

Abstract
Approximately ten million people suffer from Chagas disease worldwide, caused by Trypanosoma cruzi, with the disease burden predominately focused in Latin America. Sleeping sickness is another serious health problem, caused by Trypanosoma brucei, especially in sub-Saharan countries. Unfortunately, the drugs currently available to treat these diseases have toxic effects and are not effective against all disease phases or parasite strains. Therefore, there is a clear need for the development of novel drugs and drug targets to treat these diseases. We propose the trypanosome prereplication machinery component, Orc1/Cdc6, as a potential target for drug development. In trypanosomes, Orc1/Cdc6 is involved in nuclear DNA replication, and, despite its involvement in such a conserved process, Orc1/Cdc6 is distinct from mammalian Orc1 and Cdc6 proteins. Moreover, RNAi-mediated silencing of trypanosome Orc1/Cdc6 expression in T. brucei decreased cell survival, indicating that Orc1/Cdc6 is critical for trypanosome survival.
1. Introduction
Currently, approximately ten million people have Chagas disease worldwide, with the disease burden being centered in Latin America. In 2008, approximately ten thousand people died from this disease. Sleeping sickness is another serious health problem, particularly in sub-Saharan countries; in the first half of the twentieth century, this disease practically decimated entire communities in central Africa [1]. After many surveillance programs, 2009 marked the first time in 50 years that less than ten thousand new cases of sleeping sickness were reported in Africa (http://www.who.org/).
The causal agents of Chagas disease and sleeping sickness are, respectively, the protozoan parasites Trypanosoma cruzi and T. brucei of the Kinetoplastida order. Two subspecies of T. brucei, T. b. rhodesiense, and T. b. gambiense, are responsible for acute sleeping sickness in Eastern and Southern Africa and chronic sleeping sickness in Western and Central Africa, respectively. Chronic sleeping sickness accounts for approximately 95% of all reported cases.
These protozoa have life cycles that alternate between a mammalian host and an insect host. T. cruzi epimastigotes are a noninfective life cycle stage of the parasite that proliferate by binary fission in the guts of Triatoma infestans insects, which are more commonly known as kissing bugs. These epimastigotes then transform into the infective metacyclic form in the insect hindgut. Then, when the insect vector bites a mammalian host, they eliminate the metacyclic parasites in their feces. This allows the parasites to penetrate the wounded skin and enter into the mammalian host's circulatory system. Within the bloodstream, the metacyclic parasites transform into trypomastigotes, which then infect mammalian cells and transform into amastigotes. Amastigotes are spherically shaped and proliferate inside the infected cells until transforming into nonreplicative trypomastigotes. The life cycle is completed when an insect vector bites an infected mammalian host and takes up trypomastigotes within the blood that then transform into epimastigotes inside the insect gut.
In contrast to T. cruzi, the life cycle of T. brucei spp. is entirely extracellular. In this case, an infected tsetse fly (Glossina genus) bites a mammalian host, transferring metacyclic trypomastiogte forms into the circulatory system. Metacyclic trypomastiogtes transform into bloodstream trypomastigotes that then proliferate in the hemolymphatic system as trypomastigotes, which are slender in form; next, the parasites transform in a non-proliferative form that is stumpy in appearance. When a new tsetse fly bites an infected mammalian host, these non-proliferative, stumpy parasites are taken up by the fly and transform into proliferating procyclic forms in the fly midgut. Parasites then migrate to the salivary glands of the insect where they transform into epimastigotes that can proliferate by binary fission. Finally, the epimastigotes transform into infective metacyclic trypomastigotes, which are then injected into a new mammalian host during the tsetse fly's bite.
As shown above, Chagas disease is transmitted through the infected feces of triatomines, whereas sleeping sickness is transmitted through the infected saliva of tsetse flies. However, other transmission modes are shared by both species, and include transmission through blood transfusions, vertical or mother-to-child transmission, and accidental infections in the laboratory.
Diagnosis is based on the presentation of clinical symptoms and signs, direct parasitological testing of blood or cerebrospinal fluids (in the case of T. brucei infection), serological tests and/or by parasite DNA detection using polymerase chain reactions (PCR). To detect T. cruzi by PCR, samples are assayed for the presence of minicircle kinetoplast DNA and a 195-bp reiterated DNA sequence [2, 3]. One serological test that is used for sleeping sickness diagnosis is the card agglutination test for trypanosomiasis (CATT); it detects T. b. gambiense-specific antibodies [4]. For Chagas disease, T. cruzi-specific antibodies can be detected with assays using either crude or recombinant antigens [5, 6]. However, the use of serological tests to infer Chagas disease cure is controversial, as antibodies against parasite antigens can remain in circulation for long periods of time [7].
The absence or presence of mild symptoms is associated with the chronic phases of both diseases. However, the acute symptoms of Chagas disease can be diagnosed by a trained physician and include swelling, nausea, diarrhea, vomiting, liver or spleen enlargement, fever, headaches, and chest or abdominal pain. About 40% of patients develop chronic disease with heart or colon dilation after 10–20 years of infection. Similarly, sleeping sickness is also characterized by two distinct stages. In the acute phase or hemolymphatic stage, symptoms include headache, malaise, weight loss, fatigue, fever, and vomiting. In the second phase, also known as the neurological phase, parasites are present within the cerebral spinal fluid and brain and cause many neurological and physiatric symptoms including anxiety, disruption of the sleep-wake cycle, behavior changes, and motor features such as muscle tremors and walking difficulties [8]. If untreated, sleeping sickness is fatal. In the absence of treatment, it is estimated that T. b. gambiense infection is fatal within three years [9] and that T. b. rhodesiense infection is fatal within six to eight months [10, 11].
