- 50.Eklund CM, Kennedy RC, Hadlow WJ. Pathogenesis of scrapie virus infection in the mouse. J Infect Dis. 1967;117:15–22. doi: 10.1093/infdis/117.1.15. [DOI] [PubMed] [Google Scholar]
- 51.Hadlow WJ, Kennedy RC, Race RE. Natural infection of Suffolk sheep with scrapie virus. J Infect Dis. 1982;146:657–664. doi: 10.1093/infdis/146.5.657. [DOI] [PubMed] [Google Scholar]
- 52.Hadlow WJ, Kennedy RC, Race RE, Eklund CM. Virologic and neurohistologic findings in dairy goats affected with natural scrapie. Vet Pathol. 1980;17:187–199. doi: 10.1177/030098588001700207. [DOI] [PubMed] [Google Scholar]
- 53.Hadlow WJ, Eklund CM, Kennedy RC, Jackson TA, Whitford HW, Boyle CC. Course of experimental scrapie virus infection in the goat. J Infect Dis. 1974;129:559–567. doi: 10.1093/infdis/129.5.559. [DOI] [PubMed] [Google Scholar]
- 54.Caplazi P, O'Rourke K, Wolf C, Shaw D, Baszler TV. Biology of PrPsc accumulation in two natural scrapie-infected sheep flocks. J Vet Diagn Invest. 2004;16:489–496. doi: 10.1177/104063870401600601. [DOI] [PubMed] [Google Scholar]
- 55.Manuelidis EE, Gorgacz EJ, Manuelidis L. Viremia in experimental Creutzfeldt-Jakob disease. Science. 1978;200:1069–1071. doi: 10.1126/science.349691. [DOI] [PubMed] [Google Scholar]
- 56.Kuroda Y, Gibbs CJ, Jr, Amyx HL, Gajdusek DC. Creutzfeldt-Jakob disease in mice: persistent viremia and preferential replication of virus in low-density lymphocytes. Infect Immun. 1983;41:154–161. doi: 10.1128/iai.41.1.154-161.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brown P, Rohwer RG, Dunstan BC, MacAuley C, Gajdusek DC, Drohan WN. The distribution of infectivity in blood components and plasma derivatives in experimental models of transmissible spongiform encephalopathy. Transfusion. 1998;38:810–816. doi: 10.1046/j.1537-2995.1998.38998408999.x. [DOI] [PubMed] [Google Scholar]
- 58.Brown P, Cervenakova L, McShane LM, Barber P, Rubenstein R, Drohan WN. Further studies of blood infectivity in an experimental model of transmissible spongiform encephalopathy, with an explanation of why blood components do not transmit Creutzfeldt-Jakob disease in humans. Transfusion. 1999;39:1169–1178. doi: 10.1046/j.1537-2995.1999.39111169.x. [DOI] [PubMed] [Google Scholar]
- 59.Houston F, Foster JD, Chong A, Hunter N, Bostock CJ. Transmission of BSE by blood transfusion in sheep. Lancet. 2000;356:999–1000. doi: 10.1016/s0140-6736(00)02719-7. [DOI] [PubMed] [Google Scholar]
- 60.Hunter N, Foster J, Chong A, McCutcheon S, Parnham D, Eaton S, MacKenzie C, Houston F. Transmission of prion diseases by blood transfusion. J Gen Virol. 2002;83(Pt. 11):2897–2905. doi: 10.1099/0022-1317-83-11-2897. [DOI] [PubMed] [Google Scholar]
- 61.Houston F, McCutcheon S, Goldmann W, Chong A, Foster J, Sisó S, González L, Jeffrey M, Hunter N. Prion diseases are efficiently transmitted by blood transfusion in sheep. Blood. 2008;112:4739–4745. doi: 10.1182/blood-2008-04-152520. [DOI] [PubMed] [Google Scholar]
- 62.Taylor DM, Fernie K, Reichl HE, Somerville RA. Infectivity in the blood of mice with a BSE-derived agent. J Hosp Infect. 2000;46:78–79. doi: 10.1053/jhin.2000.0790. [DOI] [PubMed] [Google Scholar]