Drugs used to treat Chagas disease and sleeping sickness have undesired toxic side effects and are not effective against all parasite life cycle stages or parasite strains because of drug resistance. However, effective drugs exist to treat the acute phases of both diseases. Despite the presence of numerous side effects, benznidazole (BNZ) (N-benzyl-2-nitroimidazole-1-acetamide) and nifurtimox (NF) (4-([5-nitrofurfuryledene]amino)-3-methylthiomorpholine-1,1 dioxide) are effective against Chagas disease. Both were introduced into clinical use in the 1970's [12]. While NF increases the production of free radicals believed to cause trypanosome death [13, 14], BNZ disrupts protein synthesis and the respiratory chain of T. cruzi (for a review see [15]). However, these drugs are ineffective during the chronic phase of the disease. In the latter phases of Chagas disease, the course of treatment is dependent on the patient's symptoms; medicines or surgery are recommended for patients with heart complications, while diet changes and possibly surgery are suggested for patients with digestive complications.
To treat sleeping sickness, four medicines are currently commonly used. Pentamidine and suramin are considered first-stage drugs. Pentamidine has been used since 1940 and is the first-choice drug to treat the initial stages of T. b. gambiense infection; it is administered as an intramuscular injection. The exact antiprotozoal mechanisms of action of pentamidine are still unknown, perhaps because it acts against many targets including mitochondria and DNA (reviewed at [16, 17]). Suramin has six negative charges that allow it to interact electrostatically with many trypanosomal enzymes, including enzymes involved glycolysis and the pentose phosphate pathway [18]. As resistance against suramin has been observed in T. b. gambiense, suramin is currently only used for cases of T. b. rhodesiense infection (for a review see [16]).
More toxic that pentamidine and suramin, melarsoprol, eflornithine, and a combination of eflornithine and NF (for melarsoprol-refractory patients) are used to treat patients in the neurological phase. Eflornithine, alpha-difluoromethylornithine, is administered intravenously and infusion and irreversible inhibits polyamine biosynthesis acting at ornithine decarboxylase (EC 4.1.1.17). It is only effective against T. brucei gambiense infections [19]. In spite of its toxic side effects, melarsoprol is the most widely used drug during the second stage of sleeping sickness; further, it is the only drug available to treat T. b. rhodesiense infections. Melarsoprol is derived from arsenic, and it is believed that melarsen oxide is its active metabolite in vivo. Although it is unknown how melarsoprol kills parasites, parasite lysis following melarsoprol exposure was demonstrated [20].
Although there is a clear need for new drugs to treat trypanosome-induced diseases, few drugs and clinical trials have been initiated recently. The absence of standard protocols to evaluate drug efficacy and the absence of clinical parasitological markers contribute to the difficulty of launching new treatment initiatives. In the case of Chagas disease, novel drugs with less toxic effects and shorter administration times and drugs to treat chronic disease should be prioritized. Therefore, a standard protocol for drug screening against acute T. cruzi infections was proposed to evaluate drug efficacy both in in vitro and in vivo models compared to BNZ. Although it is very important to evaluate the effect of these compounds during chronic infection, it is not possible due to the difficulty in evaluating parasite clearance using current methods [21]. Most recently, studies have focused on the azolic compounds itraconazole and posaconazole that inhibit ergosterol synthesis. In mouse models, these drugs have been shown to cure both acute and chronic Chagas disease [22]. In addition, posaconazole has activity against T. cruzi both in vivo and in vivo.
Regarding sleeping sickness, new drugs should have lower toxic side effects, treat both T. b. gambiense and T. b. rhodesiense infections, and treat late-stage T. b. rhodesiense infection. Pafuramidine maleate (DB289) is a new drug in clinical trial for the treatment of sleeping sickness [23]. However, as it cannot cross the blood-brain barrier, it can only be used for early stage treatment. Despite this, it has an advantage over pentamidine because it can be orally administrated.
As illustrated above, the need for new drugs and novel drug targets to treat both Chagas disease and sleeping sickness is evident. These drugs should target fundamental pathways within these parasites. In spite of the conserved nature of essential pathways among eukaryotic organisms, we identified a trypanosome prereplication machinery component that is fundamental for replication and that is distinct from eukaryotic prereplication machinery; this component is necessary for origin selection and the establishment of the DNA replication fork [24]. These data indicate that this enzyme is a potential drug target for the treatment of both Chagas disease and sleeping sickness.
2. Eukaryotic Prereplication Machinery
DNA replication in eukaryotes begins with the assembly of the prereplication complex on regions along chromosomes known as replication origins [25, 26]. The prereplication complex assembly (depicted in Figure 1) begins when the origin recognition complex (ORC), comprised of six different subunits (Orc1–6) [25, 27], recognizes the replication origins and allows the recruitment of cell division cycle Cdc6 and Cdt1 proteins to the complex. Together, the ORC, Cdc6 and Cdt1 proteins recruit the mini-chromosome maintenance (MCM) complex, which is comprised of six subunits (Mcm2–7); the MCM complex harbors the helicase activity that is essential for DNA replication [28]. Once the prereplication machinery is bound to the replication origins, these origins are licensed, meaning that the replication process can initiate at these origins through the binding of regulatory factors and DNA replication fork components during the onset of S-phase.
Figure 1.
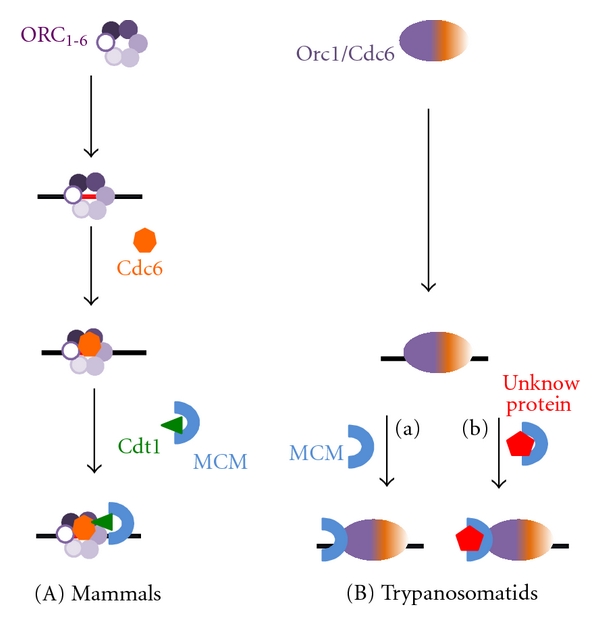
Schematic representation of the prereplication machinery from mammals (A) and trypanosomatids (B). (a) and (b) indicate the two possible mechanisms for MCM recruitment to the replication origin. (a) Orc1/Cdc6 recruits the MCM complex. (b) An unknown protein recruits the MCM complex to the replication origin.
3. Structural Aspects of the ORC and Cdc6 Prereplication Machinery Components
The ORC complex was first purified from S. cerevisiae extract as proteins that bound to replication origins [29]. S. cerevisiae ORC (413 KDa) is a heterohexamer that contains six proteins named according their relative molecular mass: Orc1 (120 KDa), Orc2 (72 KDa), Orc3 (62 KDa), Orc4 (56 KDa), Orc5 (53 KDa), and Orc6 (50 KDa). Orc1-5 orthologs were identified in a wide range of organisms, suggesting that these genes are present in all eukaryotic organisms. Of the six Orcs, Cdc6 is most notably similar to Orc1 (see Figure 2). Therefore, Orc1 is more closely related to Cdc6 than to any other ORC component [30].
Figure 2.
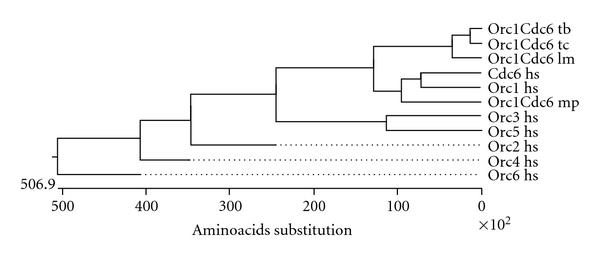
Phylogenetic tree of human Orc1 to Orc6, human Cdc6, and Orc1/Cdc6 from Trypanosoma cruzi (Orc1Cdc6 tc), Trypanosoma brucei (Orc1Cdc6 tb), Leishmania major (Orc1Cdc6 lm), and Methanoplanus petrolearius (orc1Cdc6 Mp), an Archaea species. The phylogenetic tree was generated using the ClustalW alignment method.
Among the ORC subunits, Orc1, Orc4, and Orc5 belong to the AAA+ family, proteins that exhibit ATPase activity and function in multiple cellular activities [31]. Orc2 and Orc3 contain an ATPase-like domain; Orc6 lacks the AAA+ domain and shows no structural similarity to the other Orc proteins. The AAA+ domain contains the Walker A and Walker B motifs and regions named sensor 1 and sensor 2. These regions are typical in proteins that act as “Clamp-loaders”, proteins that encircle DNA and bind other factors and serve as processivity-promoting factors in DNA replication in an ATP-dependent manner. Walker A and B motifs and sensor 1 and 2 regions are responsible for ATP binding and hydrolysis [31]; ATP binding and hydrolysis trigger a conformational change within the ORC that allows for the serial recruitment of proteins during prereplication complex assembly at replication origins [32].
Cdc6 (58 KDa in S. cerevisiae) was first isolated from thermo-sensitive mutants and identified as an important factor during the beginning of DNA replication [33]. Cdc6 also has the AAA+ domain, containing the Walker A and Walker B motifs and sensor 1 and 2 regions [30]. Similar to Orc1, Cdc6 exhibits intrinsic ATPase activity in vitro [34].
4. Recruitment of the Prereplication Machinery
The molecular bases that enable the recruitment of prereplication machinery have been described in yeast and they are dependent on ATP binding and ATP hydrolysis (Figure 3). As mentioned previously, the first step in prereplication machinery assembly is the binding of the ORC to the replication origins, a step that depends on the binding of the ORC to ATP. ORC-double-stranded DNA interactions are specific and inhibit the ATPase activity of the ORC. In this way, the high ATPase activity of the ORC inhibits stable ORC interactions with nonspecific DNA. However, in the presence of a specific replication origin, the ATPase activity of the ORC is inhibited, and it undergoes a conformational change; the conformation change stabilizes the ORC-DNA interaction and allows the ORC to bind Cdc6 (reviewed in [35]). The stability of ORC-Cdc6-DNA interactions is also regulated by the ATPase activity of Cdc6, which is high in the presence of nonspecific DNA, destabilizing the complex [34]. ORC-Cdc6-DNA interactions trigger a conformational alteration in Cdc6 that allows for the Cdc6-Cdt1 interaction to occur [36]. Therefore, the ATPase activity of both the ORC and Cdc6 work to select specific replication origins.