- 63.Gregori L, McCombie N, Palmer D, Birch P, Sowemimo-Coker SO, Giulivi A, Rohwer RG. Effectiveness of leucoreduction for removal of infectivity of transmissible spongiform encephalopathies from blood. Lancet. 2004;364:529–531. doi: 10.1016/S0140-6736(04)16812-8. [DOI] [PubMed] [Google Scholar]
- 64.Gregori L, Gurgel PV, Lathrop JT, Edwardson P, Lambert BC, Carbonell RG, Burton SJ, Hammond DJ, Rohwer RG. Reduction in infectivity of endogenous transmissible spongiform encephalopathies present in blood by adsorption to selective affinity resins. Lancet. 2006;368:2226–2230. doi: 10.1016/S0140-6736(06)69897-8. [DOI] [PubMed] [Google Scholar]
- 65.Gillies M, Chohan G, Llewelyn CA, Mackenzie J, Ward H, Hewitt PE, Will RG. A retrospective case note review of deceased recipients of vCJD-implicated blood transfusions. Vox Sang. 2009;97:211–218. doi: 10.1111/j.1423-0410.2009.01222.x. Epub 2009 Aug 6. [DOI] [PubMed] [Google Scholar]
- 66.Büeler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, Weissmann C. Mice devoid of PrP are resistant to scrapie. Cell. 1993;73:1339–1347. doi: 10.1016/0092-8674(93)90360-3. [DOI] [PubMed] [Google Scholar]
- 67.Charbord P, Oostendorp R, Pang W, Hérault O, Noel F, Tsuji T, Dzierzak E, Péault B. Comparative study of stromal cell lines derived from embryonic, fetal, and postnatal mouse blood-forming tissues. Exp Hematol. 2002;30:1202–1210. doi: 10.1016/s0301-472x(02)00895-0. [DOI] [PubMed] [Google Scholar]
- 68.Tropel P, Platet N, Platel JC, Noël D, Albrieux M, Benabid AL, Berger F. Functional neuronal differentiation of bone marrow-derived mesenchymal stem cells. Stem Cells. 2006;24:2868–2876. doi: 10.1634/stemcells.2005-0636. [DOI] [PubMed] [Google Scholar]
- 69.Vogel W, Grünebach F, Messam CA, Kanz L, Brugger W, Bühring HJ. Heterogeneity among human bone marrow-derived mesenchymal stem cells and neural progenitor cells. Haematologica. 2003;88:126–133. [PubMed] [Google Scholar]
- 70.Scott M, Peretz D, Ridley RM, Baker HF, DeArmond SJ, Prusiner SB. Prion Biology and Diseases. 2nd Ed. Cold Spring Harbor Laboratory Press; 2004. Transgenic investigations of species barrier and prion strains; pp. 435–482. [Google Scholar]
- 71.Arima K, Nishida N, Sakaguchi S, Shigematsu K, Atarashi R, Yamaguchi N, Yoshikawa D, Yoon J, Watanabe K, Kobayashi N, Mouillet-Richard S, Lehmann S, Katamine S. Biological and biochemical characteristics of prion strains conserved in persistently infected cell cultures. J Virol. 2005;79:7104–7112. doi: 10.1128/JVI.79.11.7104-7112.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang CC, Steele AD, Lindquist S, Lodish HF. Prion protein is expressed on long-term repopulating hematopoietic stem cells and is important for their self-renewal. Proc Natl Acad Sci USA. 2006;103:2184–2189. doi: 10.1073/pnas.0510577103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang CC, Lodish HF. Murine hematopoietic stem cells change their surface phenotype during ex vivo expansion. Blood. 2005;105:4314–4320. doi: 10.1182/blood-2004-11-4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.World Health Organization. WHO Guidelines on Transmissible Spongiform Encephalopathies in relation to Biological and Pharmaceutical Products. 2003 http://whqlibdoc.who.int/hq/2003/a85721.pdf.