Figure 3.
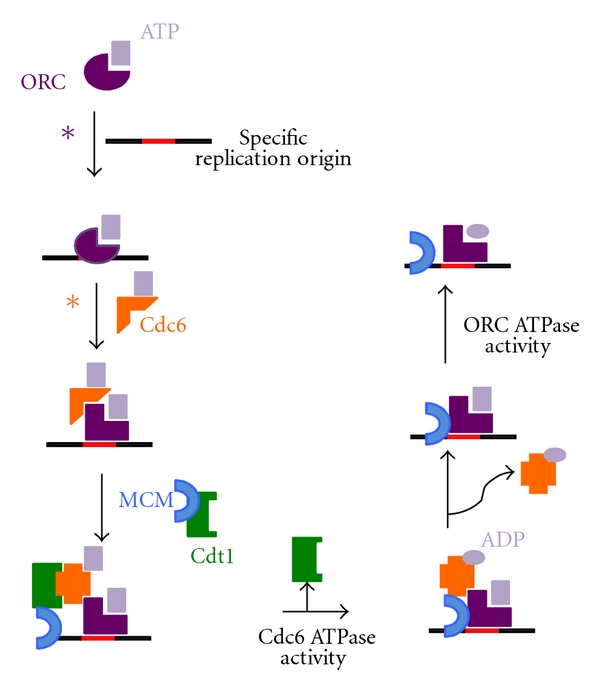
Schematic representation of prereplication machinery recruitment. Figure 3 shows that Cdc6 and ORC ATPase activities are important for the release of these molecules from the complex. Asterisks show the steps where ORC ATPase activity (purple ∗) or Cdc6 ATPase activity (orange ∗) is important for the prevention of the assembly of the prereplication machinery at nonspecific replication origins.
Once bound to DNA, the ORC and Cdc6 are able to interact with Cdt1, which brings the MCM complex to the replication origin. At this moment, the low activity of the Cdc6 ATPase that is important to establish ORC-Cdc6-DNA interaction is also required to recruit the MCM complex onto the replication origins (reviewed in [37]). After MCM complex recruitment, the Orc1 ATPase enables MCM-DNA interaction [38]. Once the MCM complex is recruited and established at the replication origins, the ORC, Cdc6 and Cdt1 do not need to be bound to replication origin sites any longer. At this point, prior to the activation of DNA replication, these proteins disassociate from the replication origins in an ATPase-dependent manner [39].
These data clearly demonstrate the extreme importance of the ATPase activities of the different components of the prereplication machinery in the stabilization and assembly of the prereplication machinery onto DNA replication origins. In the selection of replication origins, the assembly of the prereplication complex onto replication origins, the recruitment of the MCM helicase, and in the dissociation of the prereplication complex from the replication origins to avoid a new round of DNA replication in the same cell cycle, the balance between high and low ATPase activity is critical for precise DNA replication.
5. Trypanosome Orc1/Cdc6: The Prereplication Machinery Component and Its ATPase Activity
Although eukaryotic cells, trypanosomes have a prereplication machinery component that is different from eukaryotes and is instead closer to Archaea species. Genomic databases of trypanosomatids show that these organisms do not contain sequences in their genome that code for Orc1–Orc6 subunits, Cdc6 or Cdt1. Additionally, several Archaea sp. have one or more copies of proteins that exhibit high sequence homology with both Orc1 and Cdc6; these proteins are often called Orc1/Cdc6 proteins [40–42] (Figure 2). Trypanosomatids have annotated a gene for only one of the six subunits of the ORC, Orc1, which is also homologous to Cdc6. It is annotated as Orc1 [43] and we named it Orc1/Cdc6. In the TIGR Parasites Database (http://www.tigr.org/), two sequences were annotated as Orc1 (Tc00.1047053511159.20 and Tc1047053508239.10) in the genome of T. cruzi (representing gene alleles). Single sequences were annotated as Orc1 in the genomes of T. brucei (Tb11.02.5110) and T. brucei gambiense (Tbg972.11.8220). The two Orc1 protein sequences from T. cruzi are 98.2% identical. The Orc1sequences from T. cruzi and T. brucei are 77.1% and 77.8% identical. Trypanosome Orc1/Cdc6 was confirmed as a prereplication machinery component because it replaced yeast Cdc6 in a yeast phenotypic complementation assay; further, the silencing of trypanosome Orc1/Cdc6 expression by RNA interference in T. brucei impaired nuclear DNA replication [24].
Analyses of structural alignment using the Phyre server, a web-based method for protein folding recognition, showed a higher structural similarity of trypanosome Orc1/Cdc6 to archaea Orc1/Cdc6 [24]. Also, analyses of the predicted tridimensional structure of T. cruzi Orc1/Cdc6 by 3D-PSSM (http://www.sbg.bio.ic.ac.uk/~3dpssm/index2.html), which determines the most probable folds based on the occurrence of motifs present in the secondary structure, showed that the probable structural of T. cruzi Orc1/Cdc6 is homologous to Archaea Orc1/Cdc6 (Figure 4). These analyses suggest that trypanosome Orc1/Cdc6 is closer to Archaea prereplication machinery than to mammalian prereplication machinery. Primary sequences of trypanosome Orc1/Cdc6 also contain Walker A and B motifs, related to ATP/GTP binding, and sensor 1 and 2 regions that are involved in ATP hydrolysis; these features are typical of prereplication machinery components that exhibit the AAA+ ATPase fold [44, 45]. In fact, both T. cruzi and T. brucei Orc1/Cdc6 exhibit ATPase activity, which, in the presence of increased concentrations of ATP follows a Michaelis-Menten (MM) kinetic model. This ATPase activity increases in the presence of nonspecific DNA [24], suggesting that, similar to yeast, trypanosome Orc1/Cdc6 ATPase activity might be involved in the selection of specific replication origins. As mentioned previously, there is no Cdt1 homologous protein in the trypanosomatid genome database. Therefore, in these organisms, the prereplication machinery might be assembled by the recruitment of the MCM complex by Orc1/Cdc6. Alternatively, the prereplication machinery could be assembled by an unknown protein that could bind Orc1/Cdc6 and recruit the MCM complex onto the replication origins. This assembly scheme is depicted in Figure 1(b). It is important to note that further studies should confirm if this schematic representation is correct. Recently, the group of Dr. Richard McCulloch identified at least one further ORC-like factor (pers comm.) in T. brucei. Further studies should also be conducted to determine if Orc1/Cdc6 ATPase activity is important for the assembly of prereplication machinery as well as for the release of the prereplication complex proteins from the replication origins. Nevertheless, the silencing of trypanosome Orc1/Cdc6 expression by RNAi in T. brucei negative affected cell survival [24]. This data strongly indicates that Orc1/Cdc6 is extremely important for trypanosome survival and identifies Orc1/Cdc6 as a potential target for drug design.
Figure 4.

(a) Shows the archaea Orc1/Cdc6 structure together with ATP. The AAA+ and WH domains are indicated. (b) Shows the predicted structure of T. cruzi Orc1/Cdc6, obtained using the 3D-PSSM program (http://www.sbg.bio.ic.ac.uk/~3dpssm/index2.html).
6. ATPase Activity Inhibitors
Different proteins harboring ATPase activity are being studied as potential drug targets through the inhibition of their ATPase activity [46–49]. The inhibition of the ATPase activity of heat-shock protein 90 (HSP90) is a main target for cancer treatment. HSP90 is a chaperone that acts in the folding, stabilization, and assembly of several proteins that are involved in many biological processes [50, 51]; further, it is also responsible for the maintenance of cancer cells by facilitating the function of oncoproteins allowing malignant transformation [52]. Because HSP90 from tumor cells has a higher activity compared to HSP90 from normal cells [53], its inhibition effects in tumor cells are higher than in normal cells [53], making HSP90 a good target for drug design [46, 54]. Geldanamycin, a natural compound, was the first compound described with antitumor effects through the inhibition of HSP90 ATPase activity [55–57]. Geldanamycin binds to the N-terminal ATP-binding pocket of HSP90, thereby blocking its ATPase activity [58]. Since these findings have been reported, several studies have focused on HSP90 as a target, and several drugs such as 17-DMGA (alvespimycin) and 17-AAG/tanespimycin have reached phase I and phase II clinical trials, respectively [46].
Another important health problem is antibiotic resistance in the treatment of bacterial infections [59]. Different proteins with ATPase activity are being targeted for high-throughput screening in order to find new inhibitors with potential antibiotic effects. Bacterial RecA [60], DNA gyrase [48], and essential replicative DNA helicase [49] are some of the ATPase proteins upon which effort is being concentrated. RecA is a bacterial protein involved in DNA repair, which interacts with single-stranded DNA and through its ATPase activity allows for both recombinational DNA repair and horizontal gene transfer, processes that are essential for the acquisition of drug resistance genes [47]. Inhibitors of RecA ATPase activity that act by binding the ATP-binding site are being screened, indicating the potential development of antibiotics capable of preventing bacterial drug resistance [60].
DNA gyrase and the replicative DNA helicase are proteins involved in bacterial DNA replication and are essential for bacterial survival. Both have ATPase activity indispensable for their function [48]. DNA gyrase is a type II topoisomerase capable of introducing negative supercoils during DNA replication. Although it is comprised of two subunits (A and B), only B subunit has ATPase activity. The aminocoumarin antibiotic class, from which novobiocin (4-Hydroxy-3-[4-hydroxy-3-(3-methylbut-2-enyl)benzamido]-8-methylcoumarin-7-yl3-O-carbamoyl-5,5-di-C-methyl-α-l-lyxofuranoside) is licensed for treatment of human infections, binds to the B subunit inhibiting the ATP hydrolysis required for DNA supercoiling and preventing cell growth [48]. The DNA replicative helicase, DNA B, is responsible for opening double-stranded DNA through ATP hydrolysis, which makes the DNA available to the replication machinery for the duplication of the bacterial genome. Two compounds, flavonoid myricetin and triaminotriazine, are able to inhibit the ATPase activity of DNA B helicase in Escherichia coli and Pseudomonas aeruginosa, respectively, preventing cell growth [49]. However, as these drugs exhibit some cytotoxicity in mammalian cell culture, others inhibitors are being screened as possible drug candidates [49].
As discussed above, inhibitors of ATPase activity are being studied as possible drugs candidates; some, such as novobiocin, are even already being used for human treatment. This raises the possibility of searching for specific inhibitors of T. brucei and T. cruzi Orc1/Cdc6, as these proteins do not have a closely related protein in humans.
7. Final Considerations
In addition to its indispensable role as a DNA replication initiator, the ORC is known to affect diverse cell processes including chromosome segregation, cytokinesis, cell cycle regulation, and gene expression [61]. It is now time to analyze whether trypanosome Orc1/Cdc6, and its ATPase activity are also involved in non-DNA replication functions. If so, it would better justify additional efforts to search for an ATPase inhibitor against trypanosome Orc1/Cdc6.
References
- 1.Pepin J. Combination therapy for sleeping sickness: a wake-up call. Journal of Infectious Diseases. 2007;195(3):311–313. doi: 10.1086/510540. [DOI] [PubMed] [Google Scholar]
- 2.Russomando G, De Tomassone MMC, De Guillen I, et al. Treatment of congenital chagas’ disease diagnosed and followed up by the polymerase chain reaction. American Journal of Tropical Medicine and Hygiene. 1998;59(3):487–491. doi: 10.4269/ajtmh.1998.59.487. [DOI] [PubMed] [Google Scholar]
- 3.Vago AR, Andrade LO, Leite AA, et al. Genetic characterization of Trypanosoma cruzi directly from tissues of patients with chronic chagas disease: differential distribution of genetic types into diverse organs. American Journal of Pathology. 2000;156(5):1805–1809. doi: 10.1016/s0002-9440(10)65052-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Magnus E, Vervoort T, Van Meirvenne N. A card-agglutination test with stained trypanosomes (C.A.T.T.) for the serological diagnosis of T. b. gambiense trypanosomiasis. Annales de la Societe Belge de Medecine Tropicale. 1978;58(3):169–176. [PubMed] [Google Scholar]
- 5.Umezawa ES, Bastos SF, Camargo ME, et al. Evaluation of recombinant antigens for serodiagnosis of Chagas’ disease in South and Central America. Journal of Clinical Microbiology. 1999;37(5):1554–1560. doi: 10.1128/jcm.37.5.1554-1560.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pastini AC, Iglesias SR, Carricarte VC, Guerin ME, Sanchez DO, Frasch AC. Immunoassay with recombinant Trypanosoma cruzi antigens potentially useful for screening donated blood and diagnosing Chagas disease. Clinical Chemistry. 1994;40(10):1893–1897. [PubMed] [Google Scholar]
- 7.Leguizamón MS, Russomando G, Luquetti A, et al. Long-lasting antibodies detected by a trans-sialidase inhibition assay of sera from parasite-free, serologically cured chagasic patients. Journal of Infectious Diseases. 1997;175(5):1272–1275. doi: 10.1086/593697. [DOI] [PubMed] [Google Scholar]
- 8.Duggan AJ, Hutchinson MP. Sleeping sickness in Europeans: a review of 109 cases. Journal of Tropical Medicine and Hygiene. 1966;69(6):124–131. [PubMed] [Google Scholar]
- 9.Checchi F, Filipe JAN, Haydon DT, Chandramohan D, Chappuis F. Estimates of the duration of the early and late stage of gambiense sleeping sickness. BMC Infectious Diseases. 2008;8, article 16 doi: 10.1186/1471-2334-8-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Odiit M, Kansiime F, Enyaru JCK. Duration of symptoms and case fatality of sleeping sickness caused by Trypanosoma brucei rhodesiense in Tororo, Uganda. East African Medical Journal. 1997;74(12):792–795. [PubMed] [Google Scholar]
- 11.Fèvre EM, Coleman PG, Welburn SC, Maudlin I. Reanalyzing the 1900–1920 sleeping sickness epidemic in Uganda. Emerging Infectious Diseases. 2004;10(4):567–573. doi: 10.3201/eid1004.020626. [DOI] [PubMed] [Google Scholar]
- 12.Dias JCP. The treatment of Chagas disease (South American trypanosomiasis) Annals of Internal Medicine. 2006;144(10):772–774. doi: 10.7326/0003-4819-144-10-200605160-00012. [DOI] [PubMed] [Google Scholar]
- 13.Docampo R, Moreno SNJ. Free radical metabolism of antiparasitic agents. Federation Proceedings. 1986;45(10):2471–2476. [PubMed] [Google Scholar]
- 14.Enanga B, Ariyanayagam MR, Stewart ML, Barrett MP. Activity of megazol, a trypanocidal nitroimidazole, is associated with DNA damage. Antimicrobial Agents and Chemotherapy. 2003;47(10):3368–3370. doi: 10.1128/AAC.47.10.3368-3370.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Apt W. Current and developing therapeutic agents in the treatment of Chagas disease. Drug design, development and therapy. 2010;4:243–253. doi: 10.2147/dddt.s8338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barrett MP, Boykin DW, Brun R, Tidwell RR. Human African trypanosomiasis: pharmacological re-engagement with a neglected disease. British Journal of Pharmacology. 2007;152(8):1155–1171. doi: 10.1038/sj.bjp.0707354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berger BJ, Carter NS, Fairlamb AH. Polyamine and pentamidine metabolism in African trypanosomes. Acta Tropica. 1993;54(3-4):215–224. doi: 10.1016/0001-706x(93)90094-r. [DOI] [PubMed] [Google Scholar]
- 18.Hanau S, Rippa M, Bertelli M, Dallocchio F, Barrett MP. 6-Phosphogluconate dehydrogenase from Trypanosoma brucei. Kinetic analysis and inhibition by trypanocidal drugs. European Journal of Biochemistry. 1996;240(3):592–599. doi: 10.1111/j.1432-1033.1996.0592h.x. [DOI] [PubMed] [Google Scholar]
- 19.Burri C, Brun R. Eflornithine for the treatment of human African trypanosomiasis. Parasitology Research. 2003;90(1):S49–S52. doi: 10.1007/s00436-002-0766-5. [DOI] [PubMed] [Google Scholar]
- 20.Meshnick SR, Grady RW, Blobstein SH, Cerami A. Porphyrin-induced lysis of Trypanosoma brucei: a role for zinc. Journal of Pharmacology and Experimental Therapeutics. 1978;207(3):1041–1050. [PubMed] [Google Scholar]
- 21.Romanha AJ, de Castro SL, Soeiro MNC, et al. In vitro and in vivo experimental models for drug screening and development for Chagas disease. Memorias do Instituto Oswaldo Cruz. 2010;105(2):233–238. doi: 10.1590/s0074-02762010000200022. [DOI] [PubMed] [Google Scholar]
- 22.Liendo A, Lazardi K, Urbina JA. In-vitro antiproliferative effects and mechanism of action of the bis-triazole D0870 and its £(-) enantiomer against Trypanosoma cruzi. Journal of Antimicrobial Chemotherapy. 1998;41(2):197–205. doi: 10.1093/jac/41.2.197. [DOI] [PubMed] [Google Scholar]
- 23.Ansede JH, Anbazhagan M, Brun R, Easterbrook JD, Hall JE, Boykin DW. O-alkoxyamidine prodrugs of furamidine: in vitro transport and microsomal metabolism as indicators of in vivo efficacy in a mouse model of Trypanosoma brucei rhodesiense infection. Journal of Medicinal Chemistry. 2004;47(17):4335–4338. doi: 10.1021/jm030604o. [DOI] [PubMed] [Google Scholar]
- 24.De Melo Godoy PD, Nogueira-Junior LA, Paes LS, et al. Trypanosome prereplication machinery contains a single functional Orc1/Cdc6 protein, which is typical of archaea. Eukaryotic Cell. 2009;8(10):1592–1603. doi: 10.1128/EC.00161-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bell SP, Dutta A. DNA replication in eukaryotic cells. Annual Review of Biochemistry. 2002;71:333–374. doi: 10.1146/annurev.biochem.71.110601.135425. [DOI] [PubMed] [Google Scholar]
- 26.Stillman B. Origin recognition and the chromosome cycle. FEBS Letters. 2005;579(4):877–884. doi: 10.1016/j.febslet.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 27.Diffley JFX. DNA replication: building the perfect switch. Current Biology. 2001;11(9):R367–R370. doi: 10.1016/s0960-9822(01)00196-8. [DOI] [PubMed] [Google Scholar]
- 28.Labib K, Diffley JFX. Is the MCM2–7 complex the eukaryotic DNA replication fork helicase? Current Opinion in Genetics and Development. 2001;11(1):64–70. doi: 10.1016/s0959-437x(00)00158-1. [DOI] [PubMed] [Google Scholar]
- 29.Bell SP, Stillman B. ATP-dependent recognition of eukaryotic origins of DNA replication by a multiprotein complex. Nature. 1992;357(6374):128–134. doi: 10.1038/357128a0. [DOI] [PubMed] [Google Scholar]
- 30.Duncker BP, Chesnokov IN, McConkey BJ. The origin recognition complex protein family. Genome biology. 2009;10(3):p. 214. doi: 10.1186/gb-2009-10-3-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Neuwald AF, Aravind L, Spouge JL, Koonin EV. AAA: a class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Research. 1999;9(1):27–43. [PubMed] [Google Scholar]
- 32.Erzberger JP, Berger JM. Evolutionary relationships and structural mechanisms of AAA+ proteins. Annual Review of Biophysics and Biomolecular Structure. 2006;35:93–114. doi: 10.1146/annurev.biophys.35.040405.101933. [DOI] [PubMed] [Google Scholar]
- 33.Hartwell LH. Sequential function of gene products relative to DNA synthesis in the yeast cell cycle. Journal of Molecular Biology. 1976;104(4):803–817. doi: 10.1016/0022-2836(76)90183-2. [DOI] [PubMed] [Google Scholar]
- 34.Speck C, Stillman B. Cdc6 ATPase activity regulates ORC·Cdc6 stability and the selection of specific DNA sequences as origins of DNA replication. Journal of Biological Chemistry. 2007;282(16):11705–11714. doi: 10.1074/jbc.M700399200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lei M, Tye BK. Initiating DNA synthesis: from recruiting to activating the MCM complex. Journal of Cell Science. 2001;114(8):1447–1454. doi: 10.1242/jcs.114.8.1447. [DOI] [PubMed] [Google Scholar]
- 36.Chen S, De Vries MA, Bell SP. Orc6 is required for dynamic recruitment of Cdt1 during repeated Mcm2–7 loading. Genes and Development. 2007;21(22):2897–2907. doi: 10.1101/gad.1596807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Evrin C, Clarke P, Zech J, et al. A double-hexameric MCM2-7 complex is loaded onto origin DNA during licensing of eukaryotic DNA replication. Proceedings of the National Academy of Sciences of the United States of America. 2010;106(48):20240–20245. doi: 10.1073/pnas.0911500106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Randell JCW, Bowers JL, Rodríguez HK, Bell SP. Sequential ATP hydrolysis by Cdc6 and ORC directs loading of the Mcm2–7 helicase. Molecular Cell. 2006;21(1):29–39. doi: 10.1016/j.molcel.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 39.Tsakraklides V, Bell SP. Dynamics of pre-replicative complex assembly. Journal of Biological Chemistry. 2010;22:877–884. doi: 10.1074/jbc.M109.072504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.De Felice M, Esposito L, Pucci B, et al. Modular organization of a Cdc6-like protein from the crenarchaeon Sulfolobus solfataricus. Biochemical Journal. 2004;381(3):645–653. doi: 10.1042/BJ20040280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Felice M, Esposito L, Pucci B, De Falco M, Rossi M, Pisani FM. A CDC6-like factor from the archaea Sulfolobus solfataricus promotes binding of the mini-chromosome maintenance complex to DNA. Journal of Biological Chemistry. 2004;279(41):43008–43012. doi: 10.1074/jbc.M406693200. [DOI] [PubMed] [Google Scholar]
- 42.De Felice M, Esposito L, Rossi M, Pisani FM. Biochemical characterization of two Cdc6/ORC1-like proteins from the crenarchaeon Sulfolobus solfataricus. Extremophiles. 2006;10(1):61–70. doi: 10.1007/s00792-005-0473-0. [DOI] [PubMed] [Google Scholar]
- 43.El-Sayed NM, Myler PJ, Bartholomeu DC, et al. The genome sequence of Trypanosoma cruzi, etiologic agent of chagas disease. Science. 2005;309(5733):409–415. doi: 10.1126/science.1112631. [DOI] [PubMed] [Google Scholar]
- 44.Walker JE, Saraste M, Runswick MJ, Gay NJ. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. The EMBO Journal. 1982;1(8):945–951. doi: 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guenther B, Onrust R, Sali A, O’Donnell M, Kuriyan J. Crystal structure of the δ’ subunit of the clamp-loader complex of E. coli DNA polymerase III. Cell. 1997;91(3):335–345. doi: 10.1016/s0092-8674(00)80417-1. [DOI] [PubMed] [Google Scholar]
- 46.Janin YL. ATPase inhibitors of heat-shock protein 90, second season. Drug Discovery Today. 2010;15(9-10):342–353. doi: 10.1016/j.drudis.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 47.Roca AI, Cox MM. RecA protein: structure, function, and role in recombinational DNA repair. Progress in Nucleic Acid Research and Molecular Biology. 1997;56:129–223. doi: 10.1016/s0079-6603(08)61005-3. [DOI] [PubMed] [Google Scholar]
- 48.Ostrov DA, Hernández Prada JA, Corsino PE, Finton KA, Le N, Rowe TC. Discovery of novel DNA gyrase inhibitors by high-throughput virtual screening. Antimicrobial Agents and Chemotherapy. 2007;51(10):3688–3698. doi: 10.1128/AAC.00392-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aiello D, Barnes MH, Biswas EE, et al. Discovery, characterization and comparison of inhibitors of Bacillus anthracis and Staphylococcus aureus replicative DNA helicases. Bioorganic and Medicinal Chemistry. 2009;17(13):4466–4476. doi: 10.1016/j.bmc.2009.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Csermely P, Schnaider T, Soti C, Prohászka Z, Nardai G. The 90-kDa molecular chaperone family: structure, function, and clinical applications. A comprehensive review. Pharmacology and Therapeutics. 1998;79(2):129–168. doi: 10.1016/s0163-7258(98)00013-8. [DOI] [PubMed] [Google Scholar]
- 51.Pratt WB, Morishima Y, Osawa Y. The Hsp90 chaperone machinery regulates signaling by modulating ligand binding clefts. Journal of Biological Chemistry. 2008;283(34):22885–22889. doi: 10.1074/jbc.R800023200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nature Reviews Cancer. 2005;5(10):761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 53.Kamal A, Thao L, Sensintaffar J, et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425(6956):407–410. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- 54.Hahn JIS. The Hsp90 chaperone machinery: from structure to drug development. BMB Reports. 2009;42(10):623–630. doi: 10.5483/bmbrep.2009.42.10.623. [DOI] [PubMed] [Google Scholar]
- 55.Whitesell L, Mimnaugh EG, De Costa B, Myers CE, Neckers LM. Inhibition of heat shock protein HSP90-pp60(v-src) heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(18):8324–8328. doi: 10.1073/pnas.91.18.8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schnur RC, Corman ML. Preparation of 17-amino-22-(4’-azido-3’125-iodophenacyl)-17- demethoxygeldanamycin (1): an ansamycin for photoaffinity labeling. Journal of Labelled Compounds and Radiopharmaceuticals. 1994;34(6):529–535. [Google Scholar]
- 57.Miller P, Schnur RC, Barbacci E, Moyer MP, Moyer JD. Binding of benzoquinoid ansamycins to p100 correlates with their ability to deplete the erbB2 gene product p185. Biochemical and Biophysical Research Communications. 1994;201(3):1313–1319. doi: 10.1006/bbrc.1994.1847. [DOI] [PubMed] [Google Scholar]
- 58.Roe SM, Prodromou C, O’Brien R, Ladbury JE, Piper PW, Pearl LH. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. Journal of Medicinal Chemistry. 1999;42(2):260–266. doi: 10.1021/jm980403y. [DOI] [PubMed] [Google Scholar]
- 59.Boucher HW, Talbot GH, Bradley JS, et al. Bad bugs, no drugs: no ESKAPE! an update from the Infectious Diseases Society of America. Clinical Infectious Diseases. 2009;48(1):1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 60.Sexton JZ, Wigle TJ, He Q, et al. Novel inhibitors of E. coli RecA ATPase activity. Current Chemical Genomics. 2010;4(1):34–42. doi: 10.2174/1875397301004010034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Scholefield G, Veening J-W, Murray H. DnaA and ORC: more than DNA replication initiators. Trends in Cell Biology. 2011;21(3):188–194. doi: 10.1016/j.tcb.2010.10.006. [DOI] [PubMed] [Google Scholar]